Neonatal Screening for MPS Disorders in Latin America: A Survey of Pilot Initiatives

 ,
, 
 ,
,  , ,
, ,  and
and 
Abstract
1. Introduction
2. Methods and Results
2.1. Pilot Study for Six LSDs (Including MPS I) in Mexico
2.2. Targeted Newborn Screening for MPS VI in Northeast Brazil
2.3. Pilot Program for Four LSDs (Including MPS I) in a Private Laboratory in Brazil
2.4. Pilot Program for Five LSDs (Including MPS I and MPS VI) in a Research Laboratory in Brazil
2.5. Pilot Newborn Screening Program for Six LSDs in a Research Laboratory in Brazil
3. Discussion
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Howson, C.; Cedergren, B.; Giugliani, R.; Huhtinen, P.; Padilla, C.D.; Palubiak, C.; Santos, M.; Schwartz, I.; Therrell, B.L.; Umemoto, A.; et al. Universal newborn screening: A roadmap for action. Mol. Genet. Metab. 2018, 124, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Guthrie, R.; Susi, A. A simple phenylalanine method for detecting phenylketonuria in large populations of newborn infants. Pediatrics 1963, 32, 338–343. [Google Scholar] [PubMed]
- González-Irazabal, Y.; Hernandez de Abajo, G.; Martínez-Morillo, E. Identifying and overcoming barriers to harmonize newborn screening programs through consensus strategies. Crit. Rev. Clin. Lab. Sci. 2020, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Ten Great Public Health Achievements—United States, 2001–2010. Available online: https://www.cdc.gov/mmwr/preview/mmwrhtml/mm6019a5.htm (accessed on 18 August 2020).
- Neufeld, E.; Muenzer, J. ; OMMBID; McGraw-Hill Medical. The Mucopolysaccharidoses. The Online Metabolic and Molecular Bases of Inherited Disease, 8th ed.; McGraw-Hill: New York, NY, USA, 2001. [Google Scholar]
- Muenzer, J. The mucopolysaccharidoses: A heterogeneous group of disorders with variable pediatric presentations. J. Pediatr. 2004, 144, S27–S34. [Google Scholar] [CrossRef]
- Vieira, T.; Schwartz, I.; Muñoz, V.; Pinto, L.; Steiner, C.; Ribeiro, M.; Boy, R.; Ferraz, V.; de Paula, A.; Kim, C.; et al. Mucopolysaccharidoses in Brazil: What happens from birth to biochemical diagnosis? Am. J. Med. Genet. A 2008, 146A, 1741–1747. [Google Scholar] [CrossRef]
- McGill, J.J.; Inwood, A.C.; Coman, D.J.; Lipke, M.L.; de Lore, D.; Swiedler, S.J.; Hopwood, J.J. Enzyme replacement therapy for mucopolysaccharidosis VI from 8 weeks of age—a sibling control study. Clin. Genet. 2010, 77, 492–498. [Google Scholar] [CrossRef]
- Gabrielli, O.; Clarke, L.A.; Ficcadenti, A.; Santoro, L.; Zampini, L.; Volpi, N.; Coppa, G. V 12 year follow up of enzyme-replacement therapy in two siblings with attenuated mucopolysaccharidosis I: The important role of early treatment. BMC Med. Genet. 2016, 17, 19. [Google Scholar] [CrossRef]
- Kemper, A.R.; Brosco, J.; Comeau, A.M.; Green, N.S.; Prosser, L.A.; Ojodu, J.; Tanksley, S.; Jones, E.; Lam, K.K. Newborn Screening for Mucopolysaccharidosis Type 1 (MPS I): A Systematic Review of Evidence Report of Final Findings Final Version 1.1; The Condition Review Workgroup; Association of Public Health Laboratories; Duke University: Durham, NC, USA, 2015. [Google Scholar]
- Kellar-Guenther, Y.; McKasson, S.; Hale, K.; Singh, S.; Sontag, M.K.; Ojodu, J. Implementing Statewide Newborn Screening for New Disorders: U.S. Program Experiences. Int. J. Neonatal Screen. 2020, 6, 35. [Google Scholar] [CrossRef]
- Hopkins, P.V.; Campbell, C.; Klug, T.; Rogers, S.; Raburn-Miller, J.; Kiesling, J. Lysosomal storage disorder screening implementation: Findings from the first six months of full population pilot testing in Missouri. J. Pediatr. 2015, 166, 172–177. [Google Scholar] [CrossRef]
- Burton, B.K.; Hoganson, G.E.; Fleischer, J.; Grange, D.K.; Braddock, S.R.; Hickey, R.; Hitchins, L.; Groepper, D.; Christensen, K.M.; Kirby, A.; et al. Population-Based Newborn Screening for Mucopolysaccharidosis Type II in Illinois: The First Year Experience. J. Pediatr. 2019. [Google Scholar] [CrossRef]
- Burton, B.K.; Hickey, R.; Hitchins, L. Newborn Screening for Mucopolysaccharidosis Type II in Illinois: An Update. Int. J. Neonatal Screen. 2020, 6, 73. [Google Scholar] [CrossRef] [PubMed]
- Ames, E.G.; Fisher, R.; Kleyn, M.; Ahmad, A. Current Practices for U.S. Newborn Screening of Pompe Disease and MPSI. Int. J. Neonatal Screen. 2020, 6, 72. [Google Scholar] [CrossRef]
- Lin, S.-P.; Lin, H.-Y.; Wang, T.-J.; Chang, C.-Y.; Lin, C.-H.; Huang, S.-F.; Tsai, C.-C.; Liu, H.-L.; Keutzer, J.; Chuang, C.-K. A pilot newborn screening program for Mucopolysaccharidosis type I in Taiwan. Orphanet J. Rare Dis. 2013, 8, 147. [Google Scholar] [CrossRef] [PubMed]
- Chuang, C.-K.; Lin, H.-Y.; Wang, T.-J.; Huang, Y.-H.; Chan, M.-J.; Liao, H.-C.; Lo, Y.-T.; Wang, L.-Y.; Tu, R.-Y.; Fang, Y.-Y.; et al. Status of newborn screening and follow up investigations for Mucopolysaccharidoses I and II in Taiwan. Orphanet J. Rare Dis. 2018, 13, 84. [Google Scholar] [CrossRef] [PubMed]
- Chan, M.-J.; Liao, H.-C.; Gelb, M.H.; Chuang, C.-K.; Liu, M.-Y.; Chen, H.-J.; Kao, S.-M.; Lin, H.-Y.; Huang, Y.-H.; Kumar, A.B.; et al. Taiwan National Newborn Screening Program by Tandem Mass Spectrometry for Mucopolysaccharidoses Types I, II, and VI. J. Pediatr. 2019, 205, 176–182. [Google Scholar] [CrossRef]
- Chuang, C.-K.; Lin, H.-Y.; Wang, T.-J.; Huang, S.-F.; Lin, S.-P. Bio-Plex immunoassay measuring the quantity of lysosomal N-acetylgalactosamine-6-sulfatase protein in dried blood spots for the screening of mucopolysaccharidosis IVA in newborn: A pilot study. BMJ Open 2017, 7, e014410. [Google Scholar] [CrossRef]
- Donati, M.A.; Pasquini, E.; Spada, M.; Polo, G.; Burlina, A. Newborn screening in mucopolysaccharidoses. Ital. J. Pediatr. 2018, 44, 126. [Google Scholar] [CrossRef]
- Burlina, A.B.; Polo, G.; Salviati, L.; Duro, G.; Zizzo, C.; Dardis, A.; Bembi, B.; Cazzorla, C.; Rubert, L.; Zordan, R.; et al. Newborn screening for lysosomal storage disorders by tandem mass spectrometry in North East Italy. J. Inherit. Metab. Dis. 2018, 41, 209–219. [Google Scholar] [CrossRef]
- Burlina, A.B.; Polo, G.; Rubert, L.; Gueraldi, D.; Cazzorla, C.; Duro, G.; Salviati, L.; Burlina, A.P. Implementation of Second-Tier Tests in Newborn Screening for Lysosomal Disorders in North Eastern Italy. Int. J. Neonatal Screen. 2019, 5, 24. [Google Scholar] [CrossRef]
- Tanaka, A.; Sawada, T.; Suzuki, K.; Sakuraba, H.; Saito, S.; Sakabuchi, T.; Kitagawa, T. Newborn screening of mucopolysaccharidosis I and II and characterization of pseudodeficiency alleles of iduronate 2-sulfatase gene found in the screening. In Proceedings of the 12th International Symposium on MPS and Related Diseases, Noordwijkerhout, The Netherlands, 28 June–2 July 2012; pp. 92–93. [Google Scholar]
- Ruijter, G.J.G.; Goudriaan, D.A.; Boer, A.M.; Van den Bosch, J.; Van der Ploeg, A.T.; Elvers, L.H.; Weinreich, S.S.; Reuser, A.J. Newborn screening for hunter disease: A small-scale feasibility study. JIMD Rep. 2014, 14, 23–27. [Google Scholar] [CrossRef]
- The UK NSC Recommendation on Mucopolysaccharidosis Type I. Available online: https://legacyscreening.phe.org.uk/mps1 (accessed on 1 October 2020).
- Therrell, B.L.; Padilla, C.D. Newborn screening in the developing countries. Curr. Opin. Pediatr. 2018, 30, 734–739. [Google Scholar] [CrossRef] [PubMed]
- Borrajo, G.J.C. Newborn screening in Latin America at the beginning of the 21st century. J. Inherit. Metab. Dis. 2007, 30, 466–481. [Google Scholar] [CrossRef] [PubMed]
- Therrell, B.L.; Padilla, C.D.; Loeber, J.G.; Kneisser, I.; Saadallah, A.; Borrajo, G.J.C.; Adams, J. Current status of newborn screening worldwide: 2015. Semin. Perinatol. 2015, 39, 171–187. [Google Scholar] [CrossRef] [PubMed]
- Navarrete-Martínez, J.I.; Limón-Rojas, A.E.; Gaytán-García, M.D.J.; Reyna-Figueroa, J.; Wakida-Kusunoki, G.; Delgado-Calvillo, M.D.R.; Cantú-Reyna, C.; Cruz-Camino, H.; Cervantes-Barragán, D.E. Newborn screening for six lysosomal storage disorders in a cohort of Mexican patients: Three-year findings from a screening program in a closed Mexican health system. Mol. Genet. Metab. 2017, 121, 16–21. [Google Scholar] [CrossRef]
- Costa-Motta, F.M.; Acosta, A.X.; Abé-Sandes, K.; Bender, F.; Schwartz, I.V.D.; Giugliani, R.; Leistner-Segal, S. Genetic studies in a cluster of mucopolysaccharidosis type VI patients in Northeast Brazil. Mol. Genet. Metab. 2011, 104, 603–607. [Google Scholar] [CrossRef]
- Civallero, G.; Michelin, K.; de Mari, J.; Viapiana, M.; Burin, M.; Coelho, J.C.; Giugliani, R. Twelve different enzyme assays on dried-blood filter paper samples for detection of patients with selected inherited lysosomal storage diseases. Clin. Chim. Acta 2006, 372, 98–102. [Google Scholar] [CrossRef]
- Giugliani, R.; Bender, F.; Couto, R.; Bochernitsan, A.; Brusius-Facchin, A.C.; Burin, M.; Amorim, T.; Acosta, A.X.; Purificação, A.; Leistner-Segal, S.; et al. Population medical genetics: Translating science to the community. Genet. Mol. Biol. 2019, 42, 312–320. [Google Scholar] [CrossRef]
- Camargo Neto, E.; Schulte, J.; Pereira, J.; Bravo, H.; Sampaio-Filho, C.; Giugliani, R. Neonatal screening for four lysosomal storage diseases with a digital microfluidics platform: Initial results in Brazil. Genet. Mol. Biol. 2018, 41, 414–416. [Google Scholar] [CrossRef]
- Bravo, H.; Neto, E.C.; Schulte, J.; Pereira, J.; Filho, C.S.; Bittencourt, F.; Sebastião, F.; Bender, F.; de Magalhães, A.P.S.; Guidobono, R.; et al. Investigation of newborns with abnormal results in a newborn screening program for four lysosomal storage diseases in Brazil. Mol. Genet. Metab. Rep. 2017, 12, 92–97. [Google Scholar] [CrossRef]
- Bender, F.; Burin, M.G.; Tirelli, K.M.; Medeiros, F.; Bitencourt, F.H.; Civallero, G.; Kubaski, F.; Bravo, H.; Daher, A.; Carnier, V.; et al. Newborn screening for lysosomal disorders in Brazil: A pilot study using customized fluorimetric assays. Genet. Mol. Biol. 2020, 43, e20180334. [Google Scholar] [CrossRef]
- Brusius-Facchin, A.C.; Siebert, M.; Leão, D.; Malaga, D.R.; Pasqualim, G.; Trapp, F.; Matte, U.; Giugliani, R.; Leistner-Segal, S. Phenotype-oriented NGS panels for mucopolysaccharidoses: Validation and potential use in the diagnostic flowchart. Genet. Mol. Biol. 2019, 42, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef] [PubMed]
- Desmet, F.-O.; Hamroun, D.; Lalande, M.; Collod-Béroud, G.; Claustres, M.; Béroud, C. Human Splicing Finder: An online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009, 37, e67. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef]
- Venturi, N.; Rovelli, A.; Parini, R.; Menni, F.; Brambillasca, F.; Bertagnolio, F.; Uziel, G.; Gatti, R.; Filocamo, M.; Donati, M.A.; et al. Molecular analysis of 30 mucopolysaccharidosis type I patients: Evaluation of the mutational spectrum in Italian population and identification of 13 novel mutations. Hum. Mutat. 2002, 20, 231. [Google Scholar] [CrossRef]
- Polo, G.; Gueraldi, D.; Giuliani, A.; Rubert, L.; Cazzorla, C.; Salviati, L.; Marzollo, A.; Biffi, A.; Burlina, A.P.; Burlina, A.B. The combined use of enzyme activity and metabolite assays as a strategy for newborn screening of mucopolysaccharidosis type I. Clin. Chem. Lab. Med. 2020. [Google Scholar] [CrossRef]
- Tarini, B.A.; Goldenberg, A.J. Ethical issues with newborn screening in the genomics era. Annu. Rev. Genomics Hum. Genet. 2012, 13, 381–393. [Google Scholar] [CrossRef]
- Gelb, M.H.; Lukacs, Z.; Ranieri, E.; Schielen, P.C. Newborn Screening for Lysosomal Storage Disorders: Methodologies for Measurement of Enzymatic Activities in Dried Blood Spots. Int. J. Neonatal Screen. 2018, 5, 1. [Google Scholar] [CrossRef]
- Kubaski, F.; Mason, R.W.; Nakatomi, A.; Shintaku, H.; Xie, L.; van Vlies, N.N.; Church, H.; Giugliani, R.; Kobayashi, H.; Yamaguchi, S.; et al. Newborn screening for mucopolysaccharidoses: A pilot study of measurement of glycosaminoglycans by tandem mass spectrometry. J. Inherit. Metab. Dis. 2017, 40, 151–158. [Google Scholar] [CrossRef]
- Herbst, Z.M.; Urdaneta, L.; Klein, T.; Fuller, M.; Gelb, M.H. Evaluation of Multiple Methods for Quantification of Glycosaminoglycan Biomarkers in Newborn Dried Blood Spots from Patients with Severe and Attenuated Mucopolysaccharidosis-I. Int. J. Neonatal Screen. 2020, 6, 69. [Google Scholar] [CrossRef] [PubMed]
- Peck, D.S.; Lacey, J.M.; White, A.L.; Pino, G.; Studinski, A.L.; Fisher, R.; Ahmad, A.; Spencer, L.; Viall, S.; Shallow, N.; et al. Incorporation of Second-Tier Biomarker Testing Improves the Specificity of Newborn Screening for Mucopolysaccharidosis Type I. Int. J. Neonatal Screen. 2020, 6, 10. [Google Scholar] [CrossRef] [PubMed]
- Lajic, S.; Karlsson, L.; Zetterström, R.H.; Falhammar, H.; Nordenström, A. The Success of a Screening Program Is Largely Dependent on Close Collaboration between the Laboratory and the Clinical Follow-Up of the Patients. Int. J. Neonatal Screen. 2020, 6, 68. [Google Scholar] [CrossRef] [PubMed]
- Hall, P.L.; Sanchez, R.; Hagar, A.F.; Jerris, S.C.; Wittenauer, A.; Wilcox, W.R. Two-Tiered Newborn Screening with Post-Analytical Tools for Pompe Disease and Mucopolysaccharidosis Type I Results in Performance Improvement and Future Direction. Int. J. Neonatal Screen. 2020, 6. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Locus | Reference | Genotype | Coverage | cDNA | Protein | dbSNP |
|---|---|---|---|---|---|---|
| Chr4:981673 | G | G/A | 155 | c.235G > A | p.Ala79Thr | rs58037052 |
| Chr4:981734 | C | C/T | 158 | c.296C > T | p.Thr99Ile | rs147490060 |
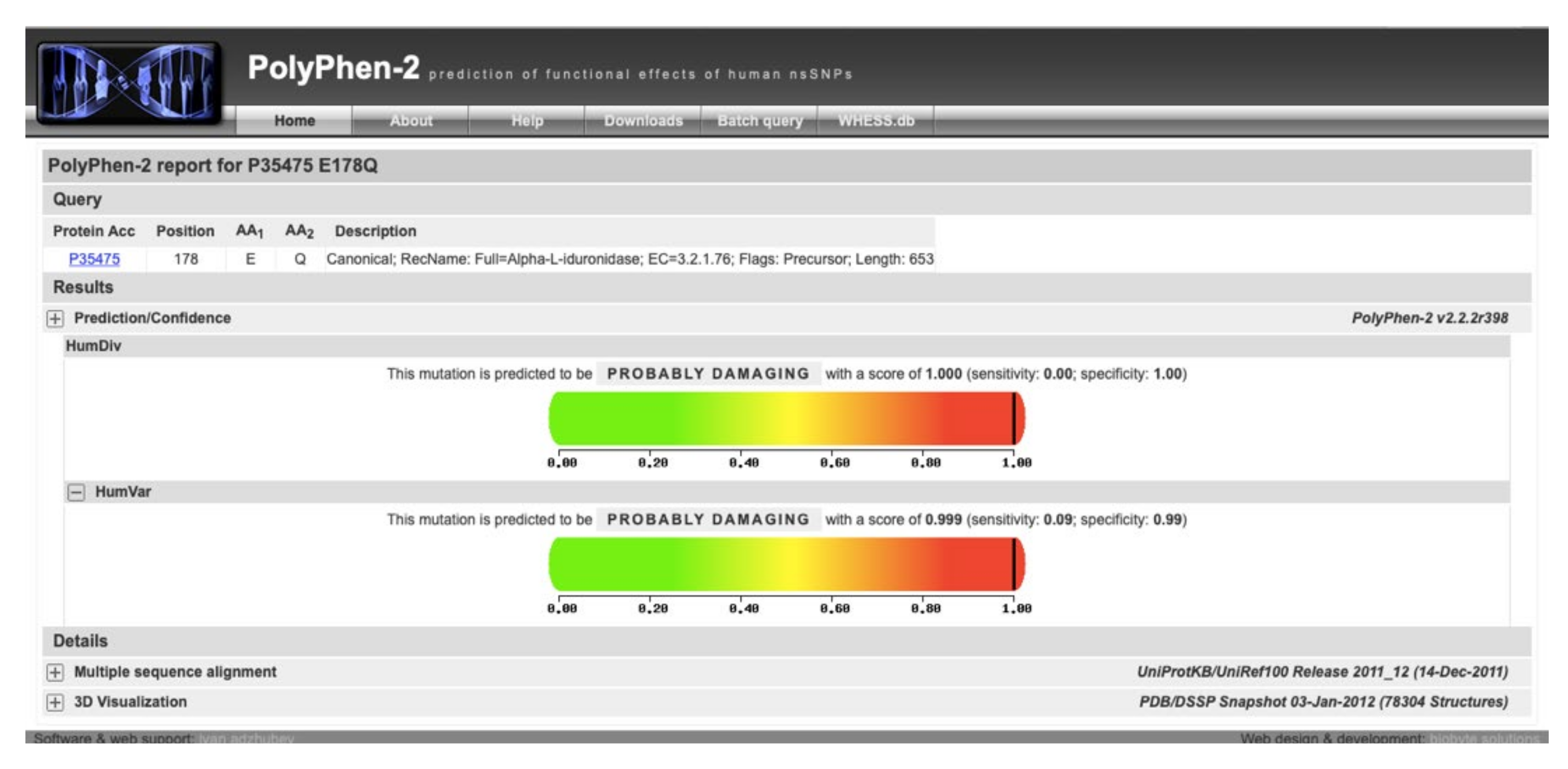
| Chr4:995294 | G | G/C | 343 | c.532G > C | p.Glu178Gln | rs992336192 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kubaski, F.; Sousa, I.; Amorim, T.; Pereira, D.; Trometer, J.; Souza, A.; Ranieri, E.; Polo, G.; Burlina, A.; Brusius-Facchin, A.C.; et al. Neonatal Screening for MPS Disorders in Latin America: A Survey of Pilot Initiatives. Int. J. Neonatal Screen. 2020, 6, 90. https://doi.org/10.3390/ijns6040090
Kubaski F, Sousa I, Amorim T, Pereira D, Trometer J, Souza A, Ranieri E, Polo G, Burlina A, Brusius-Facchin AC, et al. Neonatal Screening for MPS Disorders in Latin America: A Survey of Pilot Initiatives. International Journal of Neonatal Screening. 2020; 6(4):90. https://doi.org/10.3390/ijns6040090
Chicago/Turabian StyleKubaski, Francyne, Inês Sousa, Tatiana Amorim, Danilo Pereira, Joe Trometer, Alexandre Souza, Enzo Ranieri, Giulia Polo, Alberto Burlina, Ana Carolina Brusius-Facchin, and et al. 2020. "Neonatal Screening for MPS Disorders in Latin America: A Survey of Pilot Initiatives" International Journal of Neonatal Screening 6, no. 4: 90. https://doi.org/10.3390/ijns6040090
APA StyleKubaski, F., Sousa, I., Amorim, T., Pereira, D., Trometer, J., Souza, A., Ranieri, E., Polo, G., Burlina, A., Brusius-Facchin, A. C., Netto, A. B. O., Tomatsu, S., & Giugliani, R. (2020). Neonatal Screening for MPS Disorders in Latin America: A Survey of Pilot Initiatives. International Journal of Neonatal Screening, 6(4), 90. https://doi.org/10.3390/ijns6040090