Newborn Screening for Pompe Disease: Pennsylvania Experience


 , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
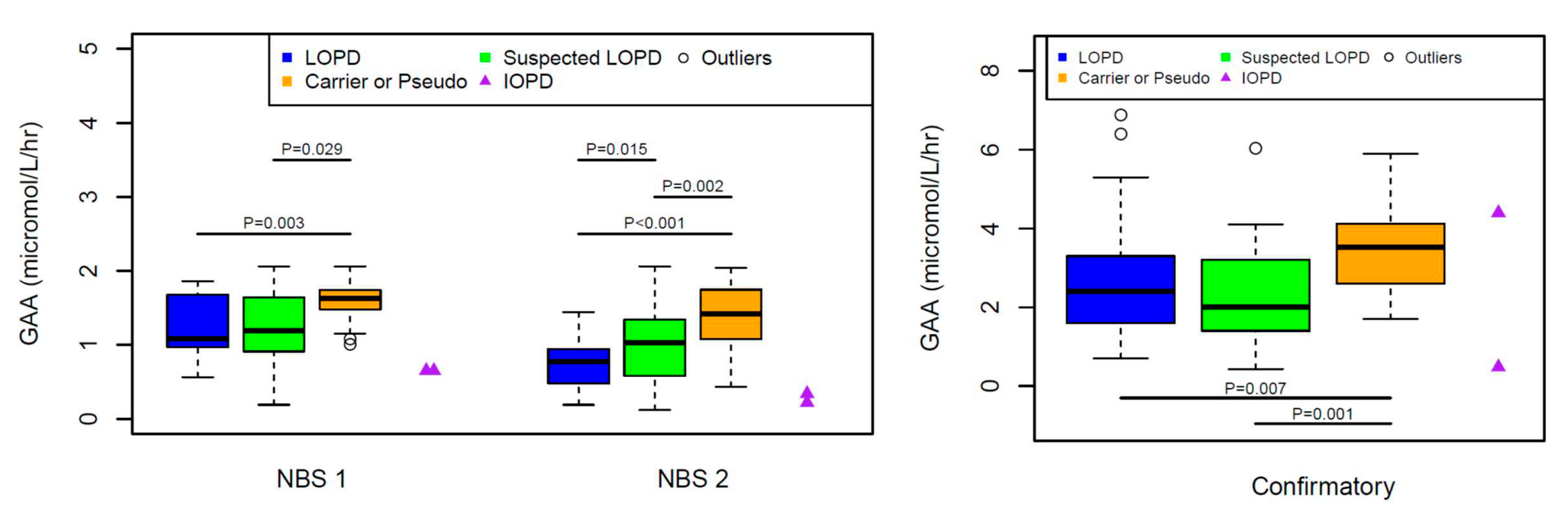
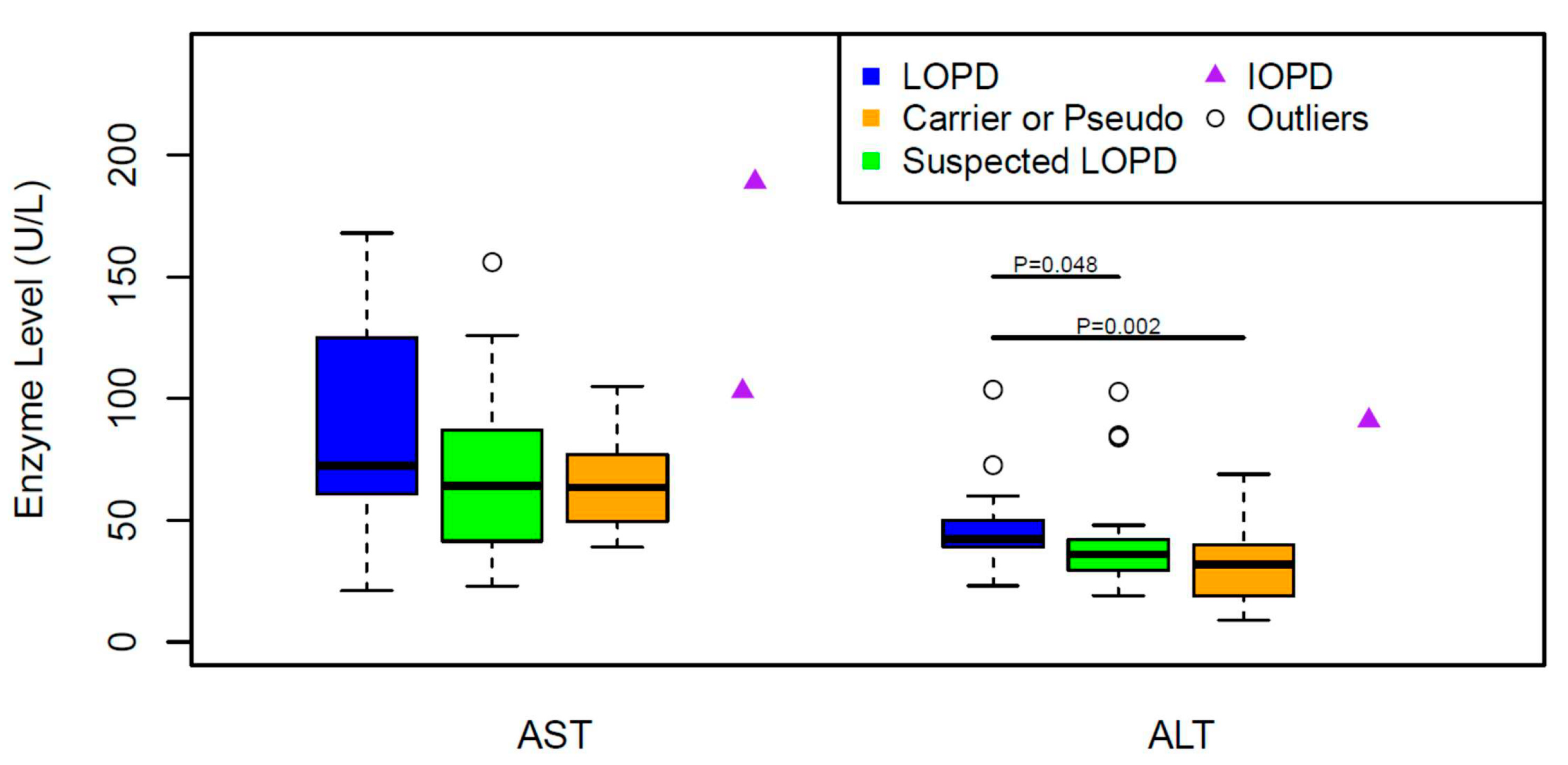
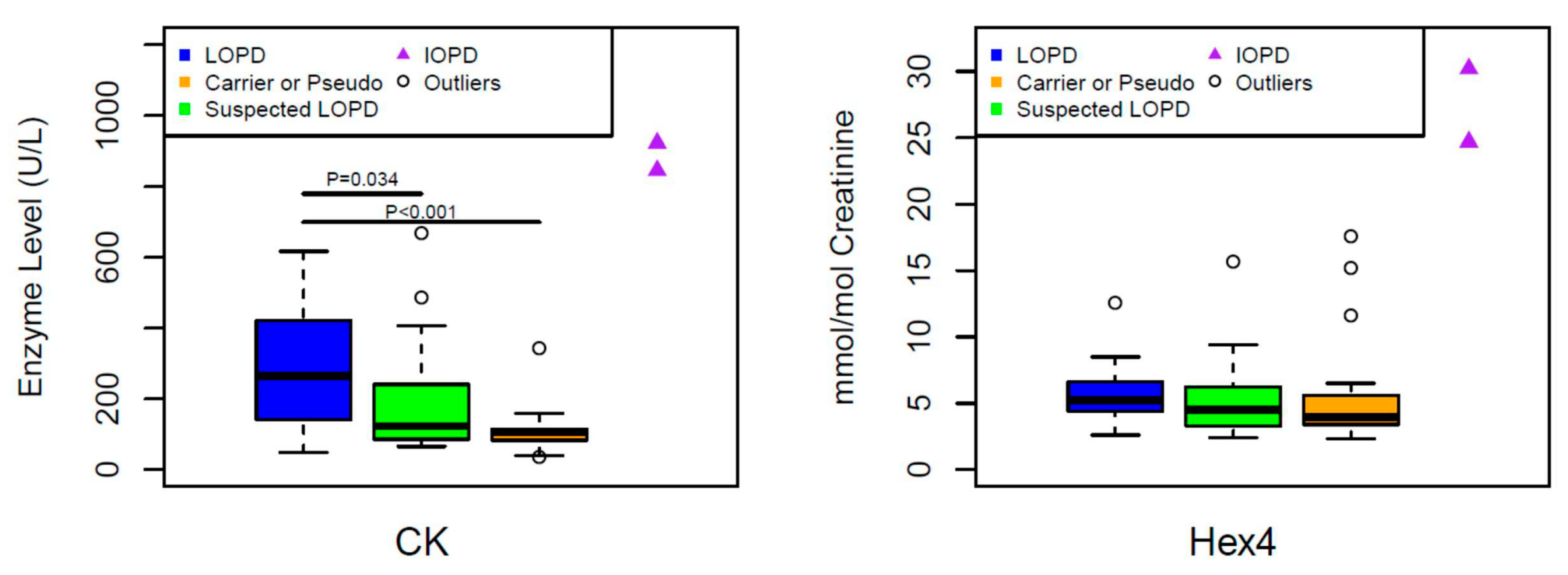
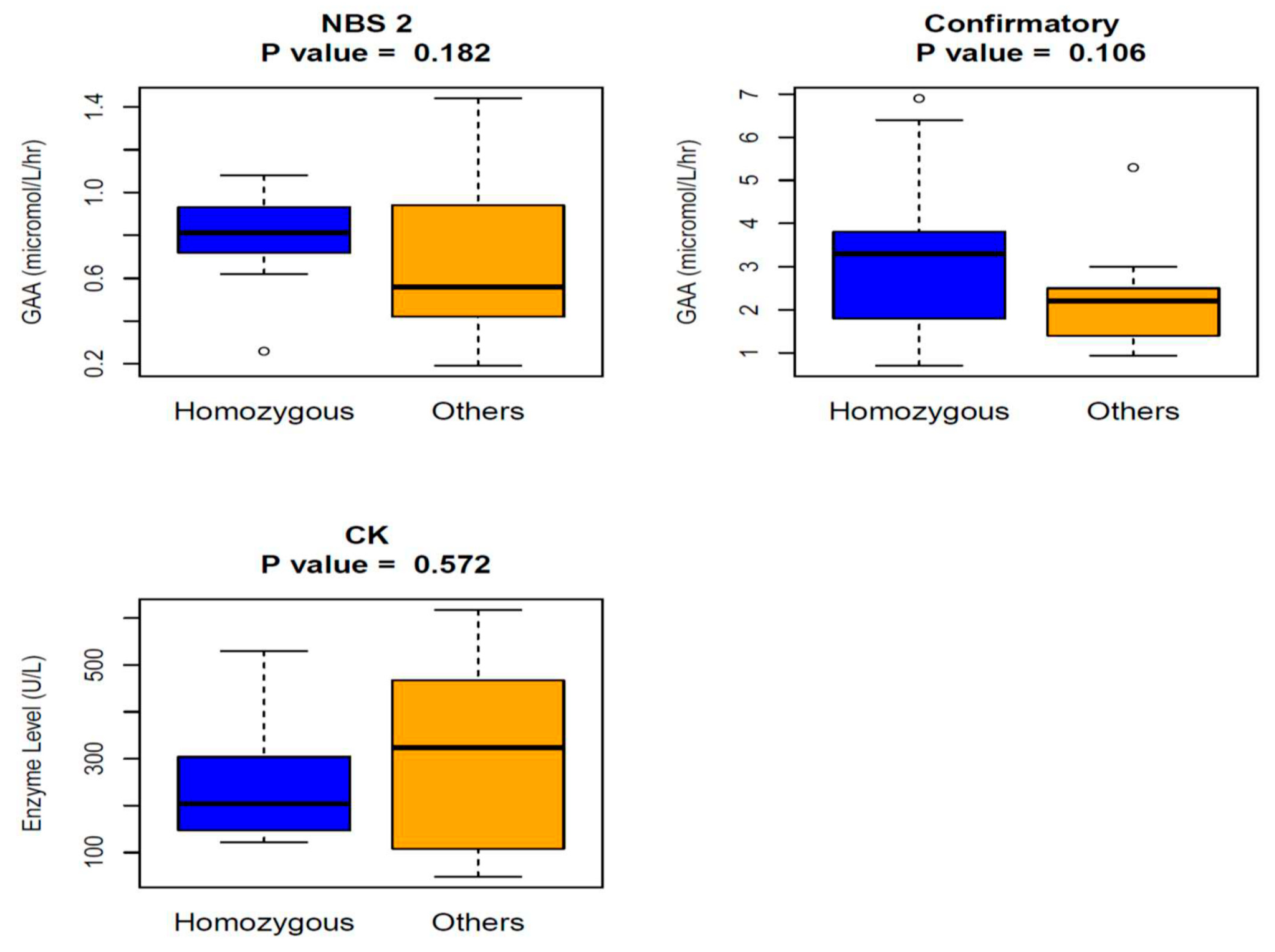
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hirschhorn, R.; Reuser, A.J. Glycogen storage disease type II: Acid alpha-glucosidase (acid maltase) deficiency. In The Metabolic and Molecular Bases of Inherited Disease, 8th ed.; Scriver, C.R., Beaudet, A.L., Sly, W.S., Childs, B., Beaudet, A., Valle, D., Kinzler, K.W., Vogelstein, B., Eds.; McGraw-Hill: New York, NY, USA, 2001; pp. 3389–3420. [Google Scholar]
- Hesselink, R.P.; Wagenmakers, A.J.; Drost, M.R.; Van der Vusse, G.J. Lysosomal dysfunction in muscle with special reference to glycogen storage disease type II. Biochim. Biophys. Acta 2003, 1637, 164–170. [Google Scholar] [CrossRef]
- Ausems, M.G.; Verbiest, J.; Hermans, M.P.; Kroos, M.A.; Beemer, F.A.; Wokke, J.H.; Sandkuijl, L.A.; Reuser, A.J.; van der Ploeg, A.T. Frequency of glycogen storage disease type II in The Netherlands: Implications for diagnosis and genetic counselling. Eur. J. Hum. Genet. 1999, 7, 713–716. [Google Scholar] [CrossRef]
- Martiniuk, F.; Chen, A.; Mack, A.; Arvanitopoulos, E.; Chen, Y.; Rom, W.N.; Codd, W.J.; Hanna, B.; Alcabes, P.; Raben, N.; et al. Carrier frequency for glycogen storage disease type II in New York and estimates of affected individuals born with the disease. Am. J. Med. Genet. 1998, 79, 69–72. [Google Scholar] [CrossRef]
- Meikle, P.J.; Hopwood, J.J.; Clague, A.E.; Carey, W.F. Prevalence of lysosomal storage disorders. JAMA 1999, 281, 249–294. [Google Scholar] [CrossRef] [PubMed]
- Pinto, R.; Caseiro, C.; Lemos, M.; Lopes, L.; Fontes, A.; Ribeiro, H.; Pinto, E.; Silva, E.; Rocha, S.; Marcão, A.; et al. Prevalence of lysosomal storage diseases in Portugal. Eur. J. Hum. Genet. 2004, 12, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Poorthuis, B.J.H.M.; Wevers, R.A.; Kleijer, W.J.; Groener, J.E.M.; de Jong, J.G.N.; van Weely, S.; Niezen-Koning, K.E.; van Diggelen, O.P. The frequency of lysosomal storage diseases in the Netherlands. Hum. Genet. 1999, 105, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Kishnani, P.S.; Howell, R.R. Pompe Disease in infants and children. J. Pediatr. 2004, 144, S35–S43. [Google Scholar] [CrossRef] [PubMed]
- Young, S.P.; Zhang, H.; Corozo, D.; Thurberg, B.L.; Bali, D.; Kishnani, P.; Millington, D. Long-term monitoring of patients with infantile-onset Pompe disease on enzyme replacement therapy using a urinary glucose tetrasaccharide biomarker. Genet. Med. 2009, 11, 536–541. [Google Scholar] [CrossRef]
- Chien, Y.H.; Goldstein, J.L.; Hwu, W.; Smith, P.B.; Lee, N.; Chiang, S.; Tolun, A.A.; Zhang, H.; Vaisnins, A.E.; Millington, D.S.; et al. Baseline Urinary Glucose Tetrasaccharide Concentrations in Patients with Infantile- and Late-Onset Pompe Disease Identified by Newborn Screening. JIMD Rep. 2015, 19, 67–73. [Google Scholar]
- Van der Ploeg, A.T.; Reuser, A.J. Pompe disease. Lancet 2008, 372, 1342–1353. [Google Scholar] [CrossRef]
- Kishnani, P.S.; Amartino, H.M.; Lindberg, C.; Miller, T.M.; Wilson, A.; Keutzer, J. Timing of diagnosis of patients with Pompe disease: Data from the Pompe registry. Am. J. Med. Genet. A 2013, 161, 2431–2443. [Google Scholar] [CrossRef] [PubMed]
- Kishnani, P.S.; Corzo, D.; Nicolino, M.; Byrne, B.; Mandel, H.; Hwu, W.L. Recombinant human acid (alpha]-glucosidase: Major clinical benefits in infantile-onset Pompe disease. Neurology 2007, 68, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Kishnani, P.S.; Goldenberg, P.C.; DeArmey, S.L.; Heller, J.; Benjamin, D.; Young, S.; Bali, D.; Smith, S.A.; Li, J.S.; Mandel, H.; et al. Cross-reactive immunologic material status affects treatment outcomes in Pompe disease infants. Mol. Genet. Metab. 2010, 99, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Mendelsohn, N.J.; Messinger, Y.H.; Rosenberg, A.S.; Kishnani, P.S. Elimination of antibodies to recombinant enzyme in Pompe’s disease. N. Engl. J. Med. 2009, 360, 194–195. [Google Scholar] [CrossRef] [PubMed]
- Messinger, Y.H.; Mendelsohn, N.J.; Rhead, W.; Dimmock, D.; Hershkovitz, E.; Champion, M.; Jones, S.A.; Olson, R.; White, A.; Wells, C.; et al. Successful immune tolerance induction to enzyme replacement therapy in CRIM-negative infantile Pompe disease. Genet. Med. 2012, 14, 135–142. [Google Scholar] [CrossRef]
- Kazi, Z.B.; Desai, A.K.; Troxler, R.B.; Kronn, D.; Packman, S.; Sabbadini, M.; Rizzo, W.B.; Scherer, K.; Abdul-Rahman, O.; Tanpaiboon, P.; et al. An immune tolerance approach using transient low-dose methotrexate in the ERT-naive setting of patients treated with a therapeutic protein: Experience in infantile-onset Pompe disease. Genet. Med. 2019, 21, 887–895. [Google Scholar] [CrossRef]
- Chien, Y.H.; Hwu, W.; Lee, N.C. Pompe Disease: Early Diagnosis and Early Treatment Make a Difference. Pediatr. Neonatol. 2013, 54, 219–227. [Google Scholar] [CrossRef]
- Chien, Y.H.; Lee, N.C.; Chen, C.A.; Tsai, F.S.; Tsai, W.H.; Shieh, J.Y.; Huang, H.J.; Hsu, W.C.; Tsai, T.H.; Hwu, W.L. Long-term prognosis of patients with infantile-onset Pompe disease diagnosed by newborn screening and treated since birth. J. Pediatr. 2015, 166, 985–991. [Google Scholar] [CrossRef]
- Chien, Y.H.; Lee, N.C.; Thurberg, B.L.; Chiang, S.C.; Zhang, X.K.; Keutzer, J.; Huang, A.C.; Wu, M.H.; Huang, P.H.; Tsai, F.J.; et al. Pompe Disease in Infants: Improving the Prognosis by Newborn Screening and Early Treatment. Pediatrics 2009, 124, e1116. [Google Scholar] [CrossRef]
- Chien, Y.H.; Lee, N.C.; Huang, H.S.; Thurberg, T.; Tsai, F.J.; Hwu, W.L. Later-Onset Pompe Disease: Early Detection and Early Treatment Initiation Enabled by Newborn Screening. J. Pediatr. 2011, 158, 1023–1027. [Google Scholar] [CrossRef]
- Chien, Y.H.; Hwu, W.L.; Lee, N.C. Newborn screening: Taiwanese experience. Ann. Transl. Med. 2019, 7, 281. [Google Scholar] [CrossRef] [PubMed]
- Bodamer, O.A.; Scott, C.R.; Giugliani, R. Newborn screening for Pompe disease. Pediatrics 2017, 140, S4–S13. [Google Scholar] [CrossRef] [PubMed]
- Kemper, A.R.; Browning, M. Evidence Review: Pompe Disease. Available online: https://www.hrsa.gov/sites/default/files/hrsa/advisory-committees/heritable-disorders/rusp/previous-nominations/pompe-evidence-review-report-2008.pdf (accessed on 12 November 2020).
- Tortorelli, S.; Turgeon, C.T.; Gavrilov, D.K.; Oglesbee, D.; Raymond, K.M.; Rinaldo, P.; Matern, D. Simultaneous Testing for 6 Lysosomal Storage Disorders and X-Adrenoleukodystrophy in Dried Blood Spots by Tandem Mass Spectrometry. Clin. Chem. 2016, 62, 1248–1254. [Google Scholar] [CrossRef] [PubMed]
- Mechtler, T.P.; Stary, S.; Metz, T.F.; de Jesús, V.R.; Greber-Platzer, S.; Pollak, A.; Herkner, K.R.; Streubel, B.; Kasper, D.C. Neonatal screening for lysosomal storage disorders: Feasibility and incidence from a nationwide study in Austria. Lancet 2012, 379, 335–341. [Google Scholar] [CrossRef]
- Burton, B.K.; Charrow, J.; Hoganson, G.E.; Fleischer, J.; Grange, D.K.; Braddock, S.R.; Hitchins, L.; Hickey, R.; Christensen, K.M.; Groepper, D.; et al. Newborn Screening for Pompe Disease in Illinois: Experience with 684,290 Infants. Int. J. Neonatal Screen. 2020, 6, 4. [Google Scholar] [CrossRef]
- Smith, L.D.; Bainbridge, M.N.; Parad, R.B.; Bhattacharjee, A. Second-Tier Molecular Testing in New-born Screening for Pompe Disease: Landscape and Challenges. Int. J. Neonatal Screen. 2020, 6, 32. [Google Scholar] [CrossRef]
- Jang, C.F.; Jang, C.C.; Liao, H.C.; Huang, L.Y.; Chiang, C.C.; Ho, H.C.; Lai, C.H.; Chu, T.H.; Yang, T.F.; Hsu, T.R.; et al. Very Early Treatment for Infantile-Onset Pompe Disease Contributes to Better Outcomes. J. Pediatr. 2016, 169, 174–180. [Google Scholar]
- Pruniski, B.; Lisi, E.; Ali, N. Newborn screening for Pompe disease: Impact on families. J. Inherit. Metab. Dis. 2018, 41, 1189–1203. [Google Scholar] [CrossRef]
- Kroos, M.A.; Pomponio, R.J.; Hagemans, M.L.; Keulemans, J.L.M.; Phipps, M.; DeRiso, M.; Palmer, R.E.; Ausems, M.G.E.M.; van der Beek, N.A.M.E.; van Diggelen, O.P.; et al. Broad spectrum of Pompe disease in patients with the same c-32-13T->G haplotype. Neurology 2007, 68, 110–115. [Google Scholar] [CrossRef]
- Rairkar, M.V.; Case, L.E.; Bailey, L.A.; Kazi, Z.B.; Desai, A.K.; Berrier, K.L.; Coats, J.; Gandy, R.; Quinones, R.; Kishnani, P. Insight into the phenotype of infants with Pompe disease identified by newborn screening with the common c.-32-13T>G “late-onset” GAA variant. Mol. Genet. Metab. 2017, 122, 99–107. [Google Scholar] [CrossRef]
- Herbert, M.; Cope, H.; Li, J.S.; Kishnani, P. Severe Cardiac Involvement Is Rare in Patients with Late-Onset Pompe Disease and the Common c.-32-13T>G Variant: Implications for Newborn Screening. J. Pediatr. 2018, 198, 308–312. [Google Scholar] [CrossRef] [PubMed]
- Musumeci, O.; Thieme, A.; Claeys, K.G.; Wenninger, S.; Kley, R.A.; Kuhn, M.; Lukacs, Z.; Deschauer, M.; Gaeta, M.; Toscano, A.; et al. Homozygosity for the common GAA gene splice site mutation c-32-13T>G in Pompe disease is associated with the classical adult phenotypical spectrum. Neuromuscul. Disord. 2015, 25, 719–724. [Google Scholar] [CrossRef] [PubMed]
- Klug, T.; Swartz, L.B.; Washburn, J.; Brannen, C.; Kiesling, J. Lessons Learned from Pompe Disease Newborn Screening and Follow-up. Int. J. Neonatal Screen. 2020, 6, 11. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pompe Disease | n | Incidence |
|---|---|---|
| IOPD | 2 | 1:265,570 |
| LOPD | 31 | 1:17,134 |
| Suspected LOPD | 30 | 1:17,705 |
| IOPD+LOPD | 33 | 1:16,095 |
| IOPD+ LOPD+ suspected LOPD | 63 | 1:8431 |
| Carriers | 35 | 1:15,175 |
| Pseudodeficiency | 15 | 1:35,409 |
| False positive (Normal confirmatory GAA activity, negative GAA full gene sequencing) | 2 | 1:265,570 |
| Newborn Screening | Confirmatory Testing | |||||||
|---|---|---|---|---|---|---|---|---|
| GAA#1 (>2.10 micromole/L/h) | GAA#2 (>2.10 micromole/L/h) | Genotype | GAA | AST and ALT (Normal Range for Lab, U/L) | CK (Normal Range for Lab, U/L) | BNP (Normal Range for Lab, pg/mL) | Hex4 (Normal Range for Lab, (mmol/mol Creatinine) | |
| IOPD Patients | ||||||||
| 1 | 0.65 | 0.22 | c.759delC/c.1551+1G>C | 4.4 (>6.7) | AST: 103 (30–100) | 923 (60–305) | 1080 (0.0–100.0) | 24.7 |
| 2 | 0.65 | 0.34 | c.525delT/c.1694_1697delTCTC | 0.4 (<3.88) | AST: 189 (22–71); ALT: 91 (7–50) | 846 (60–305) | 1232.5 (0.0–100.0) | 30.2 (<20) |
| LOPD Patients | ||||||||
| 1 | 1.86 | 0.78 | c.-32-13T>G/c.-32-13T>G | 3.1 (>6.7) | AST: 72 (24–72); ALT: 41 (17–63) | 530 (28–300) | ND | 4.9 (≤20) |
| 2 | 1.86 | 1.44 | c.2238G>C/c.1552-3C>G | 5.3 (>6.7) | AST: 21 (24–72); ALT: 23 (17–63) | 72 (28–300) | ND | 4.2 (≤20) |
| 3 | 1.73 | 1.08 | c.-32-13T>G/c.-32-13T>G | 3.6 (>6.7) | AST: 56 (24–72); ALT: 49 (17–63) | 125 (28–300) | ND | 4.5 (≤20) |
| 4 | 1.24 | 0.67 | c.-32-13T>G/c.-32-13T>G | 2.4 (>6.7) | AST: 66 (24–72); ALT: 52 (17–63) | 361 (28–300) | ND | 3.9 (≤20) |
| 5 | 1.82 | 0.94 | c.-32-13T>G/c.-32-13T>G | 6.9 (>6.7) | AST: 52 (24–72); ALT: 46 (17–63) | 333 (28–300) | ND | 4.4 (≤20) |
| 6 | 1.32 | 0.88 | c.-32-13T>G/c.-32-13T>G | 0.7 (>6.7) | AST: 61 (24–72); ALT: 39 (17–63) | 176 (28–300) | ND | 5.9 (≤20) |
| 7 | 1.05 | 0.19 | c.-32-13 T>C/c.546 G>A | 2.2 (>3.88) | 283 (30–135) | ND | 2.6 (<3.0) | |
| 8 | 0.83 | 0.85 | c.-32-13 T>G/c. 1655 T>C | 0.93 (>3.88) | 365 (60–305) | ND | 5.23 (<−8.9) | |
| 9 | 0.97 | 0.33 | c.2238G>C/c.2281delGinsAT/c.2065G>A | 0.93 (>3.88) | AST: 72 (22–71); ALT: 43 (7–50) | 145 (60–305) | 13.0 (0.0–100.0) | 5.3 (≤20) |
| 10 | 0.84 | 0.49 | c.1438-1G>C/c.-32-13T>G | 2.1 (>3.88) | AST: 168 (20–64); ALT: 104 (12–42) | 617 (60–305) | - | 6.6 (≤20) |
| 11 | - | 0.63 | c.-32-13T>G/c.2560C>T | 3.00 (>3.88) | AST: 125 (22–71); ALT: 50 (7–50) | 421 (60–305) | - | - |
| 12 | - | - | c.-32-13T>G /c.2238G>C | 2.60 (>3.88) | AST: 112 (22–71); ALT: 50 (7–50) | 464 (60–305) | 51.6 (0.0–100.0) | 6.1 (≤20) |
| 13 | 0.56 | 0.62 | c.-32-13T>G/c.-32-13T>G | 3.80 (>3.88) | AST: 78 (20–64); ALT: 42 (12–42) | 122 (60–305) | 71.8 (0.0–100.0) | 4.8 (≤20) |
| 14 | 1.68 | 0.79 | c.156_157delTC/c.-32-13T>G | 2.40 (>3.88) | AST: 166 (20–64); ALT: 73 (12–42) | 467 (60–305) | - | 7.0 (≤20) |
| 15 | 1.11 | 0.5 | c.2238G>C/c.-32-13T>G/c.2065G>A | 1.60 (>3.88) | AST: 49 (20–64); ALT: 27 (12–42) | 68 (60–305) | - | 2.8 (≤20) |
| 16 | 1.06 | 0.26 | c.-32-13T>G/c.-32-13T>G | 6.4 (>3.88) | AST: 73 (22–71); ALT: 39 (7–50) | 140 (60–305) | 99.8 (0.0–100.0) | 8.5 (≤20) |
| 17 | 1.04 | 0.77 | c.-32-13T>G/c.-32-13T>G | 1.80 (>3.88) | AST: 150 (22–71); ALT: 42 (7–50) | 275 (60–305) | 44.5 (0.0–100.0) | 5.2 (≤20) |
| 18 | 0.7 | 0.34 | c.-32-13T>G/c.2560C>T | 1.40 (>3.88) | AST: 152 (22–71); ALT: 55 (7–50) | 542 (60–305) | 16.5 (0.0–100.0) | 7.6 (≤20) |
| 19 | 1.07 | 0.38 | c.456_458insGA/c.-32-13T>G | 1.10 (>3.88) | AST: 158 (22–71); ALT: 39 (7–50) | 510 (60–305) | <10.0 (0.0–100.0) | 8.1 (≤20) |
| 20 | 0.88 | 0.77 | c.-32-13T>G/c.-32-13T>G | 3.50 (>3.88) | AST: 63 (20–64); ALT: 34 (12–42) | 204 (60–305) | 65.4 (0.0–100.0) | - |
| 21 | 1.77 | 0.84 | c.-32-13T>G/c.-32-13T>G | - | AST: 71 (22–71); ALT: 60 (7–50) | 155 (60–305) | - | 12.6 (≤20) |
| 22 | 1.07 | 0.92 | c.-32-13T>G/c.-32-13T>G | 1.60 (3.88) | AST: 92 (22–71); ALT: 46 (7–50) | 265 (60–305) | 81.5 (0.0–100.0) | 5.9 (≤20) |
| 23 | 1.12 | 0.62 | c.2238G>C;c.2065G>A/c.2238G>C;c.2065G>A | 2.5 (3.88) | AST: 73 (22–71); ALT: 28 (7–50) | 48 (60–305) | 102.3 (0.0–100.0) | - |
| 24 | 0.43 | c.1856G>A/c.2238G>C | 197 (60–305) | |||||
| 25 | 0.95 | c.2238G>C/c.2238G>C | ||||||
| 26 | 1.01 | c.2238G>C/c.2238G>C | ||||||
| 27 | 1.1 | 0.42 | c.1441T>C/c.2238G>C/c.2065G>A | 2.40 (>3.88) | AST: 32 (22–71); ALT: 29 (7–50) | 108 (60–305) | 35.7 (0.0–100.0) | 3.3 (≤20) |
| 28 | 1.06 | c.-32-13T>G/c.2238G>C/c.2065G>A | ||||||
| 29 | 0.48 | c.-32-13T>G/c.1547G>A | ||||||
| 30 | 1.07 | c.32-13T>G/c.32-13T>G | ||||||
| 31 | 0.94 | c.32-13T>G/c.2238G>C /c.2065G>A | ||||||
| Newborn Screening | Confirmatory Testing | |||||||
|---|---|---|---|---|---|---|---|---|
| Suspected LOPD Patients | GAA#1 (>2.10 micromole/L/h) | GAA#2 (>2.10 micromole/L/h) | Genotype | GAA | AST and ALT (normal range for lab, U/L) | CK (normal range for lab, U/L) | BNP (normal range for lab, pg/mL) | Hex4 (normal range for lab, (mmol/mol creatinine) |
| 1 | 0.91 | 0.2 | c.-32-13T>G/c.692+3G>C | 2.1 | AST: 30 (24–72); ALT: 30 (17–63) | 150 | ND | 3.3 |
| 2 | 2.06 | 1.99 | c.-32-13T>G/c.1594G>A | 3.8 | AST: 41 (24–72); ALT: 30 (17–63) | 134 | ND | 3.0 |
| 3 | 1.48 | 0.67 | c.-32-13T>G/c.546G>A | 1.6 | AST: 42 (24–72); ALT: 37 (17–63) | ND | ND | 6.0 |
| 4 | 1.85 | 0.98 | c.-32-13T>G/c.266G>A;c.1377C>G | 1.4 | AST: 39 (24–72); ALT: 32 (17–63) | 293 | ND | 8.3 |
| 5 | 1.04 | 0.53 | c.-32-13T>G/c.2003 A>G | 0.6 (>3.88) | 107 (26-192) | 2.4 (<20) | ||
| 6 | 0.61 | 0.41 | c.-32-13T>G/c.1721T>C | 0.8 | AST: 96 (24–72); ALT: 84 (17–63) | 377 | ND | 4.9 |
| 7 | 0.52 | 0.45 | c.-32-13T>G/c.1291_1299del9 | 1.9 | AST: 83 (20–70); ALT: 48 (17–63) | 486 | ND | 9.4 |
| 8 | 0.56 | 0.63 | c.-32-13T>G/1655T>C | 2.5 | AST: 156 (24–72); ALT: 103 (17–63) | 668 | ND | 6.0 |
| 9 | ND | 1.34 | c.-32-13T>G/c.533G>A | 3.7 | AST: 25 (24–72); ALT: 19 (17–63) | 86 | ND | 2.5 |
| 10 | 1.3 | 2.01 | c.-32-13T>G/ c.862G>A;c.271G>A | 2.0 | AST: 32 (24–72); ALT: 31 (17–63) | 406 | ND | 3.4 |
| 11 | 1.58 | 0.64 | c.-32-13T>G/ c.841C>T | 1.4 | AST: 23 (24–72); ALT: 42 (17–63) | 136 | ND | 6.4 |
| 12 | 0.58 umol/L/h | 1.84 umol/L/h | c.1A>G/c.1345C>T | 1.90 (>3.88) | AST: 44 (15–41 U/L) ALT: 26 (12–42) | 241 (15-200 U/L) | - | 8.4 (≤20 mmol/mol creatinine) |
| 13 | 1.02 umol/L/h | 1.22 umol/L/h | c.2560C>T/c.1888+5G>T;c.2065G>A | 3.80 (3.88) | AST: 112 (20–64); ALT: 29 (12–42) | 69 (60–305) | 407.3 (0.0–100.0) | 4.6 (≤20 mmol/mol creatinine) |
| 14 | <0.19 umol/L/h | 0.12 umol/L/h | c.2236T>C/c.700A>G/C | 0.90 (>3.88) | AST: 52 (22–71); ALT: 36 (7–50) | 142 (60–305) | 44.2 (0.0–100.0) | 15.7 (≤20) |
| 15 | 1.94 umol/L/h | 2.06 umol/L/h | c.1478C>T/1194+3G>C | 6.05 (>3.88) | - | 86 (60–305) | - | <4.4 (≤20) |
| 16 | 1.97 umol/L/h | 1.16 umol/L/h | c.784G>A/c.859-19G>A/c.1392G>C | 4.00 (>3.88) | AST: 92 (20–64); ALT: 42 (12–42) | 201 (60–305) | 55.3 (0.0–100.0) | 6.1 (≤20) |
| 17 | 0.98 umol/L/h | 0.58 umol/L/h | c.2105G>T/c.1124G>A | 1.20 {>3.88) | AST: 64 (22–71); ALT: 20 (7–50) | 85 (60–305) | - | 3.0 (≤20) |
| 18 | 1.59 umol/L/h | 0.95 umol/L/h | c.-32-13T>G/c.692+3G>C | 2.70 (>3.88} | AST:73 (22–71); ALT:19 (7–50) | 65 (60–305) | 52.7 (0.0–100.0) | 3.3 (≤20) |
| 19 | 1.19 | 1.27 | c.1655T>C/c.1888+5G>T/c.2065G>A | 0.43 (1.29-25.7) | AST: 60 (22–71); ALT: 25 (7–50) | 110 (60–305) | 19.3 (0.0–100.0) | - |
| 20 | - | 1.27 | c.-32-13T>G/c.705G>A/c.(1726G>A;c.2065G>A) | 1.80 (3.88) | AST 80 (22–71); ALT: 41 (7–50) | 86 (60–305) | 30.0 (0.0–100.0) | 3.3 (≤20) |
| 21 | 1.64 | 1.1 | c.-32-13T>G/c.650C>T | 4.10 (>3.88) | AST: 65 (22–71); ALT: 35 (7–50) | 68 (60–305) | 33.8 (0.0–100.0) | 5.7 (≤20) |
| 22 | 1.02 | 0.85 | c.1655T>C/c.664G>A | 2.01 (>3.88) | AST:69 (20–64); ALT: 41 (12-42) | 94 (60–305) | 57.4 (0.0–100.0) | 6.2 (≤20) |
| 23 | - | 0.42 | c.-32-13T>G/c.1103G>A | - | AST: 126 (20–64); ALT:85 (12–42) | 407 {60–305) | - | 7.2 (≤20) |
| 24 | - | 1.08 | c.258dupC/c.1909C>A/c.(1726G>A;c.2065G>A) | - | AST: 64 (22–71); ALT: 38 (7–50) | 102 (60–305) | 13.0 (0.0–100.0) | 3.4 (≤20) |
| 25 | 1.98 | 1.17 | c.1552-3C>G/c.1378G>A | - | AST: 91 (22–71); ALT: 46 (7–50) | 123 (60–305) | 29.1 (0.0–100.0) | 3.7 (≤20) |
| 26 | - | 1.49 | c.1655T>C/c.266G>A | 2.10 (>3.88) | AST: 56 (22–71); ALT: 36 (7–50) | 88 3 60–305) | - | 3.8 (≤20) |
| 27 | 0.42 | c.-32-13T>G/c.2467A>T | ||||||
| 28 | 1.63 | c.1504A>G/c.2467A>T | ||||||
| 29 | 1.36 | c.2560C>T/c.726G>A | ||||||
| 30 | 0.93 | c.2238G>C/c.2467A>T | ||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ficicioglu, C.; Ahrens-Nicklas, R.C.; Barch, J.; Cuddapah, S.R.; DiBoscio, B.S.; DiPerna, J.C.; Gordon, P.L.; Henderson, N.; Menello, C.; Luongo, N.; et al. Newborn Screening for Pompe Disease: Pennsylvania Experience. Int. J. Neonatal Screen. 2020, 6, 89. https://doi.org/10.3390/ijns6040089
Ficicioglu C, Ahrens-Nicklas RC, Barch J, Cuddapah SR, DiBoscio BS, DiPerna JC, Gordon PL, Henderson N, Menello C, Luongo N, et al. Newborn Screening for Pompe Disease: Pennsylvania Experience. International Journal of Neonatal Screening. 2020; 6(4):89. https://doi.org/10.3390/ijns6040089
Chicago/Turabian StyleFicicioglu, Can, Rebecca C. Ahrens-Nicklas, Joshua Barch, Sanmati R. Cuddapah, Brenda S. DiBoscio, James C. DiPerna, Patricia L. Gordon, Nadene Henderson, Caitlin Menello, Nicole Luongo, and et al. 2020. "Newborn Screening for Pompe Disease: Pennsylvania Experience" International Journal of Neonatal Screening 6, no. 4: 89. https://doi.org/10.3390/ijns6040089
APA StyleFicicioglu, C., Ahrens-Nicklas, R. C., Barch, J., Cuddapah, S. R., DiBoscio, B. S., DiPerna, J. C., Gordon, P. L., Henderson, N., Menello, C., Luongo, N., Ortiz, D., & Xiao, R. (2020). Newborn Screening for Pompe Disease: Pennsylvania Experience. International Journal of Neonatal Screening, 6(4), 89. https://doi.org/10.3390/ijns6040089