Two-Tiered Newborn Screening with Post-Analytical Tools for Pompe Disease and Mucopolysaccharidosis Type I Results in Performance Improvement and Future Direction

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
3. Results
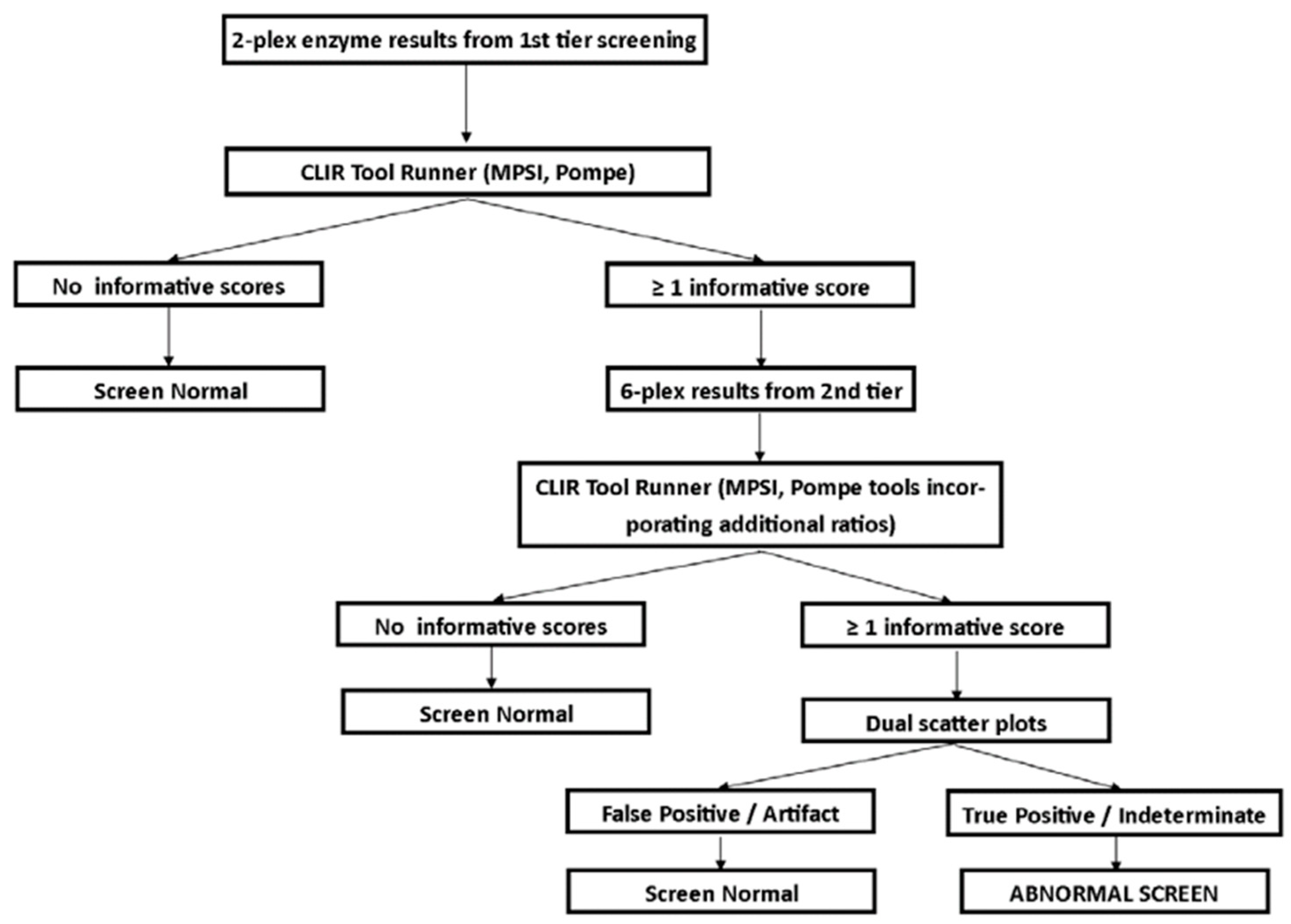
3.1. Screening Protocol
3.2. Follow-Up Testing
3.3. Screening Results
3.4. Clinical Outcomes
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Minter Baerg, M.M.; Stoway, S.D.; Hart, J.; Mott, L.; Peck, D.S.; Nett, S.L.; Eckerman, J.S.; Lacey, J.M.; Turgeon, C.T.; Gavrilov, D.; et al. Precision newborn screening for lysosomal disorders. Genet. Med. 2018, 20, 847–854. [Google Scholar] [CrossRef] [PubMed]
- Chien, Y.H.; Lee, N.C.; Chen, C.A.; Tsai, F.J.; Tsai, W.H.; Shieh, J.Y.; Huang, H.J.; Hsu, W.C.; Tsai, T.H.; Hwu, W.L. Long-term prognosis of patients with infantile-onset Pompe disease diagnosed by newborn screening and treated since birth. J. Pediatr. 2015, 166, 985–991.e1-2. [Google Scholar] [CrossRef] [PubMed]
- Bodamer, O.A.; Scott, C.R.; Giugliani, R. Pompe Disease Newborn Screening Working Group Newborn Screening for Pompe Disease. Pediatrics 2017, 140, S4–S13. [Google Scholar] [CrossRef] [PubMed]
- Kiely, B.T.; Kohler, J.L.; Coletti, H.Y.; Poe, M.D.; Escolar, M.L. Early disease progression of Hurler syndrome. Orphanet J. Rare Dis. 2017, 12, 32. [Google Scholar] [PubMed]
- Wasserstein, M.P.; Caggana, M.; Bailey, S.M.; Desnick, R.J.; Edelmann, L.; Estrella, L.; Holzman, I.; Kelly, N.R.; Kornreich, R.; Kupchik, S.G.; et al. The New York pilot newborn screening program for lysosomal storage diseases: Report of the First 65,000 Infants. Genet. Med. 2019, 21, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, P.V.; Campbell, C.; Klug, T.; Rogers, S.; Raburn-Miller, J.; Kiesling, J. Lysosomal storage disorder screening implementation: Findings from the first six months of full population pilot testing in Missouri. J. Pediatr. 2015, 166, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Burton, B.K.; Charrow, J.; Hoganson, G.E.; Waggoner, D.; Tinkle, B.; Braddock, S.R.; Schneider, M.; Grange, D.K.; Nash, C.; Shryock, H.; et al. Newborn Screening for Lysosomal Storage Disorders in Illinois: The Initial 15-Month Experience. J. Pediatr. 2017, 190, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.L.; Clinard, K.; Powell, C.M.; Rehder, C.; Young, S.P.; Bali, D.; Beckloff, S.E.; Gehtland, L.M.; Kemper, A.R.; Lee, S.; et al. The North Carolina Experience with Mucopolysaccharidosis Type I Newborn Screening. J. Pediatr. 2019, 211, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Elliott, S.; Buroker, N.; Cournoyer, J.J.; Potier, A.M.; Trometer, J.D.; Elbin, C.; Schermer, M.J.; Kantola, J.; Boyce, A.; Turecek, F.; et al. Pilot study of newborn screening for six lysosomal storage diseases using Tandem Mass Spectrometry. Mol. Genet. Metab. 2016, 118, 304–309. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.B.; Masi, S.; Ghomashchi, F.; Chennamaneni, N.K.; Ito, M.; Scott, C.R.; Turecek, F.; Gelb, M.H.; Spacil, Z. Tandem Mass Spectrometry Has a Larger Analytical Range than Fluorescence Assays of Lysosomal Enzymes: Application to Newborn Screening and Diagnosis of Mucopolysaccharidoses Types II, IVA, and VI. Clin. Chem. 2015, 61, 1363–1371. [Google Scholar] [CrossRef] [PubMed]
- Taylor, H.A.; Thomas, G.H. Pseudodeficiency of alpha-iduronidase. J. Inherit. Metab. Dis. 1993, 16, 1058–1059. [Google Scholar] [CrossRef] [PubMed]
- Aronovich, E.L.; Pan, D.; Whitley, C.B. Molecular genetic defect underlying Alpha-L-iduronidase pseudodeficiency. Am. J. Hum. Genet. 1996, 58, 75–85. [Google Scholar] [PubMed]
- Hall, P.L.; Marquardt, G.; McHugh, D.M.S.; Currier, R.J.; Tang, H.; Stoway, S.D.; Rinaldo, P. Postanalytical tools improve performance of newborn screening by tandem mass spectrometry. Genet. Med. 2014, 16, 889–895. [Google Scholar] [CrossRef] [PubMed]
- Tortorelli, S.; Eckerman, J.S.; Orsini, J.J.; Stevens, C.; Hart, J.; Hall, P.L.; Alexander, J.J.; Gavrilov, D.; Oglesbee, D.; Raymond, K.; et al. Moonlighting newborn screening markers: The incidental discovery of a second-tier test for Pompe disease. Genet. Med. 2018, 20, 840–846. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, P.V.; Klug, T.; Vermette, L.; Raburn-Miller, J.; Kiesling, J.; Rogers, S. Incidence of 4 Lysosomal Storage Disorders from 4 Years of Newborn Screening. JAMA Pediatr. 2018, 172, 696–697. [Google Scholar] [CrossRef] [PubMed]
- Niño, M.Y.; In ’t Groen, S.L.M.; Bergsma, A.J.; van der Beek, N.A.M.E.; Kroos, M.; Hoogeveen-Westerveld, M.; van der Ploeg, A.T.; Pijnappel, W.W.M.P. Extension of the Pompe mutation database by linking disease-associated variants to clinical severity. Hum. Mutat. 2019, 40, 1954–1967. [Google Scholar] [CrossRef] [PubMed]
- Bali, D.S.; Goldstein, J.L.; Banugaria, S.; Dai, J.; Mackey, J.; Rehder, C.; Kishnani, P.S. Predicting cross-reactive immunological material (CRIM) status in Pompe disease using GAA mutations: Lessons learned from 10 years of clinical laboratory testing experience. Am. J. Med. Genet. C Semin. Med. Genet. 2012, 160C, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Kroos, M.; Hoogeveen-Westerveld, M.; Michelakakis, H.; Pomponio, R.; Van der Ploeg, A.; Halley, D.; Reuser, A. GAA Database Consortium Update of the pompe disease mutation database with 60 novel GAA sequence variants and additional studies on the functional effect of 34 previously reported variants. Hum. Mutat. 2012, 33, 1161–1165. [Google Scholar] [CrossRef] [PubMed]
- ClinVar. NM_000152.5(GAA):c.868A>G (p.Asn290Asp) AND Not Provided. Available online: https://www.ncbi.nlm.nih.gov/clinvar/RCV000598383/ (accessed on 5 December 2019).
- Matern, D.; Tortorelli, S.; Oglesbee, D.; Gavrilov, D.; Rinaldo, P. Reduction of the false-positive rate in newborn screening by implementation of MS/MS-based second-tier tests: The Mayo Clinic experience (2004–2007). J. Inherit. Metab. Dis. 2007, 30, 585–592. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Region Screened | Methodology | Interpretation | Disorder | # Screened | Screen Positive | True Positives | PPV (%) | Screen Positive Rate (%) |
|---|---|---|---|---|---|---|---|---|
| Illinois [7] | MSMS (1 tier) | % daily median | Pompe | 219,973 | 139 | 10 | 7.2 | 0.06 |
| MPS I | 219,973 | 151 | 1 | 0.7 | 0.07 | |||
| Kentucky [1] | MSMS (2 tier) | Post-analytical tools | Pompe | 55,161 | 2 | 2 | 100.0 | <0.002 |
| MPS I | 55,161 | 2 | 1 | 50.0 | <0.002 | |||
| Missouri [6] | Digital microfluidics | Cutoff | Pompe | ~308,000 | 161 | 32 | 19.9 | 0.05 |
| MPS I | ~308,000 | 133 | 2 | 1.5 | 0.04 | |||
| New York [5] | MSMS (1 tier) | % daily mean | Pompe | 18,105 | 6 | 1 | 16.7 | 0.03 |
| MPS I | 35,816 | 13 | 0 | 0.0 | 0.04 | |||
| North Carolina [8] | MSMS (2 tier) + Sequencing | Cutoff (Initial) | MPS I | 62,734 | 54 | 1 | 1.9 | 0.09 |
| Georgia | MSMS (2 tier) | Post-analytical tools | Pompe | 59,332 | 6 | 4 | 66.7 | 0.01 |
| MPS I | 59,332 | 11 | 0 | 0.0 | 0.02 |
| Case ID | Birth Weight (g) | Sex | Race | Age at Collection | DBS GAA | CLIR Score (Interpretation) | GAA Confirmation * | Glucose Tetrasaccharde | Allele 1 | Allele 2 | Outcome |
|---|---|---|---|---|---|---|---|---|---|---|---|
| P1 | 3695 | F | Black | 24 | 2.555 | 37 (Possible) | 2.2 Abnormal | Normal | c.868A>G p.Asn290Asp (U) ** | c.2105G>A p.Arg702His (P) | Late onset PD |
| P2 | 3515 | F | White | 24 | 1.677 | 127 (Likely) | 3.9 Abnormal (per lab) | Normal | c.-32-13T>G (P) | c.2238G>C p.Trp746Cys (P) | Late onset PD |
| P3 | 2570 | M | Black | 114 | 1.026 | 103 (Possible) | Family refused follow-up | ||||
| P4 | 3360 | M | Black | 2 | 1.581 | 32 (Possible) | 2.7 Abnormal | Elevated | c.1755-1G>A (P) | c.2236T>C p.Trp746Arg (P) | Infantile onset PD |
| 56 | 1.103 | 127 (Likely) | |||||||||
| P5 | 2560 | M | Native Hawaiian/Pacific Islander | 33 | 1.019 | 127 (Likely) | 2.7 Abnormal | Normal | c.1726G>A/c.2065G>A p.Gly576Ser (B)/p.Glu689L (B) | c.1726G>A/c.2065G>A p.Gly576Ser/p.Glu689L | Pseudodeficiency |
| Case ID | Birth Weight (g) | Sex | Race | Age at Collection (hours) | DBS IDUA | CLIR Score (Interpretation) | IDUA Confirmation | Glycosaminoglycans | Allele 1 | Allele 2 | CNV Analysis | Outcome |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| M1 | 3412 | M | Black | 32 | 0.194 | 346 (Likely) | 0.6 nmol/h/mL (≥1.0) * | Normal | c.923T>C p.Leu308Pro (U) | None | Negative | Unresolved (Lost to Genetics Follow-up) |
| M2 | 2994 | F | Black | 24 | 0.355 | 177 (Likely) | 4.2 nmol/h/mg protein (3.57–21.40) | Essentially Normal (Repeat Normal) | Not performed | Unaffected | ||
| M3 | 3470 | M | Black | 24 | 0.238 | 337 (Likely) | 7.4 pmol/punch/h (>8.0) | Normal | C.1238_1264del (U) | None | Negative | Unresolved (Lost to Genetics Follow-up) |
| M4 ** | 2320 | M | Black | 49 | 0.186 | 266 (Likely) | Family refused follow-up | |||||
| M5 ** | 2360 | M | Black | 50 | 0.226 | 228 (Likely) | Family refused follow-up | |||||
| M6 | 3820 | F | Black | 25 | 0.597 | 112 (Likely) | 5.7 nmol/h/mg protein (3.57–21.40) | Normal | Not performed | Unaffected | ||
| M7 | 3030 | M | Black | 40 | 0.222 | 335 (Likely) | 5.1 nmol/h/mg protein (3.57–21.40) | Normal | Not performed | Unaffected | ||
| M8 | 2817 | F | Black | 25 | 0.302 | 138 (Likely) | 1.6 nmol/h/mg protein (12.0–65) * | Normal | None | None | Negative | Unresolved (Lost to Genetics Follow-up) |
| M9 | 3165 | F | White | 25 | 0.242 | 266 (Likely) | 0.3 nmol/h/mg protein (3.57–21.40) * | Family refused further follow-up | ||||
| M10 | 2906 | F | Black | 25 | 0.403 | 212 (Likely) | 0.3 nmol/h/mL (≥1.0) * | Normal | c.235G>A p.Ala79Thr (B) | c.235G>A p.Ala79Thr (B) | Not performed | Pseudodeficiency |
| M11 | 3375 | M | Black | 50 | 0.323 | 200 (Likely) | 0.2 nmol/h/mg protein (3.57–21.40) * | Normal | c.235G>A p.Ala79Thr (B) | c.235G>A p.Ala79Thr (B) | Not performed | Pseudodeficiency |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hall, P.L.; Sanchez, R.; Hagar, A.F.; Jerris, S.C.; Wittenauer, A.; Wilcox, W.R. Two-Tiered Newborn Screening with Post-Analytical Tools for Pompe Disease and Mucopolysaccharidosis Type I Results in Performance Improvement and Future Direction. Int. J. Neonatal Screen. 2020, 6, 2. https://doi.org/10.3390/ijns6010002
Hall PL, Sanchez R, Hagar AF, Jerris SC, Wittenauer A, Wilcox WR. Two-Tiered Newborn Screening with Post-Analytical Tools for Pompe Disease and Mucopolysaccharidosis Type I Results in Performance Improvement and Future Direction. International Journal of Neonatal Screening. 2020; 6(1):2. https://doi.org/10.3390/ijns6010002
Chicago/Turabian StyleHall, Patricia L., Rossana Sanchez, Arthur F. Hagar, S. Caleb Jerris, Angela Wittenauer, and William R. Wilcox. 2020. "Two-Tiered Newborn Screening with Post-Analytical Tools for Pompe Disease and Mucopolysaccharidosis Type I Results in Performance Improvement and Future Direction" International Journal of Neonatal Screening 6, no. 1: 2. https://doi.org/10.3390/ijns6010002
APA StyleHall, P. L., Sanchez, R., Hagar, A. F., Jerris, S. C., Wittenauer, A., & Wilcox, W. R. (2020). Two-Tiered Newborn Screening with Post-Analytical Tools for Pompe Disease and Mucopolysaccharidosis Type I Results in Performance Improvement and Future Direction. International Journal of Neonatal Screening, 6(1), 2. https://doi.org/10.3390/ijns6010002