Next Generation Sequencing in Newborn Screening in the United Kingdom National Health Service

 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
1.1. Next Generation Sequencing in Newborn Screening: Genome Wide or Targeted Assays?
1.2. Applications of NGS in NBS as an Adjunct Test or Primary Screening Test
1.3. Development of a Rapid Targeted NGS Assay Utilising DBS DNA
2. Materials and Methods
2.1. Sample Collection
2.2. DNA Extraction
2.3. Panel Design and Validation
2.4. Semi-Automated Library Preparation
2.5. Chip Loading and Sequencing
2.6. Data Analysis
3. Results
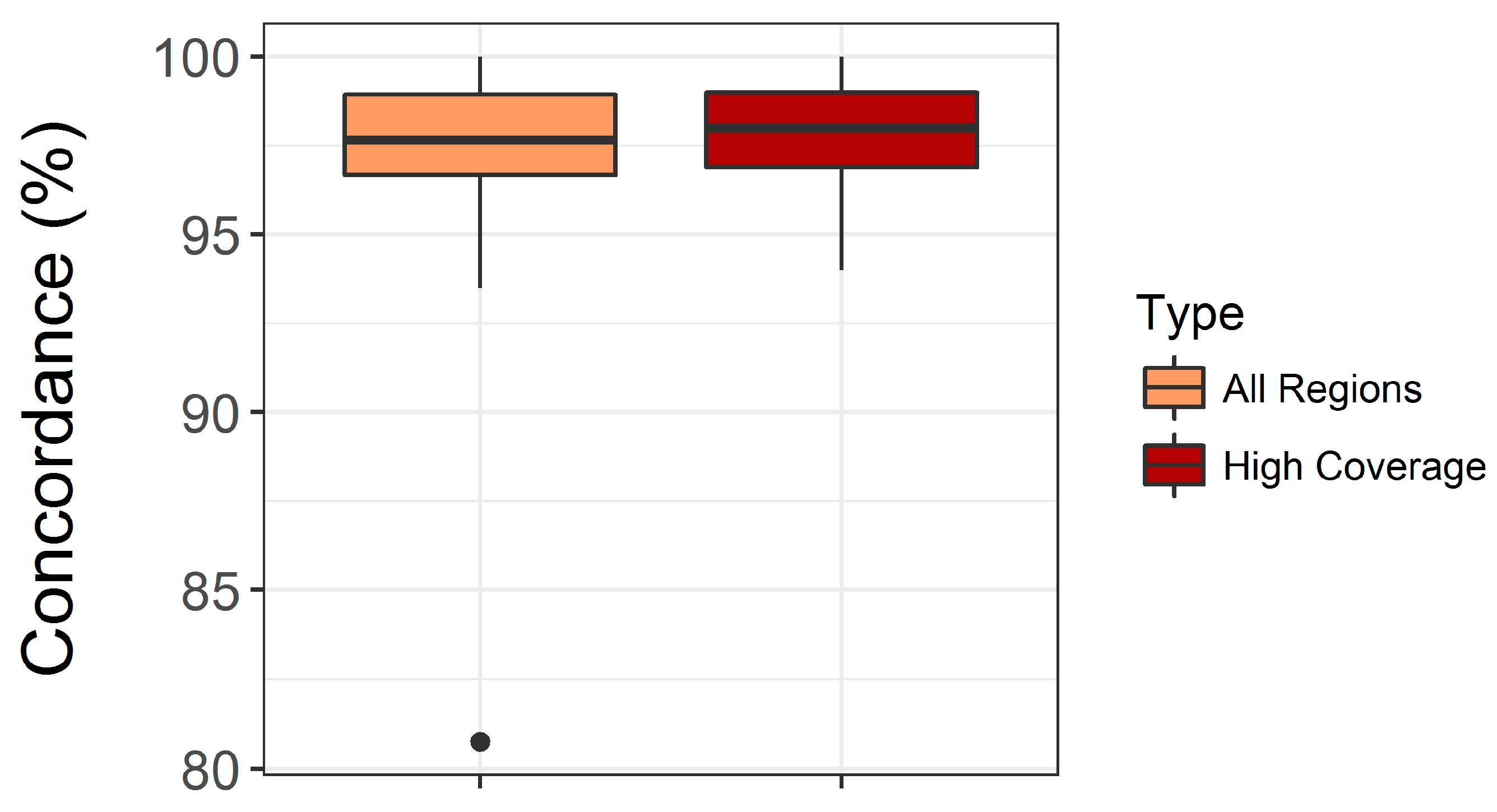
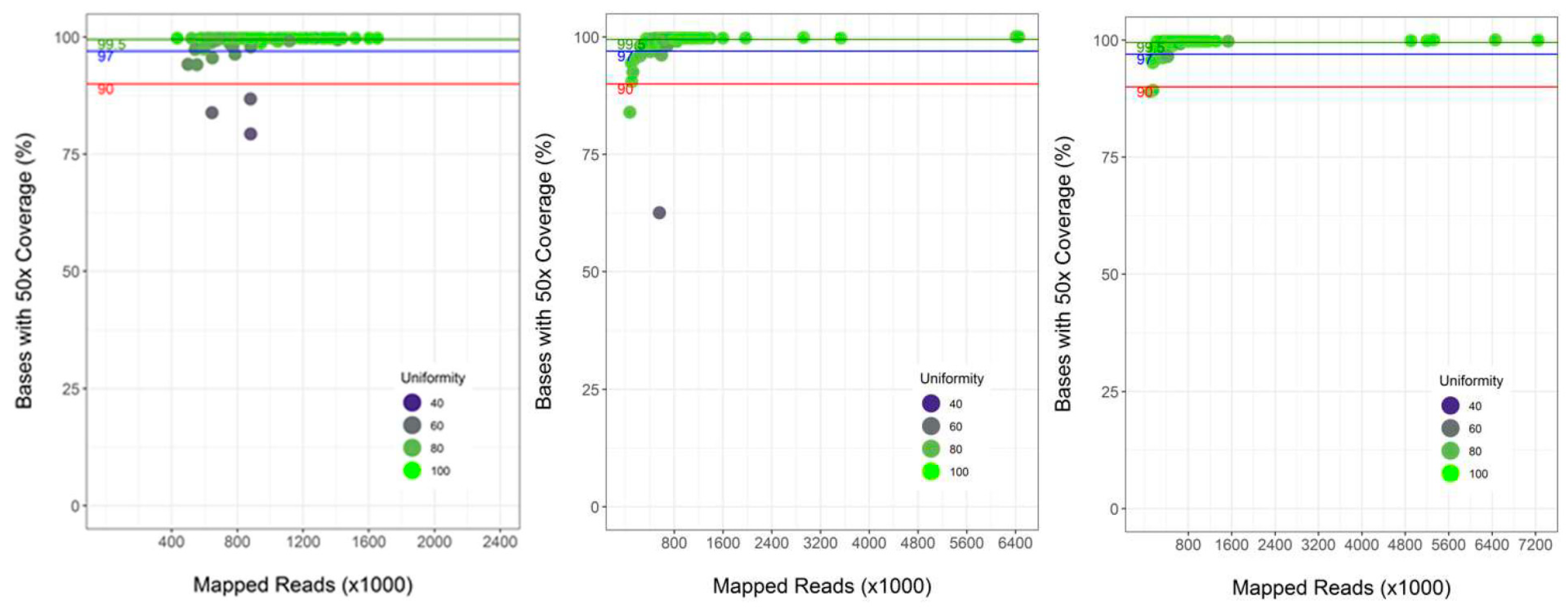
3.1. Validation of the Custom Targeted Ion AmpliSeq Panel for Use on DBS DNA
3.2. Performance of Semi-Automated Next Generation Sequencing from DBS
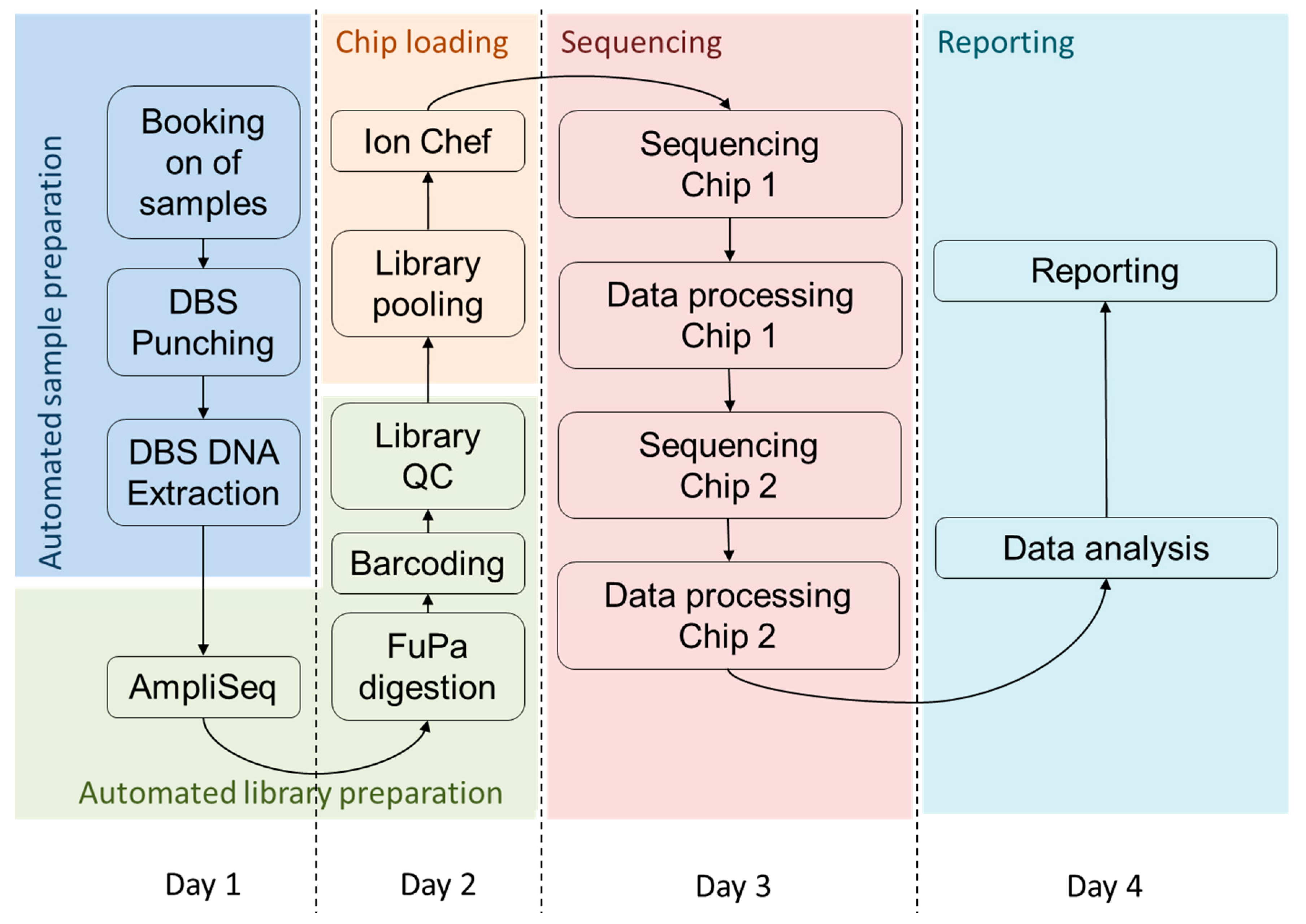
3.3. Timeline for High Throughput Sequencing from DBS
4. Discussion
4.1. Assay Performance
4.2. Turnaround Time of the Assay
4.3. Cost of High throughput NGS for NBS
4.4. Expansion of Newborn Screening through Targeted NGS as a First Line Test
5. Summary
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Almannai, M.; Marom, R.; Sutton, V.R. Newborn screening: A review of history, recent advancements, and future perspectives in the era of next generation sequencing. Curr. Opin. Pediatr. 2016, 28, 694–699. [Google Scholar] [CrossRef] [PubMed]
- Guthrie, R.; Susi, A. A Simple Phenylalanine Method for Detecting Phenylketonuria in Large Populations of Newborn Infants. Pediatrics 1963, 32, 338–343. [Google Scholar] [PubMed]
- Public Health England. A Laboratory Guide to Newborn Blood Spot Screening for Inherited Metabolic Diseases; PHE Publications: London, UK, 2017. [Google Scholar]
- Public Health England. A Laboratory Guide to Newborn Screening in the UK for Congenital Hypothyroidism; PHE Publications: London, UK, 2014. [Google Scholar]
- Public Health England. A Laboratory Guide to Newborn Screening in the UK for Cystic Fibrosis; PHE Publications: London, UK, 2014. [Google Scholar]
- Public Health England. NHS Sickle Cell and Thalassaemia Screening Programme, Handbook for Laboratories, 4th ed.; PHE Publications: London, UK, 2017. [Google Scholar]
- Bonham, J.R. Expanded newborn screening—Progress into practice. Clin. Biochem. 2014, 47, 697–698. [Google Scholar] [CrossRef] [PubMed]
- Chilcott, J.; Bessey, A.; Pandor, A.; Paisley, S. Expanded Newborn Screening for Inborn Errors of Metabolism. Health Econ. 2013, 111, 1399–1406. [Google Scholar]
- Barrett, A.N.; Saminathan, R.; Choolani, M. Thalassaemia screening and confirmation of carriers in parents. Best Pract. Res. Clin. Obstet. Gynaecol. 2017, 39, 27–40. [Google Scholar] [CrossRef]
- Smon, A.; Repic Lampret, B.; Groselj, U.; Zerjav Tansek, M.; Kovac, J.; Perko, D.; Bertok, S.; Battelino, T.; Trebusak Podkrajsek, K. Next generation sequencing as a follow-up test in an expanded newborn screening programme. Clin. Biochem. 2017, 52, 48–55. [Google Scholar] [CrossRef]
- Bhattacharjee, A.; Sokolsky, T.; Wyman, S.K.; Reese, M.G.; Puffenberger, E.; Strauss, K.; Morton, H.; Parad, R.B.; Naylor, E.W. Development of DNA confirmatory and high-risk diagnostic testing for newborns using targeted next-generation DNA sequencing. Genet. Med. 2015, 17, 337–347. [Google Scholar] [CrossRef]
- Hollegaard, M.V.; Grauholm, J.; Nielsen, R.; Grove, J.; Mandrup, S.; Hougaard, D.M. Archived neonatal dried blood spot samples can be used for accurate whole genome and exome-targeted next-generation sequencing. Mol. Genet. Metab. 2013, 110, 65–72. [Google Scholar] [CrossRef]
- Bodian, D.L.; Klein, E.; Iyer, R.K.; Wong, W.S.; Kothiyal, P.; Stauffer, D.; Huddleston, K.C.; Gaither, A.D.; Remsburg, I.; Khromykh, A.; et al. Utility of whole-genome sequencing for detection of newborn screening disorders in a population cohort of 1696 neonates. Genet. Med. 2016, 18, 221–230. [Google Scholar] [CrossRef]
- Waisbren, S.E.; Back, D.K.; Liu, C.; Kalia, S.S.; Ringer, S.A.; Holm, I.A.; Green, R.C. Parents are interested in newborn genomic testing during the early postpartum period. Genet. Med. 2015, 17, 501–504. [Google Scholar] [CrossRef]
- Dercksen, M.; Duran, M.; Ijlst, L.; Mienie, L.J.; Reinecke, C.J.; Ruiter, J.P.; Waterham, H.R.; Wanders, R.J. Clinical variability of isovaleric acidemia in a genetically homogeneous population. J. Inherit. Metab. Dis. 2012, 35, 1021–1029. [Google Scholar] [CrossRef] [PubMed]
- Barends, M.; Pitt, J.; Morrissy, S.; Tzanakos, N.; Boneh, A.; Newborn Screening Laboratory Staff. Biochemical and molecular characteristics of patients with organic acidaemias and urea cycle disorders identified through newborn screening. Mol. Genet. Metab. 2014, 113, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Grunert, S.C.; Wendel, U.; Lindner, M.; Leichsenring, M.; Schwab, K.O.; Vockley, J.; Lehnert, W.; Ensenauer, R. Clinical and neurocognitive outcome in symptomatic isovaleric acidemia. Orphanet J. Rare Dis. 2012, 7, 9. [Google Scholar] [CrossRef] [PubMed]
- Kolker, S.; Valayannopoulos, V.; Burlina, A.B.; Sykut-Cegielska, J.; Wijburg, F.A.; Teles, E.L.; Zeman, J.; Dionisi-Vici, C.; Baric, I.; Karall, D.; et al. The phenotypic spectrum of organic acidurias and urea cycle disorders. Part 2: The evolving clinical phenotype. J. Inherit. Metab. Dis. 2015, 38, 1059–1074. [Google Scholar] [CrossRef] [PubMed]
- Jacob, M.; Lopata, A.L.; Dasouki, M.; Abdel Rahman, A.M. Metabolomics toward personalized medicine. Mass Spectrom. Rev. 2019, 38, 221–238. [Google Scholar] [CrossRef]
- Olpin, S.E. Pathophysiology of fatty acid oxidation disorders and resultant phenotypic variability. J. Inherit. Metab. Dis. 2013, 36, 645–658. [Google Scholar] [CrossRef] [PubMed]
- Grunert, S.C. Clinical and genetical heterogeneity of late-onset multiple acyl-coenzyme A dehydrogenase deficiency. Orphanet J. Rare Dis. 2014, 9, 117. [Google Scholar] [CrossRef] [PubMed]
- Almannai, M.; Marom, R.; Divin, K.; Scaglia, F.; Sutton, V.R.; Craigen, W.J.; Lee, B.; Burrage, L.C.; Graham, B.H. Milder clinical and biochemical phenotypes associated with the c.482G > A(p.Arg161Gln) pathogenic variant in cobalamin C disease: Implications for management and screening. Mol. Genet. Metab. 2017, 122, 60–66. [Google Scholar] [CrossRef]
- Kang, E.; Kim, Y.M.; Kang, M.; Heo, S.H.; Kim, G.H.; Choi, I.H.; Choi, J.H.; Yoo, H.W.; Lee, B.H. Clinical and genetic characteristics of patients with fatty acid oxidation disorders identified by newborn screening. BMC Pediatr. 2018, 18, 103. [Google Scholar] [CrossRef]
- Landau, Y.E.; Waisbren, S.E.; Chan, L.M.; Levy, H.L. Long-term outcome of expanded newborn screening at Boston children’s hospital: Benefits and challenges in defining true disease. J. Inherit. Metab. Dis. 2017, 40, 209–218. [Google Scholar] [CrossRef]
- Ensenauer, R. A Common Mutation is Associated with a Mild, Potentially Asymptomatic Phenotype in Patients with Isovaleric Acidemia Diagnosed by Newborn Screening. Am. J. Hum. Genet. 2004, 75, 1136–1142. [Google Scholar] [CrossRef] [PubMed]
- Andresen, B.S.; Dobrowolski, S.F.; O’Reilly, L.; Muenzer, J.; McCandless, S.E.; Frazier, D.M.; Udvari, S.; Bross, P.; Knudsen, I.; Banas, R.; et al. Medium-chain acyl-CoA dehydrogenase (MCAD) mutations identified by MS/MS-based prospective screening of newborns differ from those observed in patients with clinical symptoms: Identification and characterization of a new, prevalent mutation that results in mild MCAD deficiency. Am. J. Hum. Genet. 2001, 68, 1408–1418. [Google Scholar] [CrossRef] [PubMed]
- Harutyunyan, M.; Huang, Y.; Mun, K.S.; Yang, F.; Arora, K.; Naren, A.P. Personalized Medicine in CF: From Modulator Development to Therapy for Cystic Fibrosis Patients with Rare CFTR Mutations. Am. J. Physiol. -Lung Cell. Mol. Physiol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Lucarelli, M.; Bruno, S.M.; Pierandrei, S.; Ferraguti, G.; Testino, G.; Truglio, G.; Strom, R.; Quattrucci, S. The Impact on Genetic Testing of Mutational Patterns of CFTR Gene in Different Clinical Macrocategories of Cystic Fibrosis. J. Mol. Diagn. 2016, 18, 554–565. [Google Scholar] [CrossRef][Green Version]
- Lucarelli, M.; Bruno, S.M.; Pierandrei, S.; Ferraguti, G.; Stamato, A.; Narzi, F.; Amato, A.; Cimino, G.; Bertasi, S.; Quattrucci, S.; et al. A Genotypic-Oriented View of CFTR Genetics Highlights Specific Mutational Patterns Underlying Clinical Macrocategories of Cystic Fibrosis. Mol. Med. 2015, 21, 257–275. [Google Scholar] [CrossRef]
- UK Department of Health. The UK Strategy for Rare Diseases; UK Department of Health: London, UK, 2013.
- Bouwman, M.G.; De Ru, M.H.; Linthorst, G.E.; Hollak, C.E.; Wijburg, F.A.; Van Zwieten, M.C. Fabry patients’ experiences with the timing of diagnosis relevant for the discussion on newborn screening. Mol. Genet. Metab. 2013, 109, 201–207. [Google Scholar] [CrossRef]
- De Ru, M.H.; Bouwman, M.G.; Wijburg, F.A.; Van Zwieten, M.C. Experiences of parents and patients with the timing of Mucopolysaccharidosis type I (MPS I) diagnoses and its relevance to the ethical debate on newborn screening. Mol. Genet. Metab. 2012, 107, 501–507. [Google Scholar] [CrossRef]
- Boycott, K.M.; Vanstone, M.R.; Bulman, D.E.; MacKenzie, A.E. Rare-disease genetics in the era of next-generation sequencing: Discovery to translation. Nat. Rev. Genet. 2013, 14, 681–691. [Google Scholar] [CrossRef]
- Schneller, J.L.; Lee, C.M.; Bao, G.; Venditti, C.P. Genome editing for inborn errors of metabolism: Advancing towards the clinic. BMC Med. 2017, 15, 43. [Google Scholar] [CrossRef]
- Schiroli, G.; Ferrari, S.; Conway, A.; Jacob, A.; Capo, V.; Albano, L.; Plati, T.; Castiello, M.C.; Sanvito, F.; Gennery, A.R.; et al. Preclinical modeling highlights the therapeutic potential of hematopoietic stem cell gene editing for correction of SCID-X1. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef]
- Diez, B.; Genovese, P.; Roman-Rodriguez, F.J.; Alvarez, L.; Schiroli, G.; Ugalde, L.; Rodriguez-Perales, S.; Sevilla, J.; Diaz de Heredia, C.; Holmes, M.C.; et al. Therapeutic gene editing in CD34(+) hematopoietic progenitors from Fanconi anemia patients. EMBO Mol. Med. 2017, 9, 1574–1588. [Google Scholar] [CrossRef] [PubMed]
- Naldini, L. Gene therapy returns to centre stage. Nature 2015, 526, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Aartsma-Rus, A.; Straub, V.; Hemmings, R.; Haas, M.; Schlosser-Weber, G.; Stoyanova-Beninska, V.; Mercuri, E.; Muntoni, F.; Sepodes, B.; Vroom, E.; et al. Development of Exon Skipping Therapies for Duchenne Muscular Dystrophy: A Critical Review and a Perspective on the Outstanding Issues. Nucleic Acid Ther. 2017, 27, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Bassaganyas, L.; Freedman, G.; Vaka, D.; Wan, E.; Lao, R.; Chen, F.; Kvale, M.; Currier, R.J.; Puck, J.M.; Kwok, P.Y. Whole exome and whole genome sequencing with dried blood spot DNA without whole genome amplification. Hum. Mutat. 2018, 39, 167–171. [Google Scholar] [CrossRef]
- Baudhuin, L.M.; Leduc, C.; Train, L.J.; Avula, R.; Kluge, M.L.; Kotzer, K.E.; Lin, P.T.; Ackerman, M.J.; Maleszewski, J.J. Technical Advances for the Clinical Genomic Evaluation of Sudden Cardiac Death: Verification of Next-Generation Sequencing Panels for Hereditary Cardiovascular Conditions Using Formalin-Fixed Paraffin-Embedded Tissues and Dried Blood Spots. Circ. Cardiovasc. Genet. 2017, 10, e001844. [Google Scholar] [CrossRef]
- Baker, M.W.; Atkins, A.E.; Cordovado, S.K.; Hendrix, M.; Earley, M.C.; Farrell, P.M. Improving newborn screening for cystic fibrosis using next-generation sequencing technology: A technical feasibility study. Genet. Med. 2016, 18, 231–238. [Google Scholar] [CrossRef]
- Rothberg, J.M.; Hinz, W.; Rearick, T.M.; Schultz, J.; Mileski, W.; Davey, M.; Leamon, J.H.; Johnson, K.; Milgrew, M.J.; Edwards, M.; et al. An integrated semiconductor device enabling non-optical genome sequencing. Nature 2011, 475, 348–352. [Google Scholar] [CrossRef]
- Cordovado, S.K. Dried Blood Spot DNA Extraction Guidelines to Ensure Robust Performance in NBS Molecular Assays. In Proceedings of the APHL Newborn Screening Symposium, Atlanta, GA, USA, 2–5 June 2013. [Google Scholar]
- Deans, Z.; Watson, C.; Charlton, R.; Ellard, S.; Wallis, Y.; Mattocks, C.; Abbs, S. Practice guidelines for Targeted Next Generation Sequencing Analysis and Interpretation; ACGS: London, UK, 2015. [Google Scholar]
- Fisher, T. Ion AmpliSeq Library Kit 2.0 User Guide. Available online: https://tools.thermofisher.com/content/sfs/manuals/MAN0006735_AmpliSeq_DNA_RNA_LibPrep_UG.pdf (accessed on 16 September 2019).
- Zook, J.M.; Chapman, B.; Wang, J.; Mittelman, D.; Hofmann, O.; Hide, W.; Salit, M. Integrating human sequence data sets provides a resource of benchmark SNP and indel genotype calls. Nat. Biotechnol. 2014, 32, 246–251. [Google Scholar] [CrossRef]
- Mu, J.C.; Tootoonchi Afshar, P.; Mohiyuddin, M.; Chen, X.; Li, J.; Bani Asadi, N.; Gerstein, M.B.; Wong, W.H.; Lam, H.Y. Leveraging long read sequencing from a single individual to provide a comprehensive resource for benchmarking variant calling methods. Sci. Rep. 2015, 5, 14493. [Google Scholar] [CrossRef]
- Tan, A.; Abecasis, G.R.; Kang, H.M. Unified representation of genetic variants. Bioinformatics 2015, 31, 2202–2204. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed]
- Tarasov, A.; Vilella, A.J.; Cuppen, E.; Nijman, I.J.; Prins, P. Sambamba: Fast processing of NGS alignment formats. Bioinformatics 2015, 31, 2032–2034. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Illumina. AmpliSeq for Illumina Targeted Resequencing Solution Data Sheet 770-2017-022-C. 2018. Available online: https://www.illumina.com/content/dam/illumina-marketing/documents/products/datasheets/ampliseq-for-illumina-targeted-resequencing-solution-data-sheet-770-2017-022.pdf (accessed on 3 October 2019).
- Tarabeux, J.; Zeitouni, B.; Moncoutier, V.; Tenreiro, H.; Abidallah, K.; Lair, S.; Legoix-Ne, P.; Leroy, Q.; Rouleau, E.; Golmard, L.; et al. Streamlined ion torrent PGM-based diagnostics: BRCA1 and BRCA2 genes as a model. Eur. J. Hum. Genet. 2014. [Google Scholar] [CrossRef] [PubMed]
- Belkadi, A.; Bolze, A.; Itan, Y.; Cobat, A.; Vincent, Q.B.; Antipenko, A.; Shang, L.; Boisson, B.; Casanova, J.L.; Abel, L. Whole-genome sequencing is more powerful than whole-exome sequencing for detecting exome variants. Proc. Natl. Acad. Sci. USA 2015, 112, 5473–5478. [Google Scholar] [CrossRef] [PubMed]
- Abel, H.J.; Duncavage, E.J. Detection of structural DNA variation from next generation sequencing data: A review of informatic approaches. Cancer Genet. 2013, 206, 432–440. [Google Scholar] [CrossRef]
- Mu, W.; Li, B.; Wu, S.; Chen, J.; Sain, D.; Xu, D.; Black, M.H.; Karam, R.; Gillespie, K.; Farwell Hagman, K.D.; et al. Detection of structural variation using target captured next-generation sequencing data for genetic diagnostic testing. Genet. Med. 2018. [Google Scholar] [CrossRef]
- Kerkhof, J.; Schenkel, L.C.; Reilly, J.; McRobbie, S.; Aref-Eshghi, E.; Stuart, A.; Rupar, C.A.; Adams, P.; Hegele, R.A.; Lin, H.; et al. Clinical Validation of Copy Number Variant Detection from Targeted Next-Generation Sequencing Panels. J. Mol. Diagn. 2017, 19, 905–920. [Google Scholar] [CrossRef]
- Feng, Y.; Ge, X.; Meng, L.; Scull, J.; Li, J.; Tian, X.; Zhang, T.; Jin, W.; Cheng, H.; Wang, X.; et al. The next generation of population-based spinal muscular atrophy carrier screening: Comprehensive pan-ethnic SMN1 copy-number and sequence variant analysis by massively parallel sequencing. Genet. Med. 2017, 19, 936–944. [Google Scholar] [CrossRef]
- Martinez-Pizarro, A.; Dembic, M.; Pérez, B.; Andresen, B.S.; Desviat, L.R. Intronic PAH gene mutations cause a splicing defect by a novel mechanism involving U1snRNP binding downstream of the 5′ splice site. PLoS Genet. 2018. [Google Scholar] [CrossRef]
- Bergougnoux, A.; Deletang, K.; Pommier, A.; Varilh, J.; Houriez, F.; Altieri, J.P.; Koenig, M.; Férec, C.; Claustres, M.; Lalau, G.; et al. Functional characterization and phenotypic spectrum of three recurrent disease-causing deep intronic variants of the CFTR gene. J. Cyst. Fibros. 2019, 18, 468–475. [Google Scholar] [CrossRef] [PubMed]
- Public Health England. Data Collection and Performance Analysis Report, Newborn Bloodspot Screening in the UK, 2016/17; PHE Publications: London, UK, 2018. [Google Scholar]
- Office of National Statistics. Birth Characteristics in England and Wales; ONS Publications: London, UK, 2016. [Google Scholar]
- Jansen, M.E.; Metternick-Jones, S.C.; Lister, K.J. International differences in the evaluation of conditions for newborn bloodspot screening: A review of scientific literature and policy documents. Eur. J. Hum. Genet. 2016, 25, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Mackie, A. UK National Screening Committee Criteria: Clarification of two misunderstandings. Eur. J. Hum. Genet. 2017, 25, 791. [Google Scholar] [CrossRef] [PubMed]
- Faulkner, A. Resisting the screening imperative: Patienthood, populations and politics in prostate cancer detection technologies for the UK. Sociol. Health Illn. 2012, 34, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Taylor-Phillips, S.; Boardman, F.; Seedat, F.; Hipwell, A.; Gale, N.K.; Clarke, A.; Slowther, A.; Sime, M.; Thomas, S.; Davis, H.; et al. The Ethical, Social and Legal Issues with Expanding the Newborn Blood Spot Test; The University of Warwick: Warwick, UK, 2014. [Google Scholar]
- Laberge, A.-M.; Burke, W. Avoiding the Technological Imperative: Criteria for Genetic Screening Programs. OBM Genet. 2017, 1. [Google Scholar] [CrossRef]
- Johnston, J.; Lantos, J.D.; Goldenberg, A.; Chen, F.; Parens, E.; Koenig, B.A.; Members of the NSIGHT Ethics and Policy Advisory Board. Sequencing Newborns: A Call for Nuanced Use of Genomic Technologies. Hastings Cent. Rep. 2018, 48, S2–S6. [Google Scholar] [CrossRef]
- Lim, E.C.; Brett, M.; Lai, A.H.; Lee, S.P.; Tan, E.S.; Jamuar, S.S.; Ng, I.S.; Tan, E.C. Next-generation sequencing using a pre-designed gene panel for the molecular diagnosis of congenital disorders in pediatric patients. Hum. Genom. 2015, 9, 33. [Google Scholar] [CrossRef]
- Green, R.C.; Berg, J.S.; Grody, W.W.; Kalia, S.S.; Korf, B.R.; Martin, C.L.; McGuire, A.L.; Nussbaum, R.L.; O’Daniel, J.M.; Ormond, K.E.; et al. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet. Med. 2013, 15, 565–574. [Google Scholar] [CrossRef]
- Abdul-Karim, R.; Berkman, B.E.; Wendler, D.; Rid, A.; Khan, J.; Badgett, T.; Hull, S.C. Disclosure of incidental findings from next-generation sequencing in pediatric genomic research. Pediatrics 2013, 131, 564–571. [Google Scholar] [CrossRef]
- Hasegawa, L.E.; Fergus, K.A.; Ojeda, N.; Au, S.M. Parental attitudes toward ethical and social issues surrounding the expansion of newborn screening using new technologies. Public Health Genom. 2011, 14, 298–306. [Google Scholar] [CrossRef]
- Waisbren, S.E.; Weipert, C.M.; Walsh, R.C.; Petty, C.R.; Green, R.C. Psychosocial Factors Influencing Parental Interest in Genomic Sequencing of Newborns. Pediatrics 2016, 137, S30–S35. [Google Scholar] [CrossRef] [PubMed]
- Ellard, S.; Baple, E.L.; Owens, M.; Eccles, D.M.; Abbs, S.; Deans, Z.C.; Newman, W.G.; McMullan, D.J. ACGS Best Practice Guidelines for Variant Classification; ACGS: London, UK, 2017. [Google Scholar]
- Yang, S.; Lincoln, S.E.; Kobayashi, Y.; Nykamp, K.; Nussbaum, R.L.; Topper, S. Sources of discordance among germ-line variant classifications in ClinVar. Genet. Med. 2017, 19, 1118–1126. [Google Scholar] [CrossRef] [PubMed]
- Whiffin, N.; Minikel, E.; Walsh, R.; O’Donnell-Luria, A.H.; Karczewski, K.; Ing, A.Y.; Barton, P.J.R.; Funke, B.; Cook, S.A.; MacArthur, D.; et al. Using high-resolution variant frequencies to empower clinical genome interpretation. Genet. Med. 2017, 19, 1151–1158. [Google Scholar] [CrossRef] [PubMed]
- Garbade, S.F.; Shen, N.; Himmelreich, N.; Haas, D.; Trefz, F.K.; Hoffmann, G.F.; Burgard, P.; Blau, N. Allelic phenotype values: A model for genotype-based phenotype prediction in phenylketonuria. Genet. Med. 2019, 21, 580–590. [Google Scholar] [CrossRef] [PubMed]
- Grauholm, J.; Khoo, S.K.; Nickolov, R.Z.; Poulsen, J.B.; Baekvad-Hansen, M.; Hansen, C.S.; Hougaard, D.M.; Hollegaard, M.V. Gene expression profiling of archived dried blood spot samples from the Danish Neonatal Screening Biobank. Mol. Genet. Metab. 2015, 116, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Gonorazky, H.D.; Naumenko, S.; Ramani, A.K.; Nelakuditi, V.; Mashouri, P.; Wang, P.; Kao, D.; Ohri, K.; Viththiyapaskaran, S.; Tarnopolsky, M.A.; et al. Expanding the Boundaries of RNA Sequencing as a Diagnostic Tool for Rare Mendelian Disease. Am. J. Hum. Genet. 2019, 104, 466–483. [Google Scholar] [CrossRef] [PubMed]
- Wai, H.; Douglas, A.G.L.; Baralle, D. RNA splicing analysis in genomic medicine. Int. J. Biochem. Cell Biol. 2019, 108, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Ferraguti, G.; Pierandrei, S.; Bruno, S.M.; Ceci, F.; Strom, R.; Lucarelli, M. A template for mutational data analysis of the CFTR gene. Clin. Chem. Lab. Med. 2011, 49, 1447–1451. [Google Scholar] [CrossRef][Green Version]
- Cutting, G.R. Cystic fibrosis genetics: From molecular understanding to clinical application. Nat. Rev. Genet. 2015, 16, 45–56. [Google Scholar] [CrossRef]
- Reinstein, E. Challenges of using next generation sequencing in newborn screening. Genet. Res. 2015, 97, e21. [Google Scholar] [CrossRef]
- Brookes, A.J.; Robinson, P.N. Human genotype-phenotype databases: Aims, challenges and opportunities. Nat. Rev. Genet. 2015, 16, 702–715. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.M.G.; Jungner, G. Principles and Practice of Screening for Disease; WHO: Geneva, Switzerland, 1968. [Google Scholar]
- Richer, J.; Laberge, A.M. Secondary findings from next-generation sequencing: What does actionable in childhood really mean? Genet. Med. 2019, 21, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Dabrowski, M.; Bukowy-Bieryllo, Z.; Zietkiewicz, E. Advances in therapeutic use of a drug-stimulated translational readthrough of premature termination codons. Mol. Med. 2018, 24, 25. [Google Scholar] [CrossRef] [PubMed]
- Welch, E.M.; Barton, E.R.; Zhuo, J.; Tomizawa, Y.; Friesen, W.J.; Trifillis, P.; Paushkin, S.; Patel, M.; Trotta, C.R.; Hwang, S.; et al. PTC124 targets genetic disorders caused by nonsense mutations. Nature 2007, 447, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Brentnall, A.R.; Evans, D.G.; Cuzick, J. Distribution of breast cancer risk from SNPs and classical risk factors in women of routine screening age in the UK. Br. J. Cancer 2014, 110, 827–828. [Google Scholar] [CrossRef]
- Zhang, L.; Bao, Y.; Riaz, M.; Tiller, J.; Liew, D.; Zhuang, X.; Amor, D.J.; Huq, A.; Petelin, L.; Nelson, M.; et al. Population genomic screening of all young adults in a health-care system: A cost-effectiveness analysis. Genet. Med. 2019. [Google Scholar] [CrossRef]
- Schuurmans, J.; Birnie, E.; Van den Heuvel, L.M.; Plantinga, M.; Lucassen, A.; Van der Kolk, D.M.; Abbott, K.M.; Ranchor, A.V.; Diemers, A.D.; Van Langen, I.M. Feasibility of couple-based expanded carrier screening offered by general practitioners. Eur. J. Hum. Genet. 2019. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
van Campen, J.C.; Sollars, E.S.A.; Thomas, R.C.; Bartlett, C.M.; Milano, A.; Parker, M.D.; Dawe, J.; Winship, P.R.; Peck, G.; Grafham, D.; et al. Next Generation Sequencing in Newborn Screening in the United Kingdom National Health Service. Int. J. Neonatal Screen. 2019, 5, 40. https://doi.org/10.3390/ijns5040040
van Campen JC, Sollars ESA, Thomas RC, Bartlett CM, Milano A, Parker MD, Dawe J, Winship PR, Peck G, Grafham D, et al. Next Generation Sequencing in Newborn Screening in the United Kingdom National Health Service. International Journal of Neonatal Screening. 2019; 5(4):40. https://doi.org/10.3390/ijns5040040
Chicago/Turabian Stylevan Campen, Julia C., Elizabeth S. A. Sollars, Rebecca C. Thomas, Clare M. Bartlett, Antonio Milano, Matthew D. Parker, Jennifer Dawe, Peter R. Winship, Gerrard Peck, Darren Grafham, and et al. 2019. "Next Generation Sequencing in Newborn Screening in the United Kingdom National Health Service" International Journal of Neonatal Screening 5, no. 4: 40. https://doi.org/10.3390/ijns5040040
APA Stylevan Campen, J. C., Sollars, E. S. A., Thomas, R. C., Bartlett, C. M., Milano, A., Parker, M. D., Dawe, J., Winship, P. R., Peck, G., Grafham, D., Kirk, R. J., Bonham, J. R., Goodeve, A. C., & Dalton, A. (2019). Next Generation Sequencing in Newborn Screening in the United Kingdom National Health Service. International Journal of Neonatal Screening, 5(4), 40. https://doi.org/10.3390/ijns5040040