Duchenne Muscular Dystrophy Newborn Screening: Evaluation of a New GSP® Neonatal Creatine Kinase-MM Kit in a US and Danish Population

 , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Populations: California and Denmark
- Location 1: PerkinElmer, Wallac Oy in Finland using anonymous DBS samples from male and female newborns born in California, USA and obtained from the California Biobank Program;
- Location 2: Danish Center for Neonatal Screening, Statens Serum Institut (SSI), in Copenhagen, Denmark using anonymized DBS specimens obtained from Danish Neonatal Screening Biobank (DNSB or DNS-Biobank).
2.2. Study Design
2.3. Bloodspot CK-MM Concentration Analysis
2.4. Bloodspot CK Activity Analysis
2.5. Calculations and Statistical Analysis
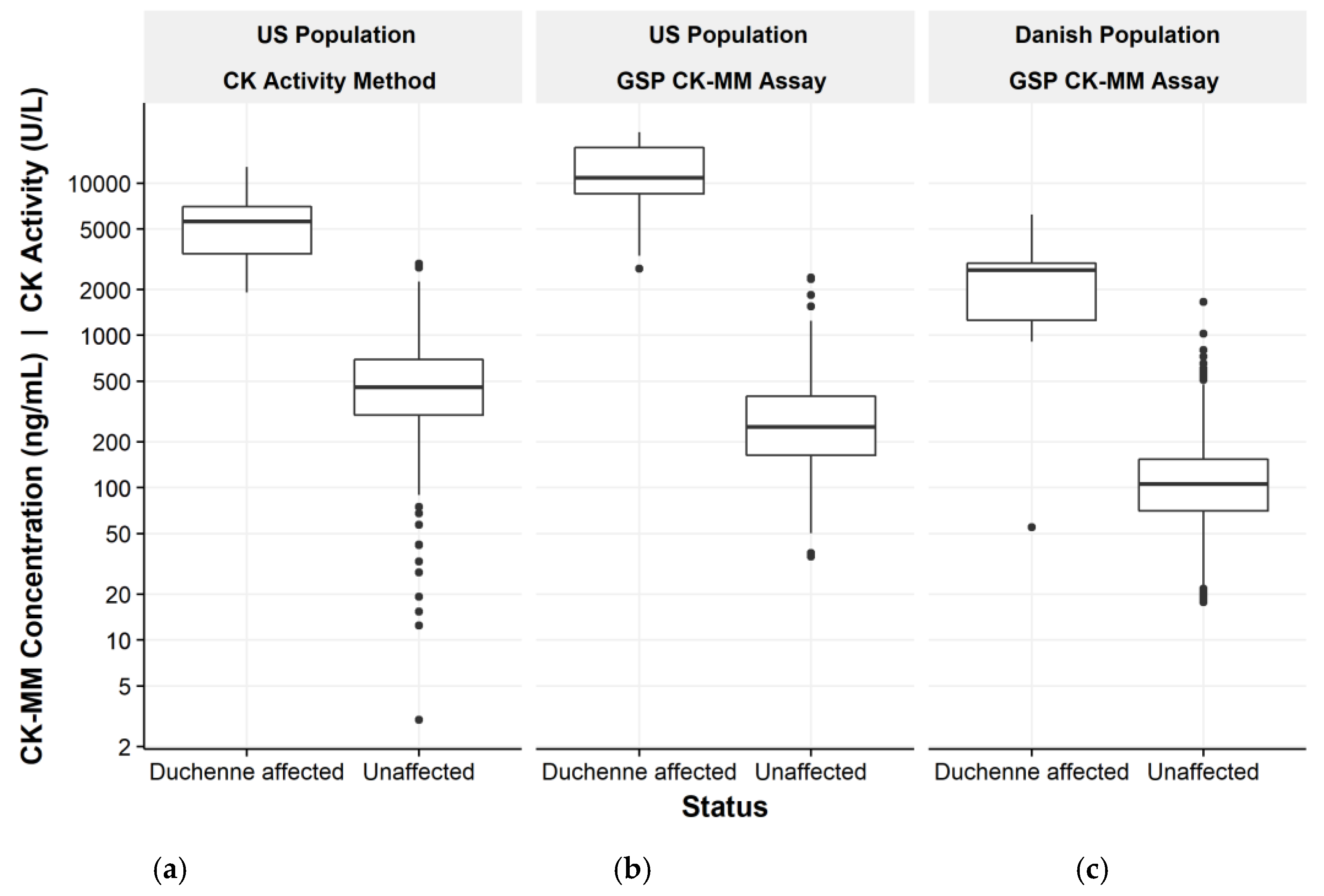
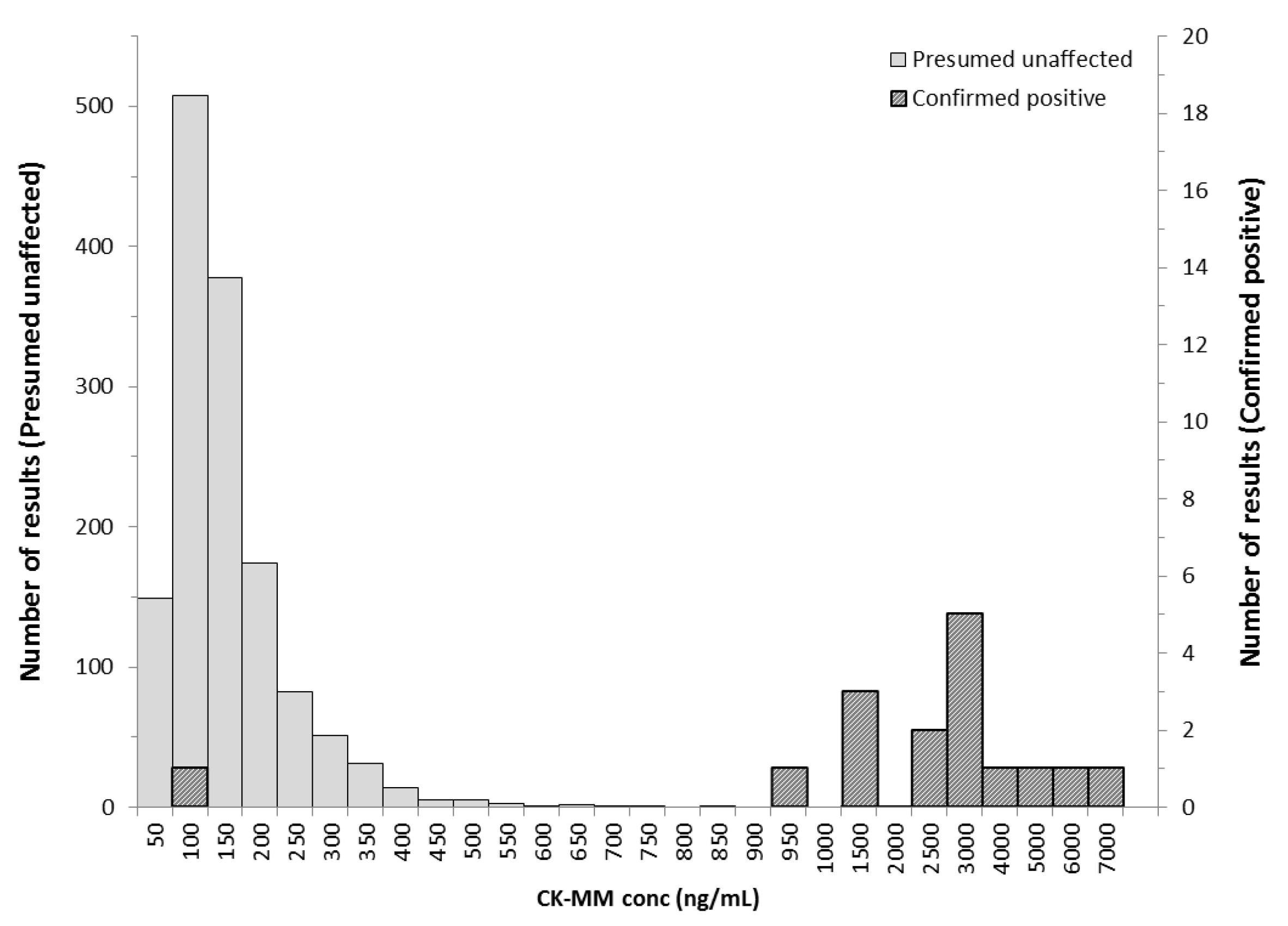
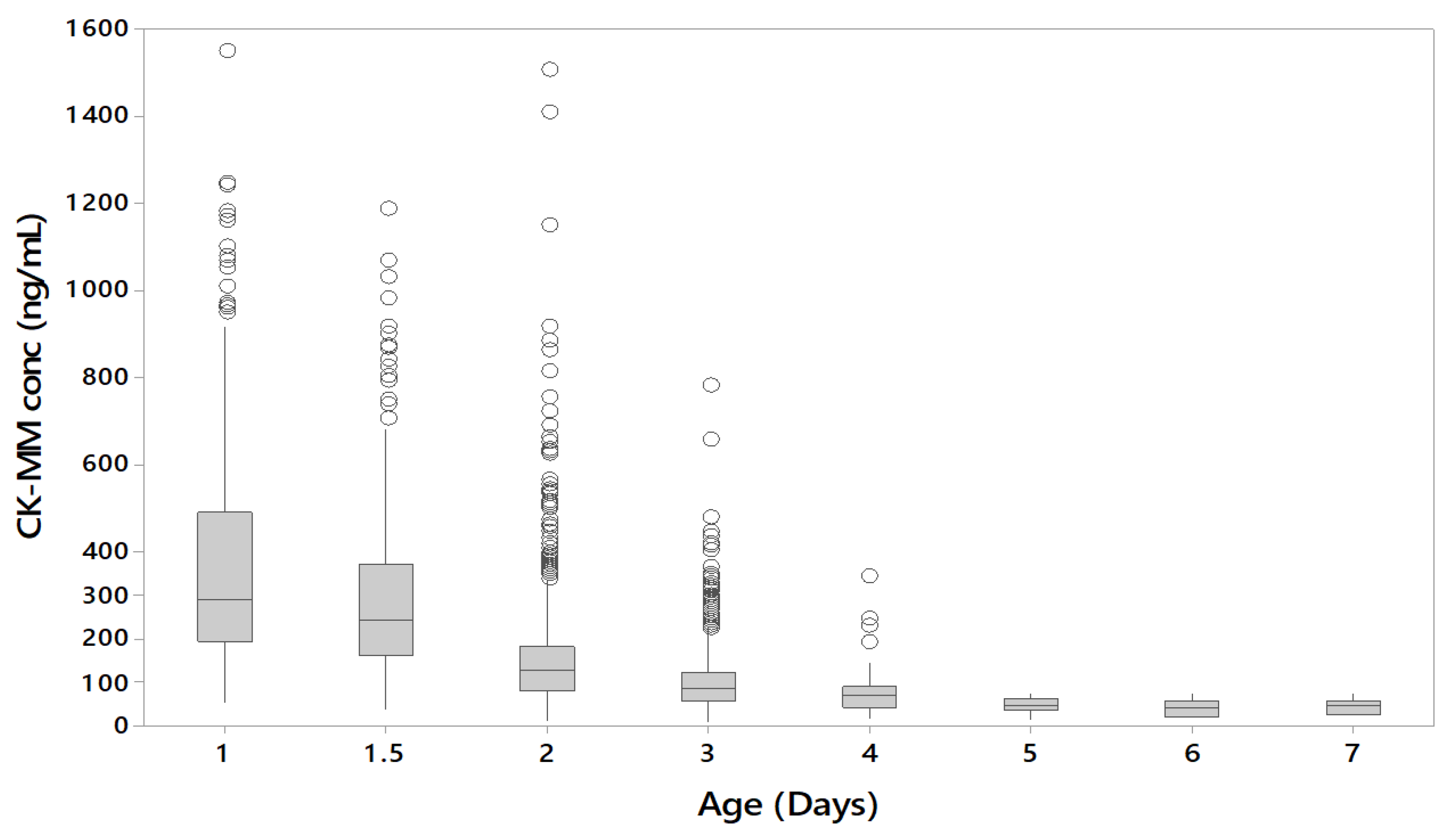
3. Results
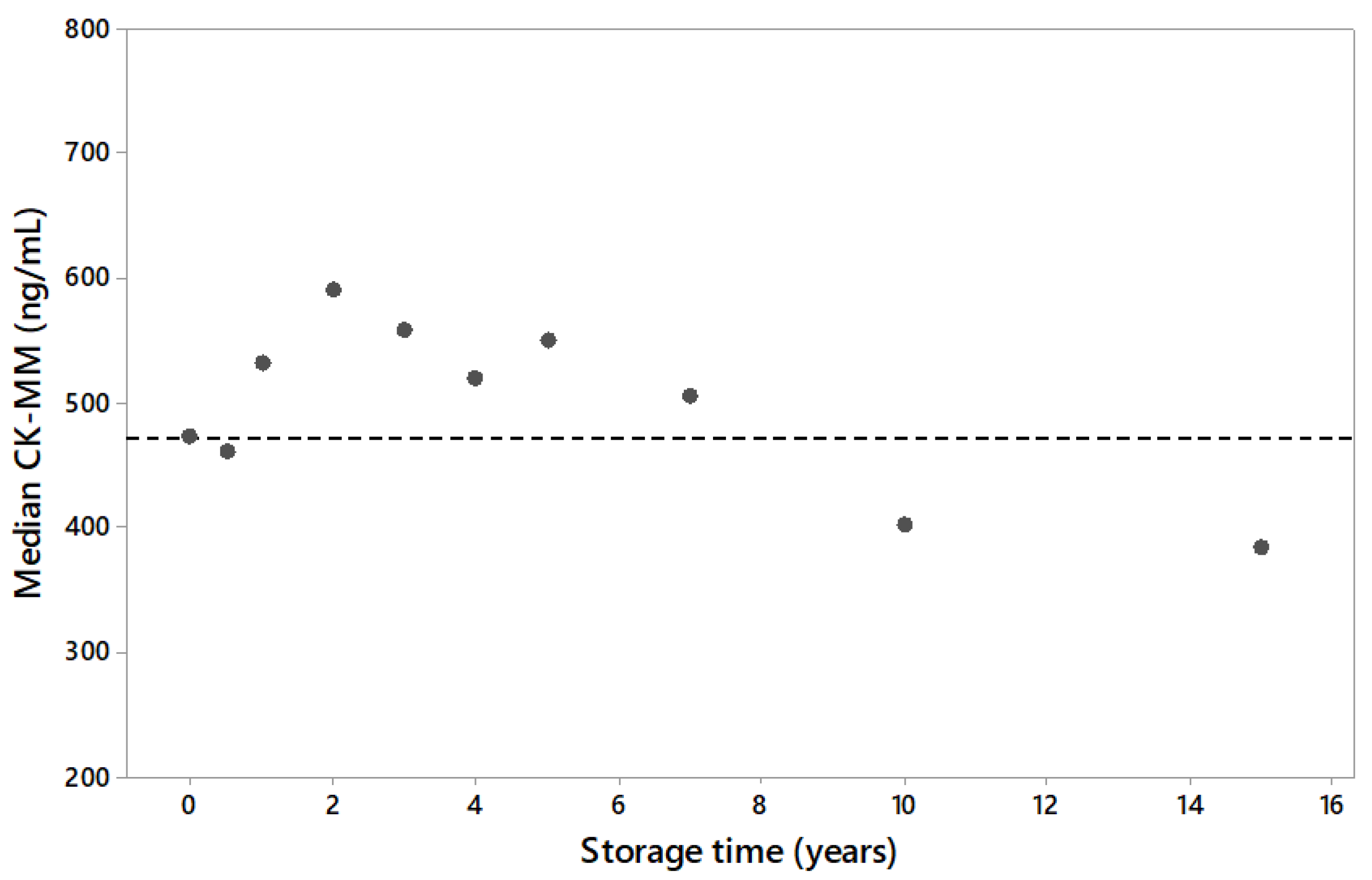
3.1. US Population and Long Term Sample Stability
3.2. Danish Population
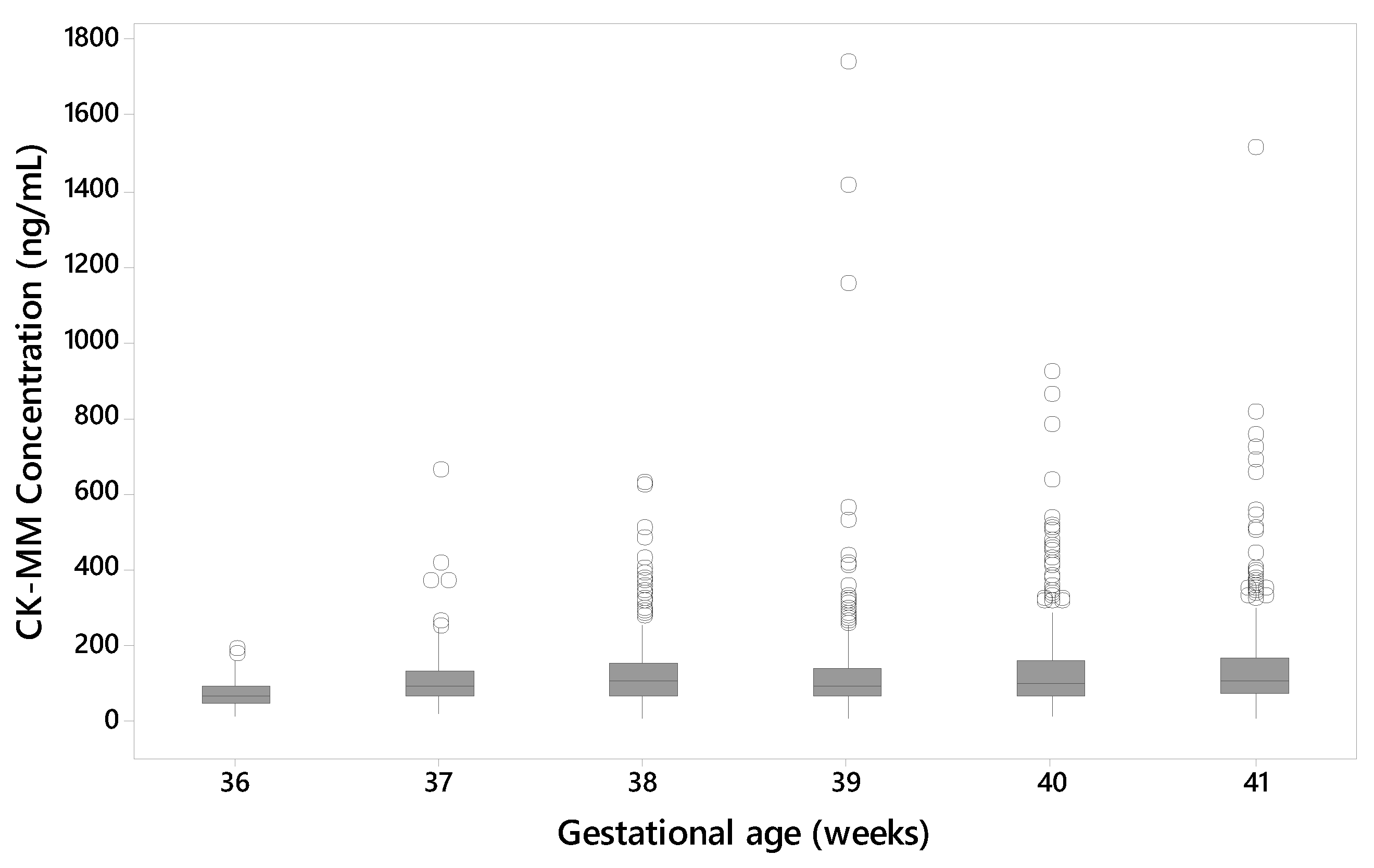
3.3. Affect of the Age and Gestational Age of the Newborn
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mendell, J.R.; Shilling, C.; Leslie, N.D.; Flanigan, K.M.; Al-Dahhak, R.; Gastier-Foster, J.; Kneile, K.; Dunn, D.M.; Duval, B.; Aoyagi, A.; et al. Evidence-based path to newborn screening for Duchenne muscular dystrophy. Ann. Neurol. 2012, 71, 304–313. [Google Scholar] [CrossRef] [PubMed]
- Emery, A.E. Population frequencies of inherited neuromuscular diseases—A world survey. Neuromuscul. Disord. 1991, 1, 19–29. [Google Scholar] [CrossRef]
- Hoffman, E.P.; Brown, R.H.; Kunkel, L.M. Dystrophin: The protein product of the Duchenne muscular dystrophy locus. Cell 1987, 51, 919–928. [Google Scholar] [CrossRef]
- Bushby, K.M.; Hill, A.; Steele, J.G. Failure of early diagnosis in symptomatic Duchenne muscular dystrophy. Lancet 1999, 353, 55–78. [Google Scholar] [CrossRef]
- Ciafaloni, E.; Fox, D.J.; Pandya, S.; Westfield, C.P.; Puzhankara, S.; Romitti, P.A.; Mathews, K.D.; Miller, T.M.; Matthews, D.J.; Miller, L.A.; et al. Delayed Diagnosis in Duchenne muscular dystrophy: Data from the Muscular Dystrophy Surveillance, Tracking, Tracking and Research Network (MD STARnet). J. Pediatr. 2009, 155, 380–385. [Google Scholar] [CrossRef] [PubMed]
- Brooke, M.H.; Fenichel, G.M.; Griggs, R.C.; Mendell, J.R.; Moxley, R.; Florence, J.; King, W.M.; Pandya, S.; Robison, J.; Schierbecker, J.; et al. Duchenne muscular dystrophy—Patterns of clinical progression and effects of supportive therapy. Neurology 1989, 39, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Birnkrant, D.J.; Bushby, K.; Bann, C.M.; Apkon, S.D.; Blackwell, A.; Brumbaugh, D.; Case, L.E.; Clemens, P.R.; Hadjiyannakis, S.; Pandya, S.; et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: Diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol. 2018, 17, 251–267. [Google Scholar] [CrossRef]
- Okinaka, S.; Kumagai, H.; Ebashi, S.; Sugita, H.; Momoi, H.; Toyokura, Y.; Fujie, Y. Serum Creatine Phosphokinase Activity in Progressive Muscular Dystrophy and Neuromuscular Diseases. Arch. Neurol. 1961, 4, 520–525. [Google Scholar] [CrossRef]
- Rudolph, N.; Gross, R.T. Creatine phosphokinase activity in serum of newborn infants as an indicator of fetal trauma during birth. Pediatrics 1966, 38, 1044–1045. [Google Scholar] [CrossRef]
- Bradley, D.; Parsons, E. Newborn screening for Duchenne muscular dystrophy. Semin. Fetal Neonatal Med. 1998, 3, 27–34. [Google Scholar] [CrossRef]
- Moat, S.J.; Korpimäki, T.; Furu, P.; Hakala, H.; Polari, H.; Meriö, L.; Mäkinen, P.; Weeks, I. Characterization of a blood spot creatine kinase skeletal muscle isoform immunoassay for high-throughput newborn screening of Duchenne muscular dystrophy. Clin. Chem. 2017, 63, 908–914. [Google Scholar] [CrossRef]
- Parent Project Muscular Dystrophy: Therapeutic Approaches. Available online: https://www.parentprojectmd.org/research/current-research/therapeutic-approaches/ (accessed on 25 February 2019).
- Drachman, D.B.; Toyka, K.V.; Myer, E. Prednisone in Duchenne muscular dystrophy. Lancet 1974, 14, 1409–1412. [Google Scholar] [CrossRef]
- Matthews, E.; Brassington, R.; Kuntzer, T.; Jichi, F.; Manzur, A.Y. Corticosteroids for the treatment of Duchenne muscular dystrophy (Review) Summary of Finding for the Main Comparison. Cochrane Database Syst. Rev. 2016. [Google Scholar] [CrossRef]
- Connolly, A.M.; Zaidman, C.M.; Golumbek, P.T.; Cradock, M.M.; Flanigan, K.M.; Kuntz, N.L.; Finkel, R.S.; McDonald, C.M.; Iannaccone, S.T.; Anand, P.; et al. Twice-weekly glucocorticosteroids in infants and young boys with Duchenne muscular dystrophy. Muscle Nerve 2019. [Google Scholar] [CrossRef]
- Lim, K.R.Q.; Maruyama, R.; Yokota, T. Eteplirsen in the treatment of Duchenne muscular dystrophy. Drug Des. Dev. Ther. 2017, 11, 533–545. [Google Scholar] [CrossRef]
- Zellweger, H.; Antonik, A. Newborn Screening for Duchenne Muscular Dystrophy. Pediatrics 1975, 55, 30–34. [Google Scholar] [CrossRef][Green Version]
- Orfanos, A.P.; Naylor, E.W. A rapid screening test for Duchenne muscular dystrophy using dried blood specimens. Clinica Chimica Acta 1984, 138, 267–274. [Google Scholar] [CrossRef]
- Moat, S.J.; Bradley, D.M.; Salmon, R.; Clarke, A.; Hartley, L. Newborn bloodspot screening for Duchenne muscular dystrophy: 21 years experience in Wales (UK). Eur. J. Hum. Genet. 2013, 21, 1049–1053. [Google Scholar] [CrossRef]
- Moat, S.J. (Wales Newborn Screening Laboratory, University Hospital of Wales, Cardiff, UK). Personal communication, 2019.
- Arenas, J.; Diaz, V.; Liras, G.; Gutierrez, E.; Santos, I.; Martinez, A.; Culebras, J.M. Activities of creatine kinase and its isoenzymes in serum in various skeletal muscle disorders. Clin. Chem. 1988, 34, 2460–2462. [Google Scholar]
- Edwards, R.J.; Watts, D.C.; Watts, R.L. Creatine kinase estimation in pure fetal blood samples for the prenatal diagnosis of Duchenne muscular dystrophy. Prenat. Diagn. 1984, 4, 267–277. [Google Scholar] [CrossRef]
- CLSI. Newborn Screening for Preterm, Low Birth Weight, and Sick Newborns; Approved Guideline; CLSI Document NBS03-A; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2009. [Google Scholar]
- Mercier, S.; Toutain, A.; Toussaint, A.; Raynaud, M.; de Barace, C.; Marcorelles, P.; Pasquier, L.; Blayau, M.; Espil, C.; Parent, P.; et al. Genetic and clinical specificity of 26 symptomatic carriers for dystrophinopathies at pediatric age. Eur. J. Hum. Genet. 2013, 21, 855–863. [Google Scholar] [CrossRef]
- Gatheridge, M.A.; Kwon, J.M.; Mendell, J.M.; Scheuerbrandt, G.; Moat, S.J.; Eyskens, F.; Rockman-Greenberg, C.; Drousiotou, A.; Griggs, R.C. Identifying Non–Duchenne Muscular Dystrophy–Positive and False Negative Results in Prior Duchenne Muscular Dystrophy Newborn Screening Programs—A Review. JAMA Neurol. 2016, 73, 111–116. [Google Scholar] [CrossRef]
- Duchenne and Becker Muscular Dystrophy. Genetics Home Reference (GHR). Available online: https://ghr.nlm.nih.gov/condition/Duchenne-and-Becker-muscular-dystrophy (accessed on 7 March 2019).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control | n | GSP CK-MM Assay | CK Activity Method | ||
|---|---|---|---|---|---|
| Mean (ng/mL) | CV% | Mean (U/L) | CV% | ||
| QC1 | 22 | 119.1 | 7 | 463.4 | 17 |
| QC2 | 22 | 417.2 | 7 | 685.9 | 14 |
| QC3 | 22 | 1863.2 | 8 | 1845.2 | 9 |
| Sample | GSP CK-MM Assay (ng/mL) | CK Activity Method (U/L) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| n | Min | Max | Mean | 95th | 99th | Min | Max | Mean | 95th | 99th | |
| Duchenne affected | 19 | 2750 | 21,600 | 12,144 | 1910 | 12,800 | 5822 | ||||
| Unaffected | 700 | 35.2 | 2390 | 328 | 867 | 1190 | 0 | 2950 | 547 | 1300 | 1980 |
| n | Mean | Median | Min | Max | 95th | 99th | 99th |
|---|---|---|---|---|---|---|---|
| 2099 | 124 | 96.6 | <6.8 | 1740 | 291 | 513 | 675 |
| Sample | n | Mean | Median | Min | Max |
|---|---|---|---|---|---|
| Duchenne affected | 16 | 2626 | 2685 | 909 1 | 6230 |
| Unaffected | 1408 | 128 | 106 | 17.7 | 1660 |
| Screening Result | Clinical Status | Total | |
|---|---|---|---|
| Duchenne Affected | Unaffected | ||
| Screening positive | 15 | 4 | 19 |
| Screening negative | 1 1 | 1404 | 1405 |
| TOTAL | 16 | 1408 | 1424 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Timonen, A.; Lloyd-Puryear, M.; Hougaard, D.M.; Meriö, L.; Mäkinen, P.; Laitala, V.; Pölönen, T.; Skogstrand, K.; Kennedy, A.; Airenne, S.; et al. Duchenne Muscular Dystrophy Newborn Screening: Evaluation of a New GSP® Neonatal Creatine Kinase-MM Kit in a US and Danish Population. Int. J. Neonatal Screen. 2019, 5, 27. https://doi.org/10.3390/ijns5030027
Timonen A, Lloyd-Puryear M, Hougaard DM, Meriö L, Mäkinen P, Laitala V, Pölönen T, Skogstrand K, Kennedy A, Airenne S, et al. Duchenne Muscular Dystrophy Newborn Screening: Evaluation of a New GSP® Neonatal Creatine Kinase-MM Kit in a US and Danish Population. International Journal of Neonatal Screening. 2019; 5(3):27. https://doi.org/10.3390/ijns5030027
Chicago/Turabian StyleTimonen, Anne, Michele Lloyd-Puryear, David M. Hougaard, Liisa Meriö, Pauliina Mäkinen, Ville Laitala, Tuukka Pölönen, Kristin Skogstrand, Annie Kennedy, Sari Airenne, and et al. 2019. "Duchenne Muscular Dystrophy Newborn Screening: Evaluation of a New GSP® Neonatal Creatine Kinase-MM Kit in a US and Danish Population" International Journal of Neonatal Screening 5, no. 3: 27. https://doi.org/10.3390/ijns5030027
APA StyleTimonen, A., Lloyd-Puryear, M., Hougaard, D. M., Meriö, L., Mäkinen, P., Laitala, V., Pölönen, T., Skogstrand, K., Kennedy, A., Airenne, S., Polari, H., & Korpimäki, T. (2019). Duchenne Muscular Dystrophy Newborn Screening: Evaluation of a New GSP® Neonatal Creatine Kinase-MM Kit in a US and Danish Population. International Journal of Neonatal Screening, 5(3), 27. https://doi.org/10.3390/ijns5030027