Expanded Newborn Screening Using Tandem Mass Spectrometry: Seven Years of Experience in Eastern Sicily

 , ,
, ,
Abstract
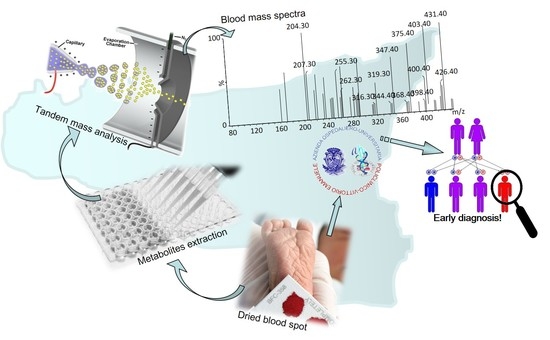
:
1. Introduction
Newborn Screening in Sicily
2. Materials and Methods
- In case of disorders at risk of metabolic decompensation during the neonatal period, a metabolic specialist immediately recalls the baby, and clinical examinations and confirmatory tests were performed.
- In case of all other disorders, the nursery was contacted to provide for a second DBS. If the repeated test showed a positive result, clinical examinations and confirmatory tests were performed.
3. Results
3.1. Amino Acid Disorders (AA)
3.2. Urea Cycle Disorders (UCD)
3.3. Mitochondrial Fatty Acidbeta-Oxidation Defects (FAO)
3.4. Organic Acidemias (OA)
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Naylor, E.W.; Chace, D.H. Automated tandem mass spectrometry for mass newborn screening for disorders in fatty acid, organic acid, and amino acid metabolism. J. Child Neurol. 1999, 14 (Suppl. S1), 4–8. [Google Scholar] [CrossRef] [PubMed]
- Wilcken, B.; Wiley, V.; Hammond, J.; Carpenter, C. Screening newborns for inborn errors of metabolism by tandem mass spectrometry. N. Engl. J. Med. 2003, 348, 2304–2312. [Google Scholar] [CrossRef] [PubMed]
- Schulze, A.; Lindner, M.; Kohlmuller, D.; Olgemoller, K.; Mayatepek, E.; Hoffman, G.F. Expanded newborn screening for inborn errors of metabolism by electrospray ionization-tandem mass spectrometry: Results, outcome, and implications. Pediatrics 2003, 111, 1399–1406. [Google Scholar] [CrossRef] [PubMed]
- Chace, D.H.; Kalas, T.A.; Naylor, E.W. Use of tandem mass spectrometry for multianalyte screening of dried blood specimens from newborns. Clin. Chem. 2003, 49 (Suppl. S11), 1797–1817. [Google Scholar] [CrossRef] [PubMed]
- Mak, C.M.; Lee, H.C.; Chan, A.Y.; Lam, C.W. Inborn errors of metabolism and expanded newborn screening: Review and update. Crit. Rev. Clin. Lab. Sci. 2013, 50 (Suppl. S6), 142–162. [Google Scholar] [CrossRef] [PubMed]
- Khoury, M.J.; McCabe, L.L.; McCabe, E.R. Population screening in the age of genomic medicine. N. Engl. J. Med. 2003, 348 (Suppl. S1), 50–58. [Google Scholar] [CrossRef] [PubMed]
- Pollitt, R.J. International perspectives on newborn screening. J. Inherit. Metab. Dis. 2006, 29, 390–396. [Google Scholar] [CrossRef] [PubMed]
- Wilcken, B. Newborn screening: How are we travelling, and where should we be going? J. Inherit. Metab. Dis. 2011, 34, 569–574. [Google Scholar] [CrossRef] [PubMed]
- Società Italiana per lo studio delle Malattie Metaboliche Ereditarie e lo Screening Neonatale (SIMMESN). Available online: https://www.simmesn.it/images/documents/glexpnbs2008.pdf (accessed on 30 July 2008).
- Alfadhel, M.; Benmeakel, M.; Hossain, M.A.; Al Mutairi, F.; Al Othaim, A.; Alfares, A.A.; Al Balwi, M.; Alzaben, A.; Eyaid, W. Thirteen year retrospective review of the spectrum of inborn errors of metabolism presenting in a tertiary center in Saudi. Orphanet J. Rare Dis. 2016, 11, 126. [Google Scholar] [CrossRef] [PubMed]
- Shrazi, N.A.; Khan, A.H.; Jafri, L.; Khan, N.A.; Afroze, B. Selective Screening for Organic Acidurias and Amino Acidopathies in Pakistani Children. J. Coll. Phys. Surg. Pak. 2017, 27 (Suppl. S4), 218–221. [Google Scholar]
- Najafi, R.; Hashemipour, M.; Mostofizadeh, N.; Ghazavi, M.; Nasiri, J.; Shahsanai, A.; Famori, F.; Najafi, F.; Moafi, F. Demographic and Clinical Findings in Pediatric Patients Affected By Organic Acidemia. Iran. J. Child Neurol. 2016, 10 (Suppl. S2), 74–81. [Google Scholar] [PubMed]
- Meldgaard Lund, A.; Hougaard, D.M.; Simonsen, H.; Andresen, B.S.; Christensen, M.; Dunø, M.; Skogstrand, K.; Olsen, R.K.; Jensen, U.G.; Cohen, A.; et al. Biochemical screening of 504,049 newborns in Denmark, the Faroe Islands and Greenland—Experience and development of a routine program for expanded newborn screening. Mol. Genet. Metab. 2012, 107, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Santer, R.; Fingerhut, R.; Lassker, U.; Wightman, P.J.; Fitzpatrick, D.R.; Olgemöller, B.; Roscher, A.A. Tandem mass Spectrometric Determination of malonyl-carnitine: Diagnosis and neonatal screening of malonyl-CoA decarboxylase deficiency. Clin. Chem. 2003, 49 (Suppl. S4), 660–662. [Google Scholar] [CrossRef] [PubMed]
- Salomons, G.S.; Jakobs, C.; Landegge Pope, L.; Errami, A.; Potter, M.; Nowaczyk, M.; Olpin, S.; Manning, N.; Raiman, J.A.; Slade, T.; et al. Clinical, enzymatic and molecular characterization of nine new patients with malonyl-coenzymeA decarboxylase deficiency. J. Inherit. Metab. Dis. 2007, 30, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Baertling, F.; Mayatepek, E.; Thimm, E.; Schlune, A.; Kovacevic, A.; Distelmaier, F.; Salomons, G.S.; Meissner, T. Malonic Aciduria: Long-term follow-up of new patients detected by newborn screening. Eur. J. Pediatr. 2014, 173, 1719–1722. [Google Scholar] [CrossRef] [PubMed]
- La Marca, G.; Malvagia, S.; Pasquini, E.; Innocenti, M.; Donati, M.A.; Zammarchi, E. Rapid 2nd-tier test for measurement of 3-OH-propionic and methylmalonic acids on dried blood spots: Reducing the false-positive rate for propionylcarnitine during expanded newborn screening by liquid chromatography—tandem mass spectrometry. Clin. Chem. 2007, 53 (Suppl. S7), 1364–1369. [Google Scholar] [CrossRef] [PubMed]
- Shigematsu, Y.; Hata, I.; Tajima, G. Useful second-tier tests in expanded newborn screening of isovaleric acidemia and methylmalonic aciduria. J. Inherit. Metab. Dis. 2010, 33 (Suppl. S2), 283–288. [Google Scholar] [CrossRef] [PubMed]
- La Marca, G.; Malvagia, S.; Casetta, B.; Pasquini, E.; Donati, M.A.; Zammarchi, E. Progress in expanded newborn screening for metabolic conditions by LC-MS/MS in Tuscany: Update on methods to reduce false tests. J. Inherit. Metab Dis 2008, 31 (Suppl. S2), 395–404. [Google Scholar] [CrossRef] [PubMed]
- Santagata, S.; Di Carlo, E.; Carducci, C.; Leuzzi, V.; Angeloni, A.; Carducci, C. Development of a new UPLC-ESI-MS/MS method for the determination of biopterin and neopterin in dried blood spot. Clin. Chim. Acta 2017, 466, 145–151. [Google Scholar] [CrossRef] [PubMed]
- La Marca, G.; Giocaliere, E.; Malvagia, S.; Funghini, S.; Ombrone, D.; Della Bona, M.L.; Canessa, C.; Lippi, F.; Romano, F.; Guerrini, R.; et al. The inclusion of ADA-SCID in expanded newborn screening by tandem mass spectrometry. J. Pharm. Biomed. Anal. 2014, 88, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Elliott, S.; Buroker, N.; Cournoyer, J.J.; Potier, A.M.; Trometer, J.D.; Elbin, C.; Schermer, M.J.; Kantola, J.; Boyce, A.; Turecek, F.; et al. Dataset and standard operating procedure for newborn screening of six lysosomal storage diseases: By tandem mass spectrometry. Data Brief 2016, 8, 915–924. [Google Scholar] [CrossRef] [PubMed]
- Lisi, E.C.; Gillespie, S.; Laney, D.; Ali, N. Patients’ perspectives on newborn screening for later-onset lysosomal storage diseases. Mol. Genet. Metab. 2016, 119, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Elliott, S.; Buroker, N.; Cournoyer, J.J.; Potier, A.M.; Trometer, J.D.; Elbin, C.; Schermer, M.J.; Kantola, J.; Boyce, A.; Turecek, F.; et al. Pilot study of newborn screening for six lysosomal storage diseases using Tandem Mass Spectrometry. Mol. Genet. Metab. 2016, 118, 304–309. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Year | Screened Babies | Preterm Babies | Total Analysis | Recall |
|---|---|---|---|---|
| 2011 | 1722 | 175 | 2052 | 2 |
| 2012 | 7315 | 740 | 9422 | 26 |
| 2013 | 8675 | 935 | 9941 | 32 |
| 2014 | 10,141 | 1050 | 11,377 | 28 |
| 2015 | 11,463 | 1323 | 13,169 | 40 |
| 2016 | 10,746 | 1343 | 12,384 | 19 |
| 2017 | 10,346 | 1244 | 11,969 | 49 |
| Class | Deficit (Acronym) | Marker Concentration * (Cut off) | Gene | Mutation |
|---|---|---|---|---|
| AA | Hypermethioninemia (MET) | Met = 207.0 (31.0) | MAT I, MAT III | in progress |
| AA | Non ketotic hyperglycinemia (NKG) | Gly = 1790 (740) | GLDC AMT | in progress |
| UCD | Argininosuccinic acid synthetase deficiency (CIT I) | Cit = 1412.8 (28.8) | ASS1 | compound Heterozygous c.323G > T exon 5 c.1022C > T exon 14 |
| FAO | Short chain acylCoA dehydrogenase deficiency (SCAD) | C4 = 1.06 (0.55) | ACADS | Homozygous c.625G > A |
| Heterozygous c.301G > A | ||||
| FAO | Medium chain acylCoA dehydrogenase deficiency (MCAD) | C6 = 2.46 (0.10) | ACADM | Homozygous c.253G > C |
| C8 = 20.82 (0.12) | ||||
| C10 = 1.68 (0.18) | ||||
| C10:1 = 0.33 (0.10) | ||||
| C8/C10 = 12.39 | ||||
| OA | Methylmalonic acidemia (MMA) with homocystinuria | C3 = 11.60 (3.84) | MMACHC | Heterozygous c.271-272dupA |
| OA | Methylmalonic acidemia (MMA) with homocystinuria | C3 = 49.79 (3.84) | MMACHC | ** |
| OA | Propionic acidemia (PA) | C3 = 7.99 (3.84) | PCCA | Homozygous c.1899_1953del |
| OA | Maternal Vitamin B12 deficiency | C3 = 7.15 (3.84) | GIF | n.a. |
| OA | Maternal 3-methyl crotonylCoA carboxylase deficiency (3MCC) | C5OH\C4DC = 4.68 (0.32) | MCCC1 MCCC2 | n.a. |
| C0 = 4.69 (8.00) | ||||
| OA | MalonylCoA decarboxilase deficiency | C4OH\C3DC = 0.41 (0.25) | MLYCD | Homozygous c.1283C > T |
| OA | 3-OH-3-methylglutarylCoA lyase | C6DC = 0.72 (0.36) C5OH\C4DC = 0.26 (0.32) | HMGCL | n.a. |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Messina, M.; Meli, C.; Raudino, F.; Pittalá, A.; Arena, A.; Barone, R.; Giuffrida, F.; Iacobacci, R.; Muccilli, V.; Sorge, G.; et al. Expanded Newborn Screening Using Tandem Mass Spectrometry: Seven Years of Experience in Eastern Sicily. Int. J. Neonatal Screen. 2018, 4, 12. https://doi.org/10.3390/ijns4020012
Messina M, Meli C, Raudino F, Pittalá A, Arena A, Barone R, Giuffrida F, Iacobacci R, Muccilli V, Sorge G, et al. Expanded Newborn Screening Using Tandem Mass Spectrometry: Seven Years of Experience in Eastern Sicily. International Journal of Neonatal Screening. 2018; 4(2):12. https://doi.org/10.3390/ijns4020012
Chicago/Turabian StyleMessina, MariaAnna, Concetta Meli, Federica Raudino, Annarita Pittalá, Alessia Arena, Rita Barone, Fortunata Giuffrida, Riccardo Iacobacci, Vera Muccilli, Giovanni Sorge, and et al. 2018. "Expanded Newborn Screening Using Tandem Mass Spectrometry: Seven Years of Experience in Eastern Sicily" International Journal of Neonatal Screening 4, no. 2: 12. https://doi.org/10.3390/ijns4020012
APA StyleMessina, M., Meli, C., Raudino, F., Pittalá, A., Arena, A., Barone, R., Giuffrida, F., Iacobacci, R., Muccilli, V., Sorge, G., & Fiumara, A. (2018). Expanded Newborn Screening Using Tandem Mass Spectrometry: Seven Years of Experience in Eastern Sicily. International Journal of Neonatal Screening, 4(2), 12. https://doi.org/10.3390/ijns4020012