Aspects of Newborn Screening in Isovaleric Acidemia



Abstract
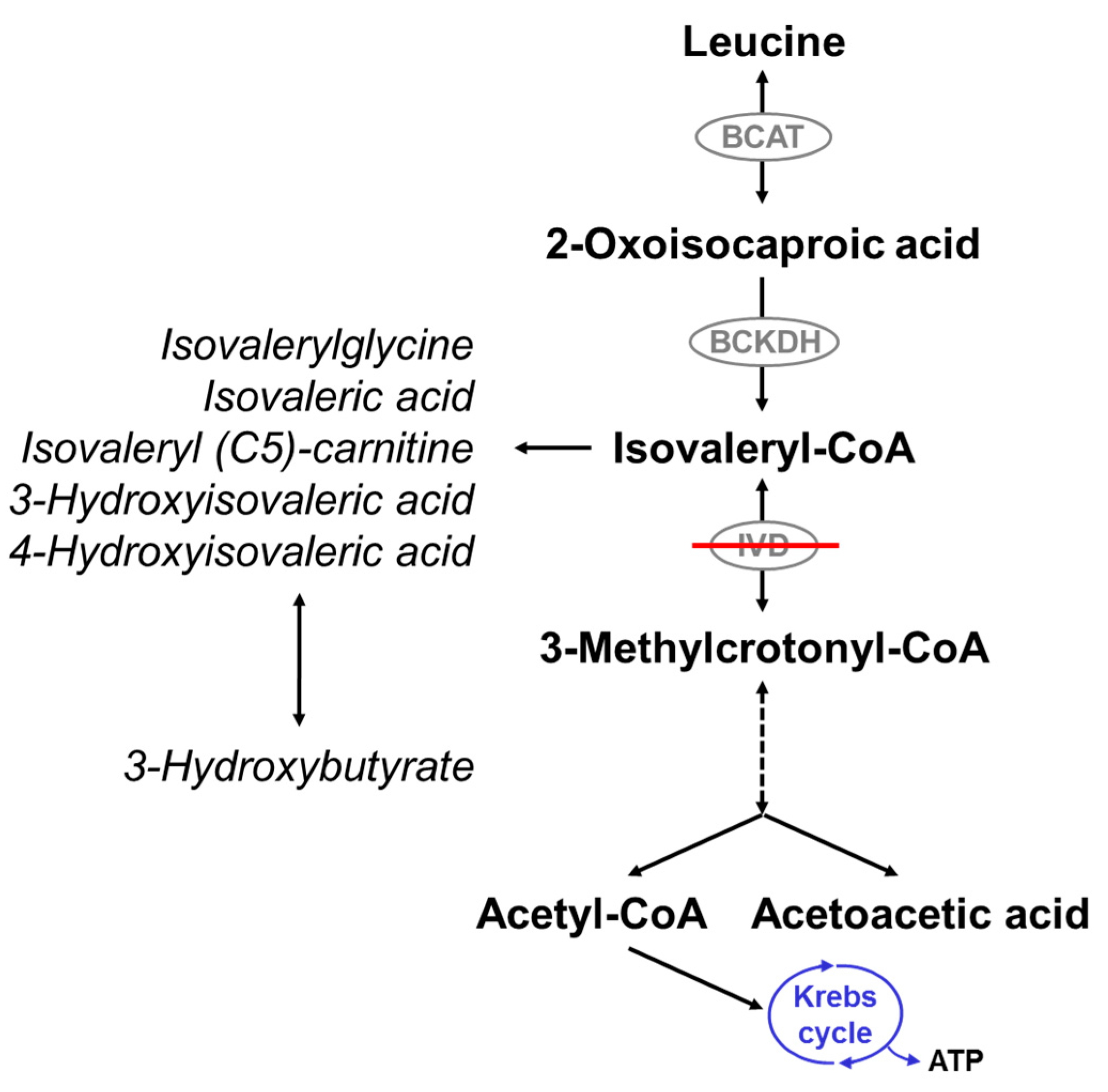
1. Introduction
2. IVA Newborn Screening: Diagnosis, Birth Prevalence and Differential Diagnosis
3. Emerging Spectrum of the Disease
3.1. Clinical Presentation
3.2. Management/Treatment
3.3. Outcome
4. Conclusions
IVA Newborn Screening—Outlook and Challenges
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hoffmann, G.F.; Lindner, M.; Loeber, J.G. 50 years of newborn screening. J. Inherit. Metab. Dis. 2014, 37, 163–164. [Google Scholar] [CrossRef] [PubMed]
- Wilcken, B. Medicine. Newborn screening: Gaps in the evidence. Science 2013, 342, 197–198. [Google Scholar] [CrossRef] [PubMed]
- Wilcken, B.; Wiley, V. Fifty years of newborn screening. J. Paediatr. Child Health 2015, 51, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Hawkes, N. Newborns in England are screened for four extra genetic conditions. BMJ 2015, 350, h10. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Therrell, B.L.; Padilla, C.D.; Loeber, J.G.; Kneisser, I.; Saadallah, A.; Borrajo, G.J.; Adams, J. Current status of newborn screening worldwide: 2015. Semin. Perinatol. 2015, 39, 171–187. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.; Knoll, D.; de Hora, M.; Kyle, C.; Glamuzina, E.; Webster, D. The risk of fatty acid oxidation disorders and organic acidemias in children with normal newborn screening. JIMD Rep. 2017, 35, 53–58. [Google Scholar] [PubMed]
- Sygdomme Som Indgår I Screeningen. Available online: https://www.ssi.dk/Diagnostik/Center%20for%20Neonatal%20Screening/Sygdomme%20som%20indgaer%20i%20screeningen.aspx (accessed on 22 February 2017).
- Ensenauer, R.; Fingerhut, R.; Maier, E.M.; Polanetz, R.; Olgemoller, B.; Roschinger, W.; Muntau, A.C. Newborn screening for isovaleric acidemia using tandem mass spectrometry: Data from 1.6 million newborns. Clin. Chem. 2011, 57, 623–626. [Google Scholar] [CrossRef] [PubMed]
- Federal Ministry of Health and Social Security. Announcement of a Decision of the Federal Joint Committee on an Amendment to the Guidelines of the Federal Committee of Physicians and Health Insurance Funds on the Early Detection of Diseases in Children up to the Age of 6 (Children’s Guidelines) for the Introduction of Extended Neonatal Screening. 2005. Available online: https://www.g-ba.de/downloads/39-261-170/2004-12-21-Kinder-TMS.pdf (accessed on 29 January 2018). (In German).
- Javaher, P.; Nyoungui, E.; Kaariainen, H.; Kristoffersson, U.; Nippert, I.; Sequeiros, J.; Schmidtke, J. Genetic screening in Europe. Public Health Genom. 2010, 13, 524–537. [Google Scholar] [CrossRef] [PubMed]
- Newborn Screening in Canada Status Report. Available online: https://www.raredisorders.ca/content/uploads/Canada-NBS-status-updated-Sept.-3-2015.pdf (accessed on 22 February 2017).
- Patchchat 6: The Case for Newborn Screening in South Africa: A Personal Perspective. Available online: https://www.ampath.co.za/pathchat/newborn-screening/ (accessed on 22 February 2017).
- Abdel-Hamid, M.; Tisocki, K.; Sharaf, L.; Ramadan, D. Development, validation and application of tandem mass spectrometry for screening of inborn metabolic disorders in Kuwaiti infants. Med. Princ. Pract. 2007, 16, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Khneisser, I.; Adib, S.; Assaad, S.; Megarbane, A.; Karam, P. Cost-benefit analysis: Newborn screening for inborn errors of metabolism in Lebanon. J. Med. Screen. 2015, 22, 182–186. [Google Scholar] [CrossRef] [PubMed]
- Krotoski, D.; Namaste, S.; Raouf, R.K.; El Nekhely, I.; Hindi-Alexander, M.; Engelson, G.; Hanson, J.W.; Howell, R.R.; Committee, M.N.S. Conference report: Second conference of the Middle East and North Africa newborn screening initiative: Partnerships for sustainable newborn screening infrastructure and research opportunities. Genet. Med. 2009, 11, 663–668. [Google Scholar] [CrossRef] [PubMed]
- Lindner, M.; Abdoh, G.; Fang-Hoffmann, J.; Shabeck, N.; Al-Sayrafi, M.; Al-Janahi, M.; Ho, S.; Abdelrahman, M.O.; Ben-Omran, T.; Bener, A.; et al. Implementation of extended neonatal screening and a metabolic unit in the state of Qatar: Developing and optimizing strategies in cooperation with the neonatal screening center in Heidelberg. J. Inherit. Metab. Dis. 2007, 30, 522–529. [Google Scholar] [CrossRef] [PubMed]
- Al Hosani, H.; Salah, M.; Osman, H.M.; Farag, H.M.; El-Assiouty, L.; Saade, D.; Hertecant, J. Expanding the comprehensive national neonatal screening programme in the United Arab Emirates from 1995 to 2011. East. Mediterr. Health J. 2014, 20, 17–23. [Google Scholar] [PubMed]
- Mohsen, A.W.; Anderson, B.D.; Volchenboum, S.L.; Battaile, K.P.; Tiffany, K.; Roberts, D.; Kim, J.J.; Vockley, J. Characterization of molecular defects in isovaleryl-CoA dehydrogenase in patients with isovaleric acidemia. Biochemistry 1998, 37, 10325–10335. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Ikeda, Y.; Matsubara, Y.; Hyman, D.B. Molecular basis of isovaleric acidemia and medium-chain acyl-CoA dehydrogenase deficiency. Enzyme 1987, 38, 91–107. [Google Scholar] [CrossRef] [PubMed]
- Vockley, J.; Parimoo, B.; Nagao, M.; Tanaka, K. Identification of the molecular defects responsible for the various genotypes of isovaleric acidemia. Prog. Clin. Biol. Res. 1992, 375, 533–540. [Google Scholar] [PubMed]
- Vockley, J.; Parimoo, B.; Tanaka, K. Molecular characterization of four different classes of mutations in the isovaleryl-CoA dehydrogenase gene responsible for isovaleric acidemia. Am. J. Hum. Genet. 1991, 49, 147–157. [Google Scholar] [PubMed]
- Vockley, J.; Rogan, P.K.; Anderson, B.D.; Willard, J.; Seelan, R.S.; Smith, D.I.; Liu, W. Exon skipping in IVD RNA processing in isovaleric acidemia caused by point mutations in the coding region of the IVD gene. Am. J. Hum. Genet. 2000, 66, 356–367. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.D.; Wang, C.H.; Lee, C.C.; Lai, C.C.; Tsai, Y.; Tsai, F.J. Genetic mutation profile of isovaleric acidemia patients in Taiwan. Mol. Genet. Metab. 2007, 90, 134–139. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.W.; Lee, D.H.; Vockley, J.; Kim, N.D.; Lee, Y.K.; Ki, C.S. Different spectrum of mutations of isovaleryl-CoA dehydrogenase (IVD) gene in Korean patients with isovaleric acidemia. Mol. Genet. Metab. 2007, 92, 71–77. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Vatanavicharn, N.; Liammongkolkul, S.; Sakamoto, O.; Sathienkijkanchai, A.; Wasant, P. Phenotypic and mutation spectrums of Thai patients with isovaleric acidemia. Pediatr. Int. 2011, 53, 990–994. [Google Scholar] [CrossRef] [PubMed]
- Hertecant, J.L.; Ben-Rebeh, I.; Marah, M.A.; Abbas, T.; Ayadi, L.; Ben Salem, S.; Al-Jasmi, F.A.; Al-Gazali, L.; Al-Yahyaee, S.A.; Ali, B.R. Clinical and molecular analysis of isovaleric acidemia patients in the United Arab Emirates reveals remarkable phenotypes and four novel mutations in the IVD gene. Eur. J. Med. Genet. 2012, 55, 671–676. [Google Scholar] [CrossRef] [PubMed]
- Bei, F.; Sun, J.H.; Yu, Y.G.; Jia, J.; Zheng, Z.J.; Fu, Q.H.; Cai, W. Two novel isovaleryl-CoA dehydrogenase gene mutations in a Chinese infant. Gene 2013, 524, 396–400. [Google Scholar] [CrossRef] [PubMed]
- Ozgul, R.K.; Karaca, M.; Kilic, M.; Kucuk, O.; Yucel-Yilmaz, D.; Unal, O.; Hismi, B.; Aliefendioglu, D.; Sivri, S.; Tokatli, A.; et al. Phenotypic and genotypic spectrum of Turkish patients with isovaleric acidemia. Eur. J. Med. Genet. 2014, 57, 596–601. [Google Scholar] [CrossRef] [PubMed]
- Kaya, N.; Colak, D.; Al-Bakheet, A.; Al-Younes, B.; Tulbah, S.; Daghestani, M.; Al-Mutairi, F.; Al-Amoudi, M.; Al-Odaib, A.; Al-Aqeel, A.I. Identification of a novel IVD mutation in a consanguineous family with isovaleric acidemia. Gene 2013, 513, 297–300. [Google Scholar] [CrossRef] [PubMed]
- Ensenauer, R.; Vockley, J.; Willard, J.M.; Huey, J.C.; Sass, J.O.; Edland, S.D.; Burton, B.K.; Berry, S.A.; Santer, R.; Grunert, S.; et al. A common mutation is associated with a mild, potentially asymptomatic phenotype in patients with isovaleric acidemia diagnosed by newborn screening. Am. J. Hum. Genet. 2004, 75, 1136–1142. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.M.; Lee, B.H.; Kim, G.H.; Kim, Y.M.; Choi, J.H.; Yoo, H.W. Chronic intermittent form of isovaleric aciduria in a 2-year-old boy. Korean J. Pediatr. 2013, 56, 351–354. [Google Scholar] [CrossRef] [PubMed]
- Couce, M.L.; Aldamiz-Echevarria, L.; Bueno, M.A.; Barros, P.; Belanger-Quintana, A.; Blasco, J.; Garcia-Silva, M.T.; Marquez-Armenteros, A.M.; Vitoria, I.; Vives, I.; et al. Genotype and phenotype characterization in a Spanish cohort with isovaleric acidemia. J. Hum. Genet. 2017, 62, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, O.; Arai-Ichinoi, N.; Mitsubuchi, H.; Chinen, Y.; Haruna, H.; Maruyama, H.; Sugawara, H.; Kure, S. Phenotypic variability and newly identified mutations of the IVD gene in Japanese patients with isovaleric acidemia. Tohoku J. Exp. Med. 2015, 236, 103–106. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Solano, A.F.; Leipnitz, G.; De Bortoli, G.M.; Seminotti, B.; Amaral, A.U.; Fernandes, C.G.; Latini, A.S.; Dutra-Filho, C.S.; Wajner, M. Induction of oxidative stress by the metabolites accumulating in isovaleric acidemia in brain cortex of young rats. Free Radic. Res. 2008, 42, 707–715. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, C.A.; Balestro, F.; Grando, V.; Wajner, M. Isovaleric acid reduces Na+, K+-ATPase activity in synaptic membranes from cerebral cortex of young rats. Cell. Mol. Neurobiol. 2007, 27, 529–540. [Google Scholar] [CrossRef] [PubMed]
- Schriever, S.C.; Deutsch, M.J.; Adamski, J.; Roscher, A.A.; Ensenauer, R. Cellular signaling of amino acids towards mTORC1 activation in impaired human leucine catabolism. J. Nutr. Biochem. 2013, 24, 824–831. [Google Scholar] [CrossRef] [PubMed]
- Dionisi-Vici, C.; Deodato, F.; Roschinger, W.; Rhead, W.; Wilcken, B. ‘Classical’ organic acidurias, propionic aciduria, methylmalonic aciduria and isovaleric aciduria: Long-term outcome and effects of expanded newborn screening using tandem mass spectrometry. J. Inherit. Metab. Dis. 2006, 29, 383–389. [Google Scholar] [CrossRef] [PubMed]
- Heringer, J.; Valayannopoulos, V.; Lund, A.M.; Wijburg, F.A.; Freisinger, P.; Baric, I.; Baumgartner, M.R.; Burgard, P.; Burlina, A.B.; Chapman, K.A.; et al. Impact of age at onset and newborn screening on outcome in organic acidurias. J. Inherit. Metab. Dis. 2016, 39, 341–353. [Google Scholar] [CrossRef] [PubMed]
- Grunert, S.C.; Wendel, U.; Lindner, M.; Leichsenring, M.; Schwab, K.O.; Vockley, J.; Lehnert, W.; Ensenauer, R. Clinical and neurocognitive outcome in symptomatic isovaleric acidemia. Orphanet J. Rare Dis. 2012, 7, 9. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K. Isovaleric acidemia: Personal history, clinical survey and study of the molecular basis. Prog. Clin. Biol. Res. 1990, 321, 273–290. [Google Scholar] [PubMed]
- Tanaka, K.; Budd, M.A.; Efron, M.L.; Isselbacher, K.J. Isovaleric acidemia: A new genetic defect of leucine metabolism. Proc. Natl. Acad. Sci. USA 1966, 56, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Isselbacher, K.J. The isolation and identification of N-isovalerylglycine from urine of patients with isovaleric acidemia. J. Biol. Chem. 1967, 242, 2966–2972. [Google Scholar] [PubMed]
- Fingerhut, R.; Olgemoller, B. Newborn screening for inborn errors of metabolism and endocrinopathies: An update. Anal. Bioanal. Chem. 2009, 393, 1481–1497. [Google Scholar] [CrossRef] [PubMed]
- Vockley, J.; Ensenauer, R. Isovaleric acidemia: New aspects of genetic and phenotypic heterogeneity. Am. J. Med. Genet. C Semin. Med. Genet. 2006, 142c, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Estrella, J.; Wilcken, B.; Carpenter, K.; Bhattacharya, K.; Tchan, M.; Wiley, V. Expanded newborn screening in New South Wales: Missed cases. J. Inherit. Metab. Dis. 2014, 37, 881–887. [Google Scholar] [CrossRef] [PubMed]
- Niu, D.M.; Chien, Y.H.; Chiang, C.C.; Ho, H.C.; Hwu, W.L.; Kao, S.M.; Chiang, S.H.; Kao, C.H.; Liu, T.T.; Chiang, H.; et al. Nationwide survey of extended newborn screening by tandem mass spectrometry in Taiwan. J. Inherit. Metab. Dis. 2010, 33, S295–S305. [Google Scholar] [CrossRef] [PubMed]
- Vilarinho, L.; Rocha, H.; Sousa, C.; Marcao, A.; Fonseca, H.; Bogas, M.; Osorio, R.V. Four years of expanded newborn screening in Portugal with tandem mass spectrometry. J. Inherit. Metab. Dis. 2010, 33 (Suppl. 3), S133–S138. [Google Scholar] [CrossRef] [PubMed]
- Wilcken, B.; Haas, M.; Joy, P.; Wiley, V.; Bowling, F.; Carpenter, K.; Christodoulou, J.; Cowley, D.; Ellaway, C.; Fletcher, J.; et al. Expanded newborn screening: Outcome in screened and unscreened patients at age 6 years. Pediatrics 2009, 124, e241–e248. [Google Scholar] [CrossRef] [PubMed]
- Van Calcar, S.C.; Baker, M.W.; Williams, P.; Jones, S.A.; Xiong, B.; Thao, M.C.; Lee, S.; Yang, M.K.; Rice, G.M.; Rhead, W.; et al. Prevalence and mutation analysis of short/branched chain acyl-CoA dehydrogenase deficiency (SBCADD) detected on newborn screening in Wisconsin. Mol. Genet. Metab. 2013, 110, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Gibson, K.M.; Sacks, M.; Kiss, D.; Pohowalla, P.; Linck, L.; Steiner, R.D.; Burlingame, T. 2-methylbutyrylglycinuria in a neonate with CNS dysfunction: Evidence for isolated 2-methylbutyryl-CoA dehydrogenase deficiency, an inborn error of L-leucine metabolism. J. Inherit. Metab. Dis. 1999, 22 (Suppl. 1), 16. [Google Scholar]
- Andresen, B.S.; Christensen, E.; Corydon, T.J.; Bross, P.; Pilgaard, B.; Wanders, R.J.; Ruiter, J.P.; Simonsen, H.; Winter, V.; Knudsen, I.; et al. Isolated 2-methylbutyrylglycinuria caused by short/branched-chain acyl-CoA dehydrogenase deficiency: Identification of a new enzyme defect, resolution of its molecular basis, and evidence for distinct acyl-CoA dehydrogenases in isoleucine and valine metabolism. Am. J. Hum. Genet. 2000, 67, 1095–1103. [Google Scholar] [PubMed]
- Alfardan, J.; Mohsen, A.W.; Copeland, S.; Ellison, J.; Keppen-Davis, L.; Rohrbach, M.; Powell, B.R.; Gillis, J.; Matern, D.; Kant, J.; et al. Characterization of new ACADSB gene sequence mutations and clinical implications in patients with 2-methylbutyrylglycinuria identified by newborn screening. Mol. Genet. Metab. 2010, 100, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Madsen, P.P.; Kibaek, M.; Roca, X.; Sachidanandam, R.; Krainer, A.R.; Christensen, E.; Steiner, R.D.; Gibson, K.M.; Corydon, T.J.; Knudsen, I.; et al. Short/branched-chain acyl-CoA dehydrogenase deficiency due to an IVS3+3A>G mutation that causes exon skipping. Hum. Genet. 2006, 118, 680–690. [Google Scholar] [CrossRef] [PubMed]
- Gibson, K.M.; Burlingame, T.G.; Hogema, B.; Jakobs, C.; Schutgens, R.B.; Millington, D.; Roe, C.R.; Roe, D.S.; Sweetman, L.; Steiner, R.D.; et al. 2-methylbutyryl-coenzyme A dehydrogenase deficiency: A new inborn error of L-isoleucine metabolism. Pediatr. Res. 2000, 47, 830–833. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kanavin, O.J.; Woldseth, B.; Jellum, E.; Tvedt, B.; Andresen, B.S.; Stromme, P. 2-methylbutyryl-CoA dehydrogenase deficiency associated with autism and mental retardation: A case report. J. Med. Case Rep. 2007, 1, 98. [Google Scholar] [CrossRef] [PubMed]
- Knerr, I.; Weinhold, N.; Vockley, J.; Gibson, K.M. Advances and challenges in the treatment of branched-chain amino/keto acid metabolic defects. J. Inherit. Metab. Dis. 2012, 35, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Abdenur, J.E.; Chamoles, N.A.; Guinle, A.E.; Schenone, A.B.; Fuertes, A.N. Diagnosis of isovaleric acidaemia by tandem mass spectrometry: False positive result due to pivaloylcarnitine in a newborn screening programme. J. Inherit. Metab. Dis. 1998, 21, 624–630. [Google Scholar] [CrossRef] [PubMed]
- Boemer, F.; Schoos, R.; de Halleux, V.; Kalenga, M.; Debray, F.G. Surprising causes of C5-carnitine false positive results in newborn screening. Mol. Genet. Metab. 2014, 111, 52–54. [Google Scholar] [CrossRef] [PubMed]
- Cloppenborg, T.; Janzen, N.; Wagner, H.; Steuerwald, U.; Peter, M.; Das, A. Application of a second-tier newborn screening assay for C5 isoforms. JIMD Rep. 2014, 13, 23–26. [Google Scholar] [PubMed]
- Minkler, P.E.; Stoll, M.S.K.; Ingalls, S.T.; Hoppel, C.L. Selective and accurate C5 acylcarnitine quantitation by UHPLC-MS/MS: Distinguishing true isovaleric acidemia from pivalate derived interference. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2017, 1061–1062, 128–133. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, H.; Fukuda, S.; Yamada, K.; Hasegawa, Y.; Takahashi, T.; Purevsuren, J.; Yamaguchi, S. Clinical features of carnitine deficiency secondary to pivalate-conjugated antibiotic therapy. J. Pediatr. 2016, 173, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Kobayashi, H.; Bo, R.; Takahashi, T.; Hasegawa, Y.; Nakamura, M.; Ishige, N.; Yamaguchi, S. Elevation of pivaloylcarnitine by sivelestat sodium in two children. Mol. Genet. Metab. 2015, 116, 192–194. [Google Scholar] [CrossRef] [PubMed]
- Shigematsu, Y.; Hata, I.; Tajima, G. Useful second-tier tests in expanded newborn screening of isovaleric acidemia and methylmalonic aciduria. J. Inherit. Metab. Dis. 2010, 33, S283–S288. [Google Scholar] [CrossRef] [PubMed]
- Shigematsu, Y.; Hata, I.; Tanaka, Y. Stable-isotope dilution measurement of isovalerylglycine by tandem mass spectrometry in newborn screening for isovaleric acidemia. Clin. Chim. Acta 2007, 386, 82–86. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Janzen, N.; Steuerwald, U.; Sander, S.; Terhardt, M.; Peter, M.; Sander, J. UPLC-MS/MS analysis of C5-acylcarnitines in dried blood spots. Clin. Chim. Acta 2013, 421, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Kolker, S.; Cazorla, A.G.; Valayannopoulos, V.; Lund, A.M.; Burlina, A.B.; Sykut-Cegielska, J.; Wijburg, F.A.; Teles, E.L.; Zeman, J.; Dionisi-Vici, C.; et al. The phenotypic spectrum of organic acidurias and urea cycle disorders. Part 1: The initial presentation. J. Inherit. Metab. Dis. 2015, 38, 1041–1045. [Google Scholar] [CrossRef] [PubMed]
- Dercksen, M.; Duran, M.; Ijlst, L.; Mienie, L.J.; Reinecke, C.J.; Ruiter, J.P.; Waterham, H.R.; Wanders, R.J. Clinical variability of isovaleric acidemia in a genetically homogeneous population. J. Inherit. Metab. Dis. 2012, 35, 1021–1029. [Google Scholar] [CrossRef] [PubMed]
- Tokatli, A.; Coskun, T.; Ozalp, I. Isovaleric acidemia. Clinical presentation of 6 cases. Turk. J. Pediatr. 1998, 40, 111–119. [Google Scholar] [PubMed]
- Feinstein, J.A.; O’Brien, K. Acute metabolic decompensation in an adult patient with isovaleric acidemia. South. Med. J. 2003, 96, 500–503. [Google Scholar] [CrossRef] [PubMed]
- Kimmoun, A.; Abboud, G.; Strazeck, J.; Merten, M.; Gueant, J.L.; Feillet, F. Acute decompensation of isovaleric acidemia induced by Graves’ disease. Intensive Care Med. 2008, 34, 2315–2316. [Google Scholar] [CrossRef] [PubMed]
- Kolker, S.; Valayannopoulos, V.; Burlina, A.B.; Sykut-Cegielska, J.; Wijburg, F.A.; Teles, E.L.; Zeman, J.; Dionisi-Vici, C.; Baric, I.; Karall, D.; et al. The phenotypic spectrum of organic acidurias and urea cycle disorders. Part 2: The evolving clinical phenotype. J. Inherit. Metab. Dis. 2015, 38, 1059–1074. [Google Scholar] [CrossRef] [PubMed]
- Sezer, T.; Balci, O. Infantile spasms during acute metabolic decompensation in an infant with isovaleric acidemia. J. Clin. Neurol. 2016, 12, 376–377. [Google Scholar] [CrossRef] [PubMed]
- Marquard, J.; El Scheich, T.; Klee, D.; Schmitt, M.; Meissner, T.; Mayatepek, E.; Oh, J. Chronic pancreatitis in branched-chain organic acidurias—A case of methylmalonic aciduria and an overview of the literature. Eur. J. Pediatr. 2011, 170, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Kahler, S.G.; Sherwood, W.G.; Woolf, D.; Lawless, S.T.; Zaritsky, A.; Bonham, J.; Taylor, C.J.; Clarke, J.T.; Durie, P.; Leonard, J.V. Pancreatitis in patients with organic acidemias. J. Pediatr. 1994, 124, 239–243. [Google Scholar] [CrossRef]
- Mantadakis, E.; Chrysafis, I.; Tsouvala, E.; Evangeliou, A.; Chatzimichael, A. Acute pancreatitis with rapid clinical improvement in a child with isovaleric acidemia. Case Rep. Pediatr. 2013, 2013, 721871. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sag, E.; Cebi, A.H.; Kaya, G.; Karaguzel, G.; Cakir, M. A rare cause of recurrent acute pancreatitis in a child: Isovaleric acidemia with novel mutation. Pediatr. Gastroenterol. Hepatol. Nutr. 2017, 20, 61–64. [Google Scholar] [CrossRef] [PubMed]
- Castelnovi, C.; Moseley, K.; Yano, S. Maternal isovaleric acidemia: Observation of distinctive changes in plasma amino acids and carnitine profiles during pregnancy. Clin. Chim. Acta 2010, 411, 2101–2103. [Google Scholar] [CrossRef] [PubMed]
- Habets, D.D.; Schaper, N.C.; Rogozinski, H.; van Spronsen, F.J.; van Rijn, M.; Bierau, J.; Bakker, J.A. Biochemical monitoring and management during pregnancy in patients with isovaleric acidaemia is helpful to prevent metabolic decompensation. JIMD Rep. 2012, 3, 83–89. [Google Scholar] [PubMed]
- Shih, V.E.; Aubry, R.H.; DeGrande, G.; Gursky, S.F.; Tanaka, K. Maternal isovaleric acidemia. J. Pediatr. 1984, 105, 77–78. [Google Scholar] [CrossRef]
- Roe, C.R.; Millington, D.S.; Maltby, D.A.; Kahler, S.G.; Bohan, T.P. L-carnitine therapy in isovaleric acidemia. J. Clin. Investig. 1984, 74, 2290–2295. [Google Scholar] [CrossRef] [PubMed]
- Yudkoff, M.; Cohn, R.M.; Puschak, R.; Rothman, R.; Segal, S. Glycine therapy in isovaleric acidemia. J. Pediatr. 1978, 92, 813–817. [Google Scholar] [CrossRef]
- Pinto, A.; Daly, A.; Evans, S.; Almeida, M.F.; Assoun, M.; Belanger-Quintana, A.; Bernabei, S.; Bollhalder, S.; Cassiman, D.; Champion, H.; et al. Dietary practices in isovaleric acidemia: A European survey. Mol. Genet. Metab. Rep. 2017, 12, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Ogier de Baulny, H. Management and emergency treatments of neonates with a suspicion of inborn errors of metabolism. Semin. Neonatol. 2002, 7, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Prietsch, V.; Lindner, M.; Zschocke, J.; Nyhan, W.L.; Hoffmann, G.F. Emergency management of inherited metabolic diseases. J. Inherit. Metab. Dis. 2002, 25, 531–546. [Google Scholar] [CrossRef] [PubMed]
- Berry, G.T.; Yudkoff, M.; Segal, S. Isovaleric acidemia: Medical and neurodevelopmental effects of long-term therapy. J. Pediatr. 1988, 113, 58–64. [Google Scholar] [CrossRef]
- Kasapkara, C.S.; Ezgu, F.S.; Okur, I.; Tumer, L.; Biberoglu, G.; Hasanoglu, A. N-carbamylglutamate treatment for acute neonatal hyperammonemia in isovaleric acidemia. Eur. J. Pediatr. 2011, 170, 799–801. [Google Scholar] [CrossRef] [PubMed]
- Schulze-Bergkamen, A.; Okun, J.G.; Spiekerkotter, U.; Lindner, M.; Haas, D.; Kohlmuller, D.; Mayatepek, E.; Schulze-Bergkamen, H.; Greenberg, C.R.; Zschocke, J.; et al. Quantitative acylcarnitine profiling in peripheral blood mononuclear cells using in vitro loading with palmitic and 2-oxoadipic acids: Biochemical confirmation of fatty acid oxidation and organic acid disorders. Pediatr. Res. 2005, 58, 873–880. [Google Scholar] [CrossRef] [PubMed]
- Lindner, M.; Gramer, G.; Haege, G.; Fang-Hoffmann, J.; Schwab, K.O.; Tacke, U.; Trefz, F.K.; Mengel, E.; Wendel, U.; Leichsenring, M.; et al. Efficacy and outcome of expanded newborn screening for metabolic diseases—Report of 10 years from South-West Germany. Orphanet J. Rare Dis. 2011, 6, 44. [Google Scholar] [CrossRef] [PubMed]
- Nizon, M.; Ottolenghi, C.; Valayannopoulos, V.; Arnoux, J.B.; Barbier, V.; Habarou, F.; Desguerre, I.; Boddaert, N.; Bonnefont, J.P.; Acquaviva, C.; et al. Long-term neurological outcome of a cohort of 80 patients with classical organic acidurias. Orphanet J. Rare Dis. 2013, 8, 148. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Region | Country | Local Specifics | IVA Targeted by NBS Since (as Available) | Reference/Source |
|---|---|---|---|---|
| Asia Pacific | Australia | [5] | ||
| China | No full population screening | [5] | ||
| India | No full population screening; program not government funded | [5] | ||
| Japan | [5] | |||
| Malaysia | No full population screening | [5] | ||
| New Zealand | 2006 | [6] | ||
| Philippines | No full population screening | [5] | ||
| Singapore | [5] | |||
| South Korea | [5] | |||
| Thailand | No full population screening | [5] | ||
| Taiwan | [5] | |||
| Europe | Austria | 2002 a | [5] | |
| Belgium | 2009 (Pilot 2007) b | [5] | ||
| Czech Republic | 2010 c | [5] | ||
| Denmark | 2012 | [5,7] | ||
| Estonia | No full population screening | [5] | ||
| Germany | Bavaria 1999 Nationwide 2005 | [8,9] | ||
| Greece | [10] | |||
| Hungary | [5] | |||
| Iceland | 2008 d | [5] | ||
| Italy | No full population screening | [5] | ||
| Liechtenstein | [5] | |||
| Macedonia | No full population screening | 2013 e | Personal communication e | |
| Netherlands | [5] | |||
| Norway | [5] | |||
| Poland | [5] | |||
| Portugal | [5] | |||
| Russia | [5] | |||
| San Marino | No full population screening | [5] | ||
| Spain | [5] | |||
| Sweden | [5] | |||
| Switzerland | [5] | |||
| United Kingdom | Not in Scotland and Northern Ireland | 2015 (Pilot 2012) f | [5] | |
| North America | United States | IVA included in all states but District of Columbia and Massachusetts | [5] | |
| Canada | IVA included in all provinces/territories but Newfoundland & Labrador; IVA screened by urine in Quebec | [11] | ||
| South America | Argentina | Offered exclusively in the private sector | [5] | |
| Brazil | Offered exclusively in the private sector | [5] | ||
| Chile | Offered as selective screening | [5] | ||
| Colombia | No full population screening; offered in the private sector | [5] | ||
| Costa Rica | [5] | |||
| Dominican Republic | Offered exclusively in the private sector | [5] | ||
| Mexico | No full population screening | [5] | ||
| Uruguay | No full population screening | [5] | ||
| Venezuela | Offered exclusively in the private sector | [5] | ||
| Africa | South Africa | Offered exclusively in the private sector | [12] | |
| Middle East | Kuwait | Pilot 2004–2006 | [13] Personal communication g | |
| Lebanon | Offered exclusively in the private sector | 2006 | [14,15] | |
| Saudi Arabia | [15] | |||
| Qatar | 2004 | [15,16] | ||
| United Arab Emirates | 2011 | [17] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schlune, A.; Riederer, A.; Mayatepek, E.; Ensenauer, R. Aspects of Newborn Screening in Isovaleric Acidemia. Int. J. Neonatal Screen. 2018, 4, 7. https://doi.org/10.3390/ijns4010007
Schlune A, Riederer A, Mayatepek E, Ensenauer R. Aspects of Newborn Screening in Isovaleric Acidemia. International Journal of Neonatal Screening. 2018; 4(1):7. https://doi.org/10.3390/ijns4010007
Chicago/Turabian StyleSchlune, Andrea, Anselma Riederer, Ertan Mayatepek, and Regina Ensenauer. 2018. "Aspects of Newborn Screening in Isovaleric Acidemia" International Journal of Neonatal Screening 4, no. 1: 7. https://doi.org/10.3390/ijns4010007
APA StyleSchlune, A., Riederer, A., Mayatepek, E., & Ensenauer, R. (2018). Aspects of Newborn Screening in Isovaleric Acidemia. International Journal of Neonatal Screening, 4(1), 7. https://doi.org/10.3390/ijns4010007