Newborn Screening for Primary Immunodeficiency Diseases: The Past, the Present and the Future



Abstract
:1. Introduction
2. The Past: Identification of Severe Combined Immunodeficiency as a Priority for Newborn Screening
3. The Present: Screening for T and B Cell Lymphopenia
4. The Future: Screening for Other Forms of PID
5. The Future: The Role of Next-Generation Sequencing in Newborn Screening for PID
5.1. Identification of Candidate Diseases and Genes to Be Evaluated in Newborn Genomic Screening Programs
5.2. Establishment of Robust and Cost-Effective Genomic Testing Systems
5.3. Ethical, Legal and Social Implications
5.4. A New Model for Newborn Screening
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of interest
References
- Itan, Y.; Shang, L.; Boisson, B.; Patin, E.; Bolze, A.; Moncada-Velez, M.; Scott, E.; Ciancanelli, M.J.; Lafaille, F.G.; Markle, J.G.; et al. The human gene damage index as a gene-level approach to prioritizing exome variants. Proc. Natl. Acad. Sci. USA 2015, 112, 13615–13620. [Google Scholar] [CrossRef] [PubMed]
- Pai, S.Y.; Logan, B.R.; Griffith, L.M.; Buckley, R.H.; Parrott, R.E.; Dvorak, C.C.; Kapoor, N.; Hanson, I.C.; Filipovich, A.H.; Jyonouchi, S.; et al. Transplantation outcomes for severe combined immunodeficiency, 2000–2009. N. Engl. J. Med. 2014, 371, 434–446. [Google Scholar] [CrossRef] [PubMed]
- Guthrie, R.; Susi, A. A simple phenylalanine method for detecting phenylketonuria in large populations of newborn infants. Pediatrics 1963, 32, 338–343. [Google Scholar] [PubMed]
- Wilson, J.; Jungner, G. The principles and Practice of Screening for Disease; World Health Organization: Geneva, Switzerland, 1968. [Google Scholar]
- Kwan, A.; Abraham, R.S.; Currier, R.; Brower, A.; Andruszewski, K.; Abbott, J.K.; Baker, M.; Ballow, M.; Bartoshesky, L.E.; Bonilla, F.A.; et al. Newborn screening for severe combined immunodeficiency in 11 screening programs in the United States. JAMA 2014, 312, 729–738. [Google Scholar] [CrossRef] [PubMed]
- Barbaro, M.; Ohlsson, A.; Borte, S.; Jonsson, S.; Zetterström, R.H.; King, J.; Winiarski, J.; von Döbeln, U.; Hammarström, L. Newborn screening for severe primary immunodeficiency diseases in sweden-a 2-year pilot trec and krec screening study. J. Clin. Immunol. 2017, 37, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.; Puck, J.M. Development of population-based newborn screening for severe combined immunodeficiency. J. Allergy Clin. Immunol. 2005, 115, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Dorsey, M.; Puck, J. Newborn screening for severe combined immunodeficiency in the US: Current status and approach to management. Int. J. Neonatal Screen. 2017, 3, 15. [Google Scholar] [CrossRef]
- Chien, Y.-H.; Yu, H.-H.; Lee, N.-C.; Ho, H.-C.; Kao, S.-M.; Lu, M.-Y.; Jaing, T.-H.; Lee, W.-I.; Chang, K.-W.; Shieh, C.-C.; et al. Newborn screening for severe combined immunodeficiency in Taiwan. Int. J. Neonatal Screen. 2017, 3, 16. [Google Scholar] [CrossRef]
- Rechavi, E.; Lev, A.; Saraf-Levy, T.; Etzioni, A.; Almashanu, S.; Somech, R. Newborn screening for severe combined immunodeficiency in Israel. Int. J. Neonatal Screen. 2017, 3, 13. [Google Scholar] [CrossRef]
- Blom, M.; Pico-Knijnenburg, I.; Sijne-van Veen, M.; Boelen, A.; Bredius, R.G.M.; van der Burg, M.; Schielen, P.C.J.I. An evaluation of the trec assay with regard to the integration of scid screening into the Dutch newborn screening program. Clin. Immunol. 2017, 180, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Audrain, M.; Thomas, C.; Mirallie, S.; Bourgeois, N.; Sebille, V.; Rabetrano, H.; Durand-Zaleski, I.; Boisson, R.; Persyn, M.; Pierres, C.; et al. Evaluation of the T-cell receptor excision circle assay performances for severe combined immunodeficiency neonatal screening on guthrie cards in a french single centre study. Clin. Immunol. 2014, 150, 137–139. [Google Scholar] [CrossRef] [PubMed]
- De Felipe, B.; Olbrich, P.; Lucenas, J.M.; Delgado-Pecellin, C.; Pavon-Delgado, A.; Marquez, J.; Salamanca, C.; Soler-Palacin, P.; Gonzalez-Granado, L.I.; Antolin, L.F.; et al. Prospective neonatal screening for severe T- and B-lymphocyte deficiencies in Seville. Pediatr. Allergy Immunol. 2016, 27, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Kanegae, M.P.; Barreiros, L.A.; Mazzucchelli, J.T.; Hadachi, S.M.; de Figueiredo Ferreira Guilhoto, L.M.; Acquesta, A.L.; Genov, I.R.; Holanda, S.M.; Di Gesu, R.S.; Goulart, A.L.; et al. Neonatal screening for severe combined immunodeficiency in Brazil. J. Pediatr. (Rio J) 2016, 92, 374–380. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, N.; Imai, K.; Kanegane, H.; Sato, H.; Yamada, M.; Kondoh, K.; Okada, S.; Kobayashi, M.; Agematsu, K.; Takada, H.; et al. Quantification of kappa-deleting recombination excision circles in Guthrie cards for the identification of early B-cell maturation defects. J. Allergy Clin. Immunol. 2011, 128, 223–225. [Google Scholar] [CrossRef] [PubMed]
- Borte, S.; von Döbeln, U.; Fasth, A.; Wang, N.; Janzi, M.; Winiarski, J.; Sack, U.; Pan-Hammarström, Q.; Borte, M.; Hammarström, L. Neonatal screening for severe primary immunodeficiency diseases using high-throughput triplex real-time PCR. Blood 2012, 119, 2552–2555. [Google Scholar] [CrossRef] [PubMed]
- Bruton, O.C. Agammaglobulinemia. Pediatrics 1952, 9, 722–728. [Google Scholar] [PubMed]
- Bruton, O.C. Agammaglobulinemia (congenital absence of gamma globulin); report of a case. Med. Ann. Dist. Columbia 1953, 22, 648–650. [Google Scholar] [PubMed]
- Vetrie, D.; Vorechovsky, I.; Sideras, P.; Holland, J.; Davies, A.; Flinter, F.; Hammarstrom, L.; Kinnon, C.; Levinsky, R.; Bobrow, M.; et al. The gene involved in X-linked agammaglobulinaemia is a member of the src family of protein-tyrosine kinases. Nature 1993, 361, 226–233. [Google Scholar] [CrossRef] [PubMed]
- Picard, C.; Al-Herz, W.; Bousfiha, A.; Casanova, J.L.; Chatila, T.; Conley, M.E.; Cunningham-Rundles, C.; Etzioni, A.; Holland, S.M.; Klein, C.; et al. Primary immunodeficiency diseases: An update on the classification from the international union of immunological societies expert committee for primary immunodeficiency 2015. J. Clin. Immunol. 2015, 35, 696–726. [Google Scholar] [CrossRef] [PubMed]
- Grazioli, S.; Bennett, M.; Hildebrand, K.J.; Vallance, H.; Turvey, S.E.; Junker, A.K. Limitation of TREC-based newborn screening for ZAP70 severe combined immunodeficiency. Clin. Immunol. 2014, 153, 209–210. [Google Scholar] [CrossRef] [PubMed]
- Hauck, F.; Blumenthal, B.; Fuchs, S.; Lenoir, C.; Martin, E.; Speckmann, C.; Vraetz, T.; Mannhardt-Laakmann, W.; Lambert, N.; Gil, M.; et al. Syk expression endows human ZAP70-deficient CD8 T cells with residual TCR signaling. Clin. Immunol. 2015, 161, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.Y.; Chase, J.; Garcia Lloret, M.; Stiehm, E.R.; Moore, T.; Aguilera, M.J.; Lopez Siles, J.; Church, J.A. Newborn screening for severe combined immunodeficiency does not identify bare lymphocyte syndrome. J. Allergy Clin. Immunol. 2013, 131, 1693–1695. [Google Scholar] [CrossRef] [PubMed]
- Lev, A.; Simon, A.J.; Broides, A.; Levi, J.; Garty, B.Z.; Rosenthal, E.; Amariglio, N.; Rechavi, G.; Somech, R. Thymic function in mhc class II-deficient patients. J. Allergy Clin. Immunol. 2013, 131, 831–839. [Google Scholar] [CrossRef] [PubMed]
- Lyttle, A.; Roifman, C.; Dadi, H.; Wright, N.; Kavadas, F. MHC class II deficiency in the Dene native population: A case report highlighting pitfalls in diagnosis and treatment. Allergy Asthma Clin. Immunol. 2014, 10, A1. [Google Scholar] [CrossRef]
- Speckmann, C.; Neumann, C.; Borte, S.; la Marca, G.; Sass, J.O.; Wiech, E.; Fisch, P.; Schwarz, K.; Buchholz, B.; Schlesier, M.; et al. Delayed-onset adenosine deaminase deficiency: Strategies for an early diagnosis. J. Allergy Clin. Immunol. 2012, 130, 991–994. [Google Scholar] [CrossRef] [PubMed]
- Janzi, M.; Sjöberg, R.; Wan, J.; Fischler, B.; von Döbeln, U.; Isaac, L.; Nilsson, P.; Hammarström, L. Screening for C3 deficiency in newborns using microarrays. PLoS ONE 2009, 4, e5321. [Google Scholar] [CrossRef] [PubMed]
- Hamsten, C.; Skattum, L.; Truedsson, L.; von Döbeln, U.; Uhlén, M.; Schwenk, J.M.; Hammarström, L.; Nilsson, P.; Neiman, M. Heat differentiated complement factor profiling. J. Proteom. 2015, 126, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, M.; Sontag, M.K.; Ren, C.L. Cystic fibrosis diagnosis and newborn screening. Pediatr. Clin. N. Am. 2016, 63, 599–615. [Google Scholar] [CrossRef] [PubMed]
- Borte, S.; Meeths, M.; Liebscher, I.; Krist, K.; Nordenskjold, M.; Hammarstrom, L.; von Dobeln, U.; Henter, J.I.; Bryceson, Y.T. Combined newborn screening for familial hemophagocytic lymphohistiocytosis and severe T- and B-cell immunodeficiencies. J. Allergy Clin. Immunol. 2014, 134, 226–228. [Google Scholar] [CrossRef] [PubMed]
- Stranneheim, H.; Wedell, A. Exome and genome sequencing: A revolution for the discovery and diagnosis of monogenic disorders. J. Intern. Med. 2016, 279, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Pavey, A.R.; Bodian, D.L.; Vilboux, T.; Khromykh, A.; Hauser, N.S.; Huddleston, K.; Klein, E.; Black, A.; Kane, M.S.; Iyer, R.K.; et al. Utilization of genomic sequencing for population screening of immunodeficiencies in the newborn. Genet. Med. 2017. [Google Scholar] [CrossRef] [PubMed]
- Boyle, J.M.; Buckley, R.H. Population prevalence of diagnosed primary immunodeficiency diseases in the United States. J. Clin. Immunol. 2007, 27, 497–502. [Google Scholar] [CrossRef] [PubMed]
- Berg, J.S.; Agrawal, P.B.; Bailey, D.B.; Beggs, A.H.; Brenner, S.E.; Brower, A.M.; Cakici, J.A.; Ceyhan-Birsoy, O.; Chan, K.; Chen, F.; et al. Newborn sequencing in genomic medicine and public health. Pediatrics 2017, 139, e20162252. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.M.; Cornel, M.C.; Goldenberg, A.J.; Lister, K.J.; Sénécal, K.; Vears, D.F.; The Global Alliance for Genomics and Health Regulatory and Ethics Working Group Paediatric Task Team. Genomic newborn screening: Public health policy considerations and recommendations. BMC Med. Genom. 2017, 10, 9. [Google Scholar] [CrossRef] [PubMed]
- Tambe, P.; Sammons, H.M.; Choonara, I. Child mortality of children aged 5–15 years in the UK and Sweden: A comparison. Arch. Dis. Child. 2016, 101, 409–410. [Google Scholar] [CrossRef] [PubMed]
- Tambe, P.; Sammons, H.M.; Choonara, I. Why do young children die in the UK? A comparison with sweden. Arch. Dis. Child. 2015, 100, 928–931. [Google Scholar] [CrossRef] [PubMed]
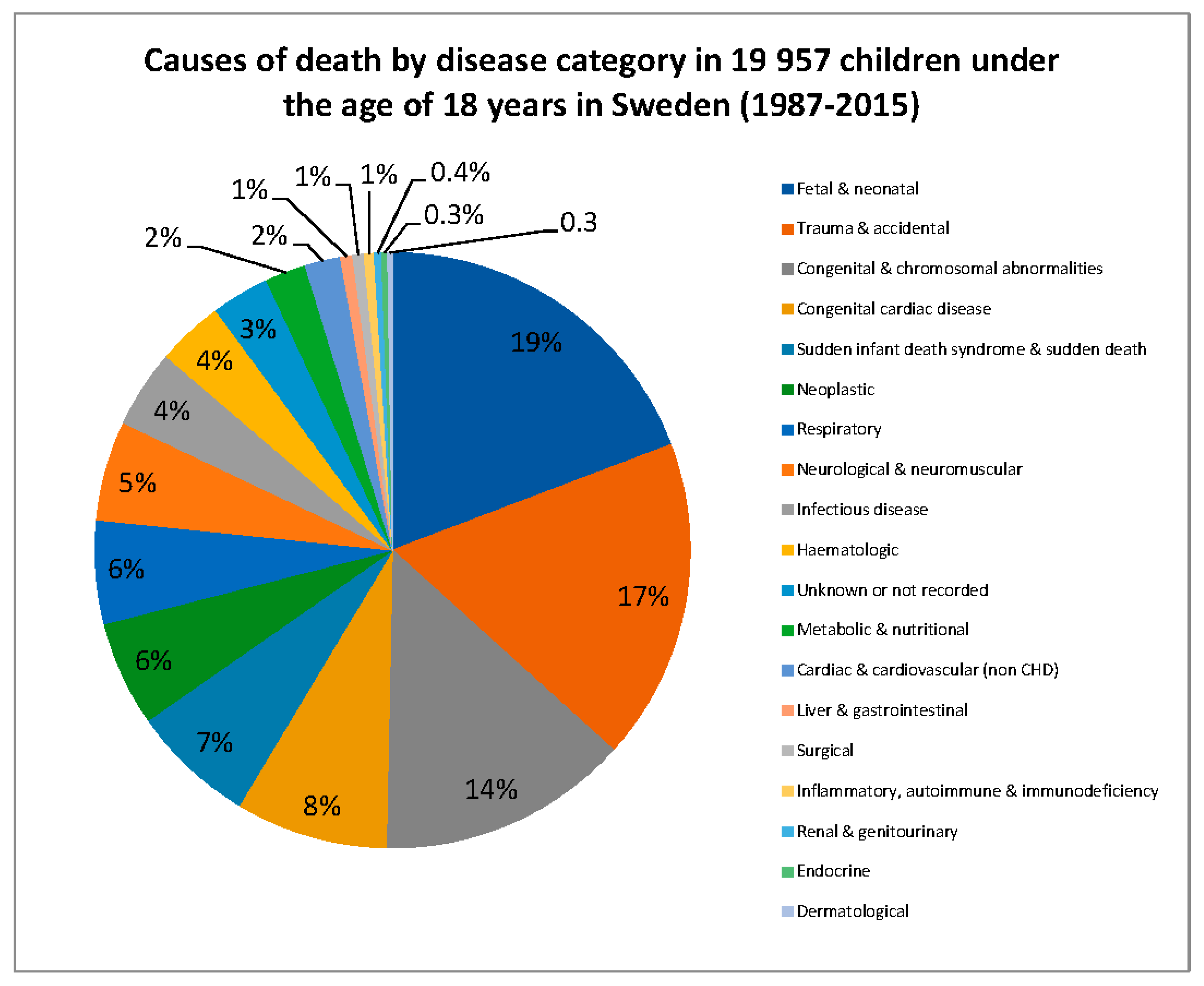

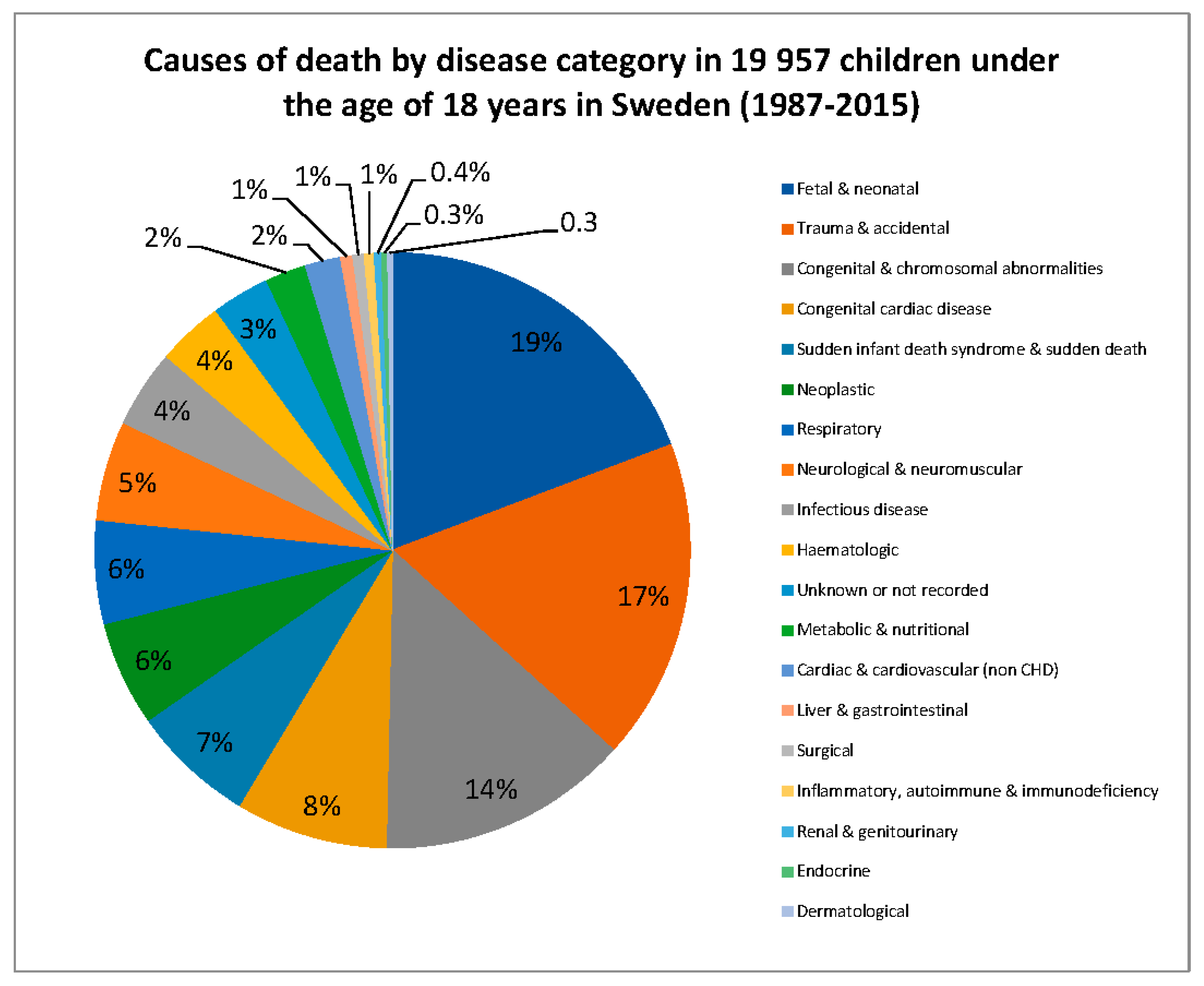{kind=link}
| Country | Screening Strategy | Date of Commencement | Reference |
|---|---|---|---|
| United States of America All 50 States District of Columbia Navajo Nation | TREC | 2008 (Wisconsin) National implementation | Dorsey & Puck 2017 [8] http://www.info4pi.org/ |
| Italy Tuscany Umbria Florence | TREC/ADA | 2010: Pilot study 2010: Pilot study 2013: Pilot study | http://ipopi.org/ |
| Taiwan | TREC | 2010: Pilot study 2012: National implementation | Chien YH et al., 2017 [9] |
| Israel | TREC | 2011: Pilot study 2015: National implementation | Rechavi et al., 2017 [10] |
| The Netherlands | TREC | 2012: Pilot study 2015: Application approved | Blom et al., 2017 [11] http://ipopi.org/ |
| Qatar | TREC | 2012: National implementation | http://ipopi.org/ |
| Germany | TREC/KREC | 2013: Pilot study Application in progress | http://ipopi.org/ |
| Sweden | TREC/KREC | 2013: Pilot study | Barbaro et al., 2016 [6] |
| Japan | TREC/KREC | 2014: Pilot study | http://ipopi.org/ |
| France | TREC | 2014: Pilot study | Audrain et al., 2014 [12] http://ipopi.org/ |
| Spain Andalucia | TREC/KREC | 2014: Pilot study | de Felipe et al., 2016 [13] http://ipopi.org/ |
| Norway | TREC | 2015: Pilot study | http://ipopi.org/ |
| Puerto Rico | TREC | 2016: National implementation | Dorsey & Puck 2017 [8] http://www.info4pi.org/ |
| New Zealand | TREC | 2017: Due to commence | http://ipopi.org/ |
| Canada Ontario British Columbia Yukon Prince Edward Island Nova Scotia New Brunswick | TREC | Screening underway Approved, pending funding Approved, pending funding Approved, pending funding Approved, pending commencement Approved, pending commencement | http://ipopi.org/ |
| Brazil | TREC | Pilot study | Kanegae et al., 2016 [14] |
| Denmark | TREC | Application in progress Pilot study | http://ipopi.org/ |
| Iceland | TREC/KREC | Application in progress Pilot study | http://ipopi.org/ |
| Iran | TREC/KREC | Pilot study | Personal communication |
| Saudi Arabia | TREC/KREC | Pilot study | http://ipopi.org/ |
| Slovenia | Pilot study Application in progress | http://ipopi.org/ | |
| Turkey | TREC/KREC | Pilot study | Personal communication |
| United Kingdom | TREC | Application in progress Pilot study | http://ipopi.org/ |
| Australia | Application in progress | http://ipopi.org/ | |
| Austria | TREC/KREC | Application in progress | http://ipopi.org/ |
| Belgium Flanders | TREC/KREC | Application in progress Application in progress | Personal communication http://ipopi.org/ |
| Czech Republic | TREC/KREC | Application in progress | Personal communication |
| Poland | Application in progress | http://ipopi.org/ | |
| Portugal | Application in progress | http://ipopi.org/ | |
| Romania | Application in progress | http://ipopi.org/ | |
| Switzerland | TREC/KREC | Application in progress | http://ipopi.org/ |
| United Arab Emirates | Applications in progress | http://ipopi.org/ |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
King, J.; Ludvigsson, J.F.; Hammarström, L. Newborn Screening for Primary Immunodeficiency Diseases: The Past, the Present and the Future. Int. J. Neonatal Screen. 2017, 3, 19. https://doi.org/10.3390/ijns3030019
King J, Ludvigsson JF, Hammarström L. Newborn Screening for Primary Immunodeficiency Diseases: The Past, the Present and the Future. International Journal of Neonatal Screening. 2017; 3(3):19. https://doi.org/10.3390/ijns3030019
Chicago/Turabian StyleKing, Jovanka, Jonas F. Ludvigsson, and Lennart Hammarström. 2017. "Newborn Screening for Primary Immunodeficiency Diseases: The Past, the Present and the Future" International Journal of Neonatal Screening 3, no. 3: 19. https://doi.org/10.3390/ijns3030019
APA StyleKing, J., Ludvigsson, J. F., & Hammarström, L. (2017). Newborn Screening for Primary Immunodeficiency Diseases: The Past, the Present and the Future. International Journal of Neonatal Screening, 3(3), 19. https://doi.org/10.3390/ijns3030019