
Newborn Screening for Severe Combined Immunodeficiency in the US: Current Status and Approach to Management


Abstract
:1. Introduction
2. Biology of SCID
3. History and Definitions of SCID
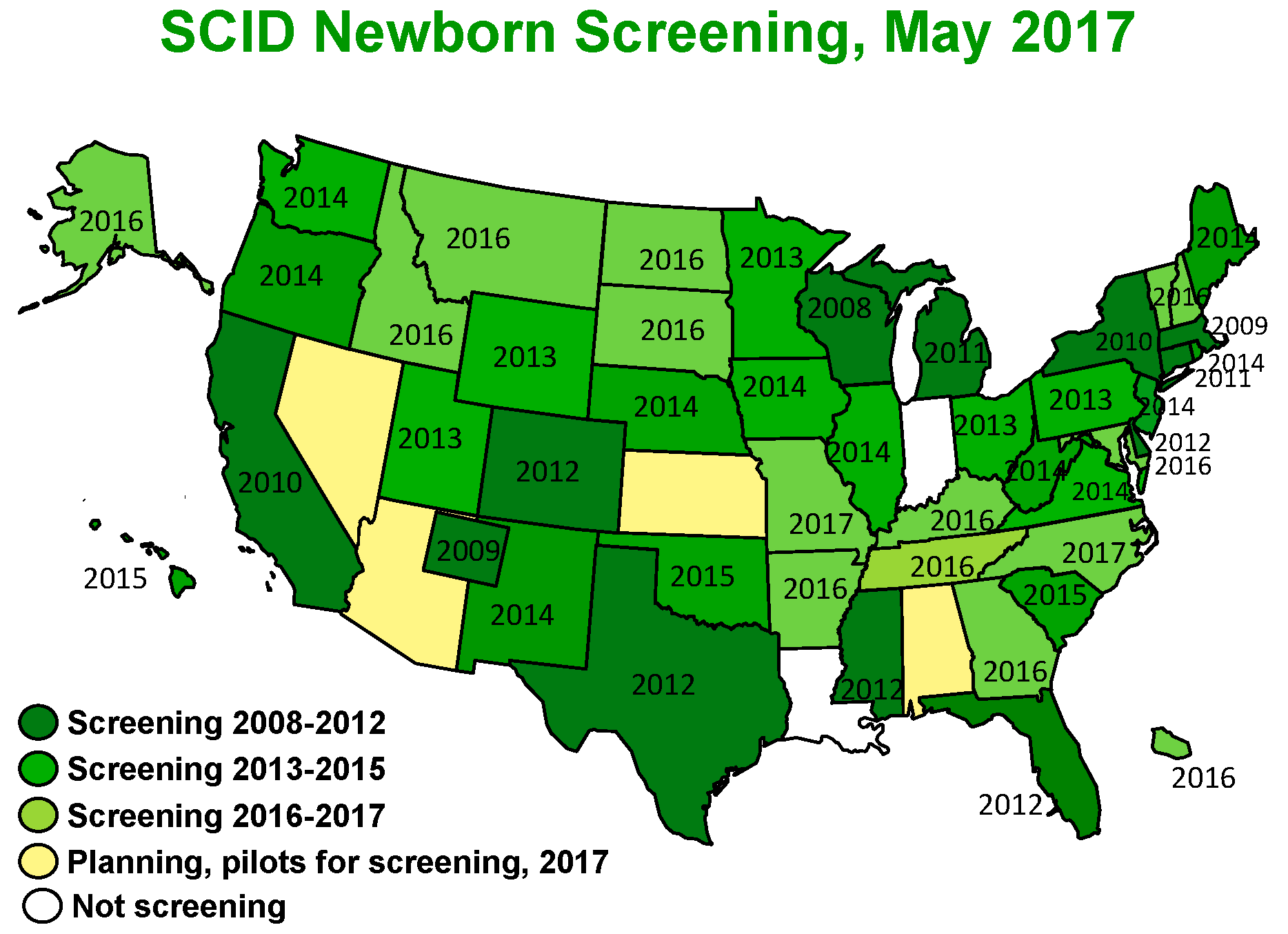
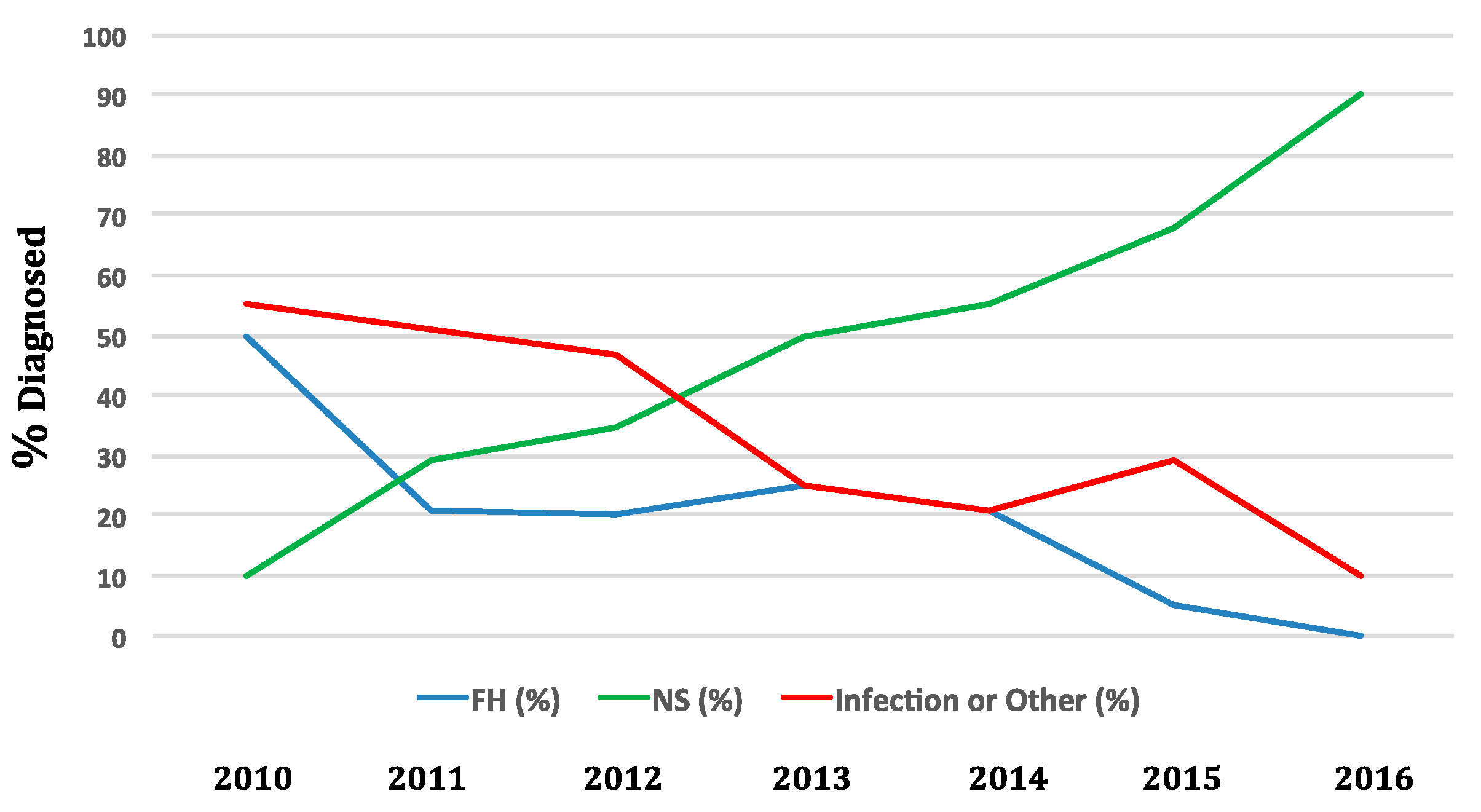
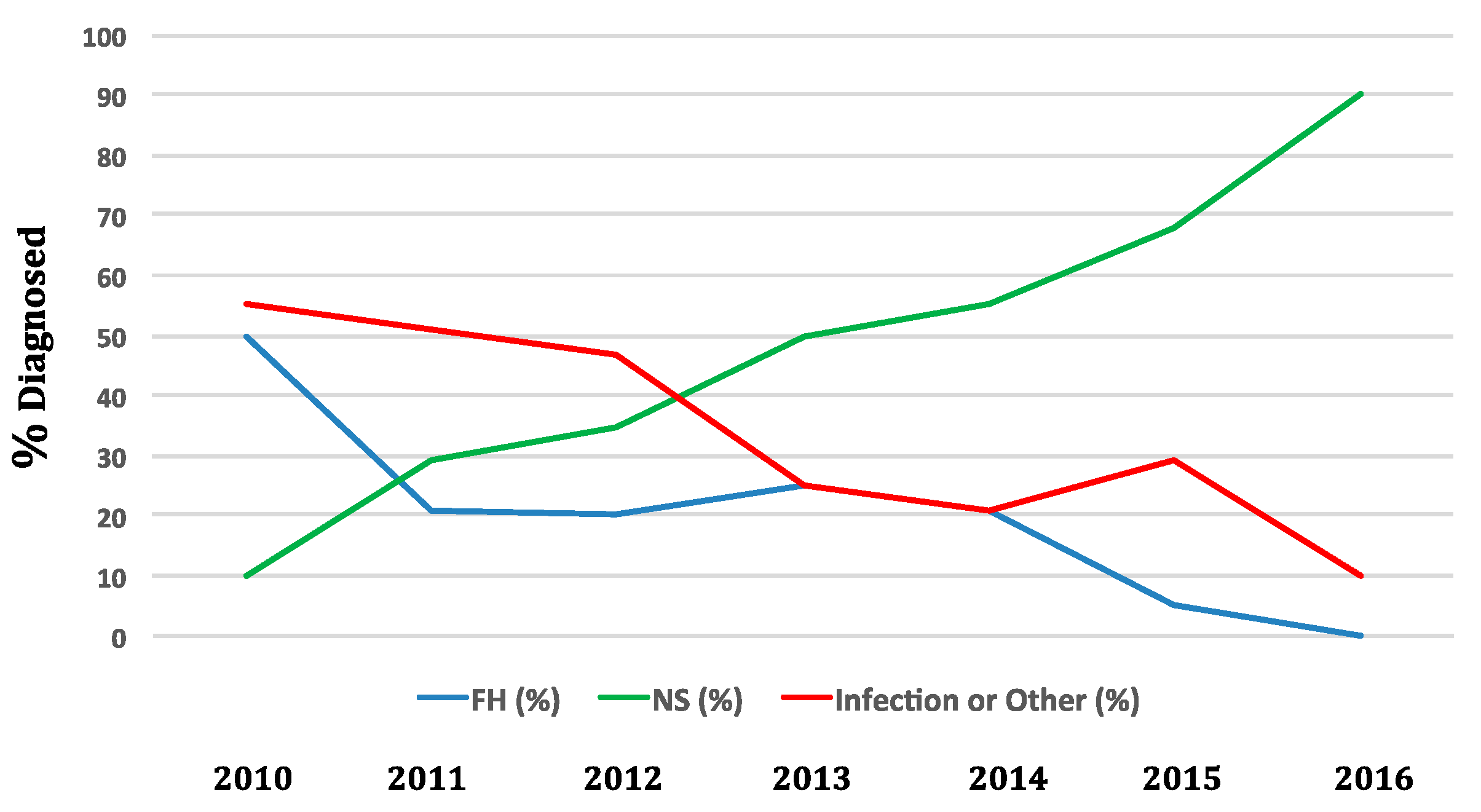
4. Epidemiology
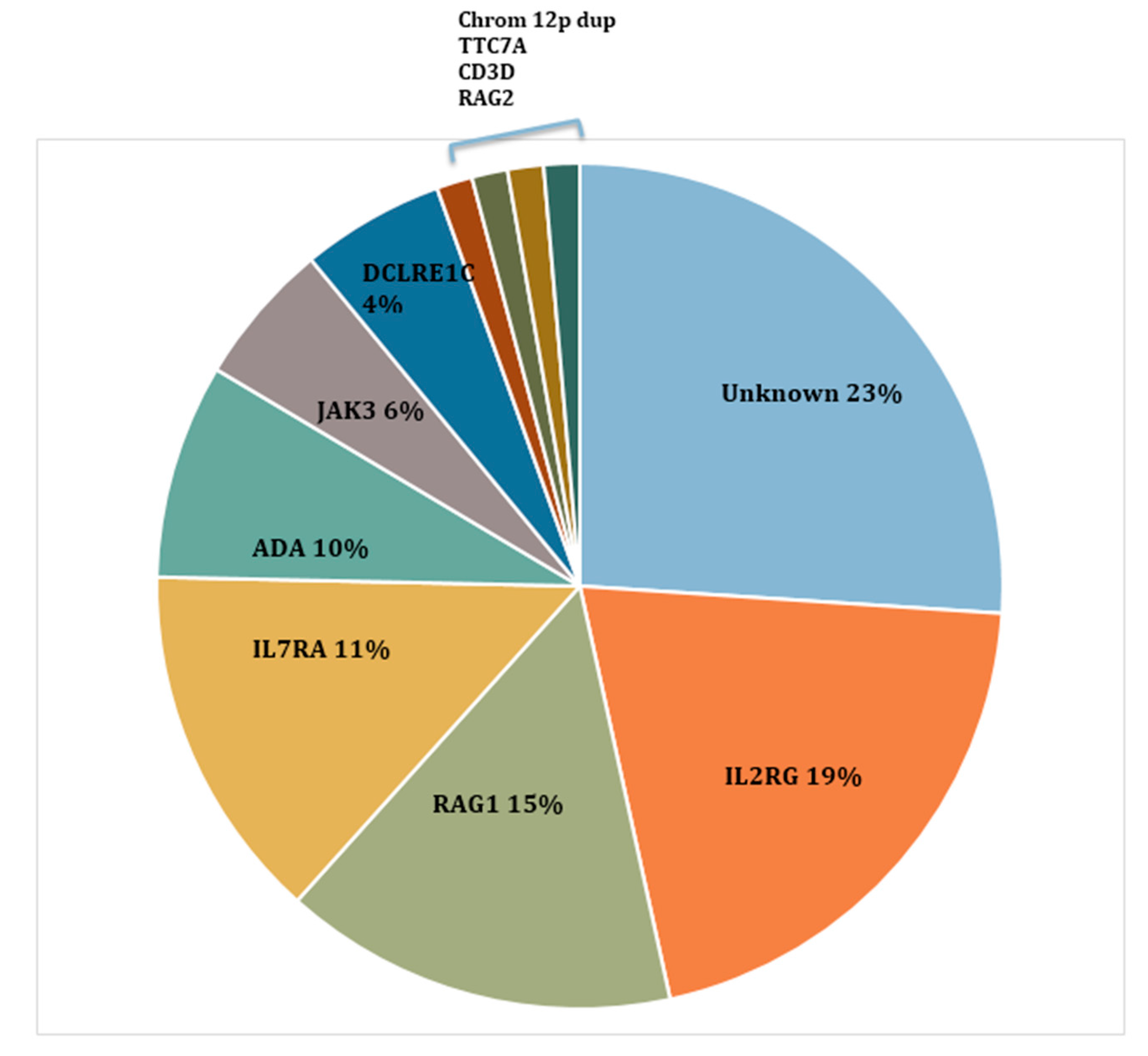
5. Different SCID Conditions Identified by SCID NBS
6. Different Non-SCID Conditions Identified by SCID NBS
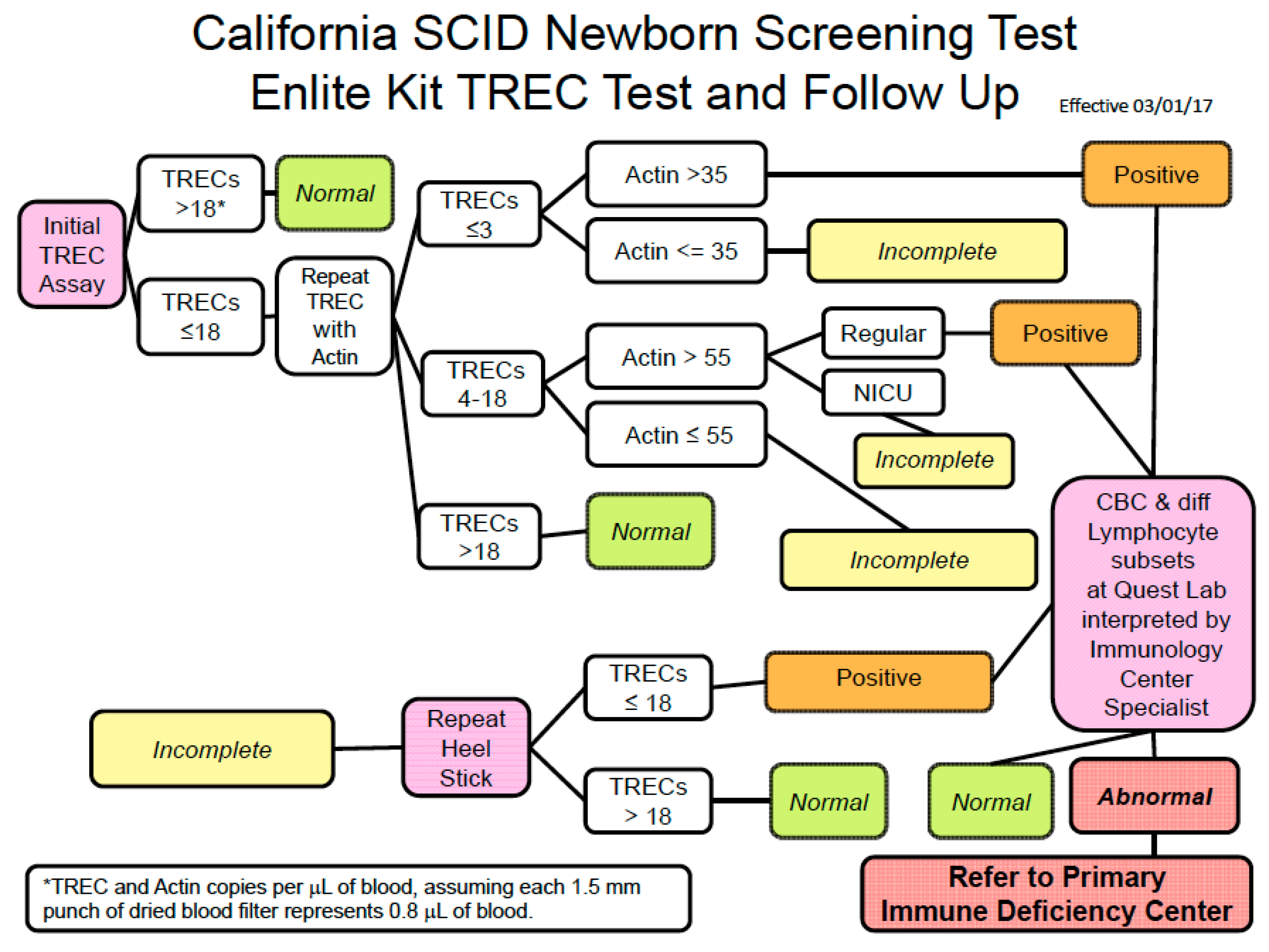
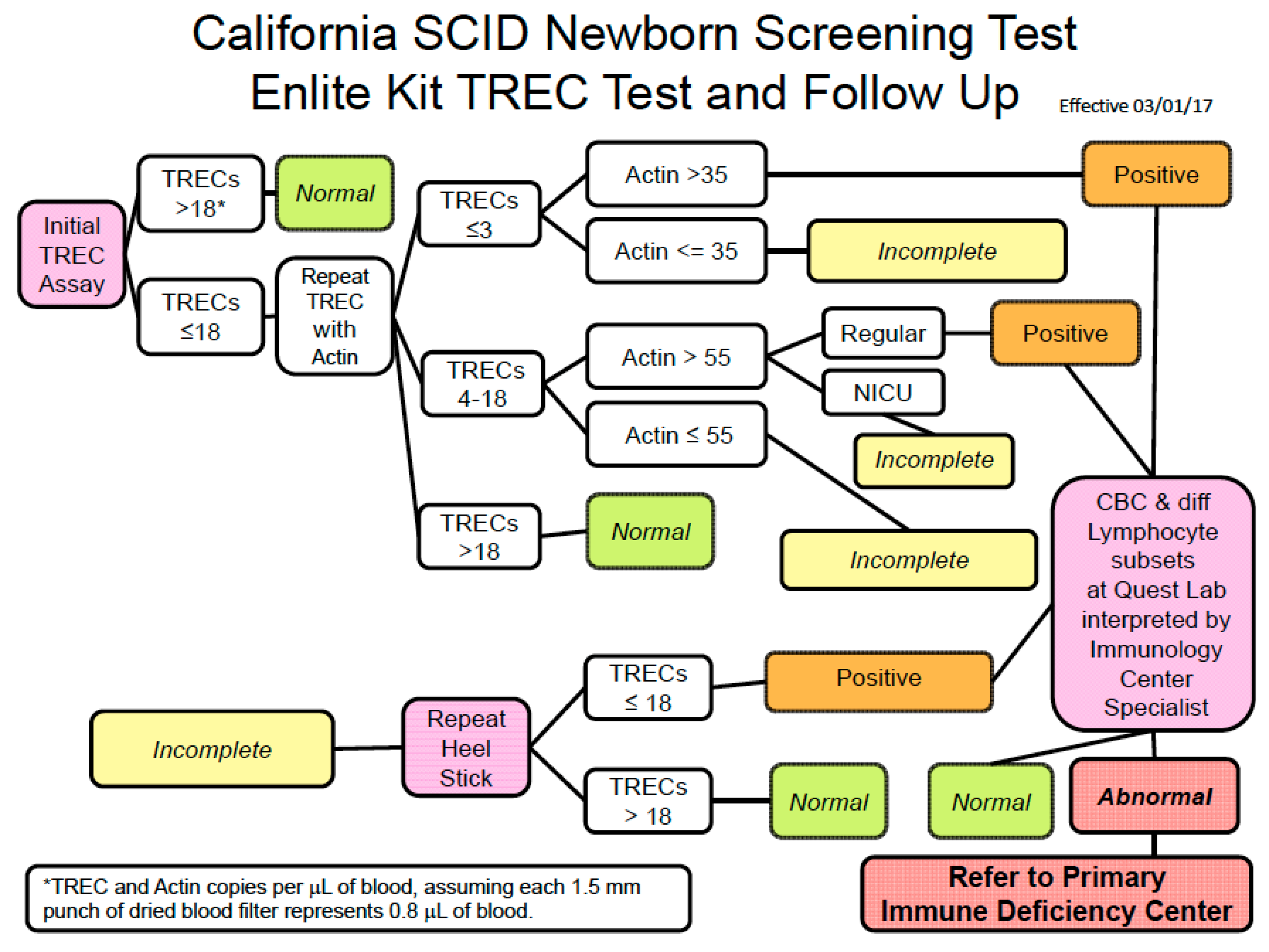
7. How Screening Is Conducted
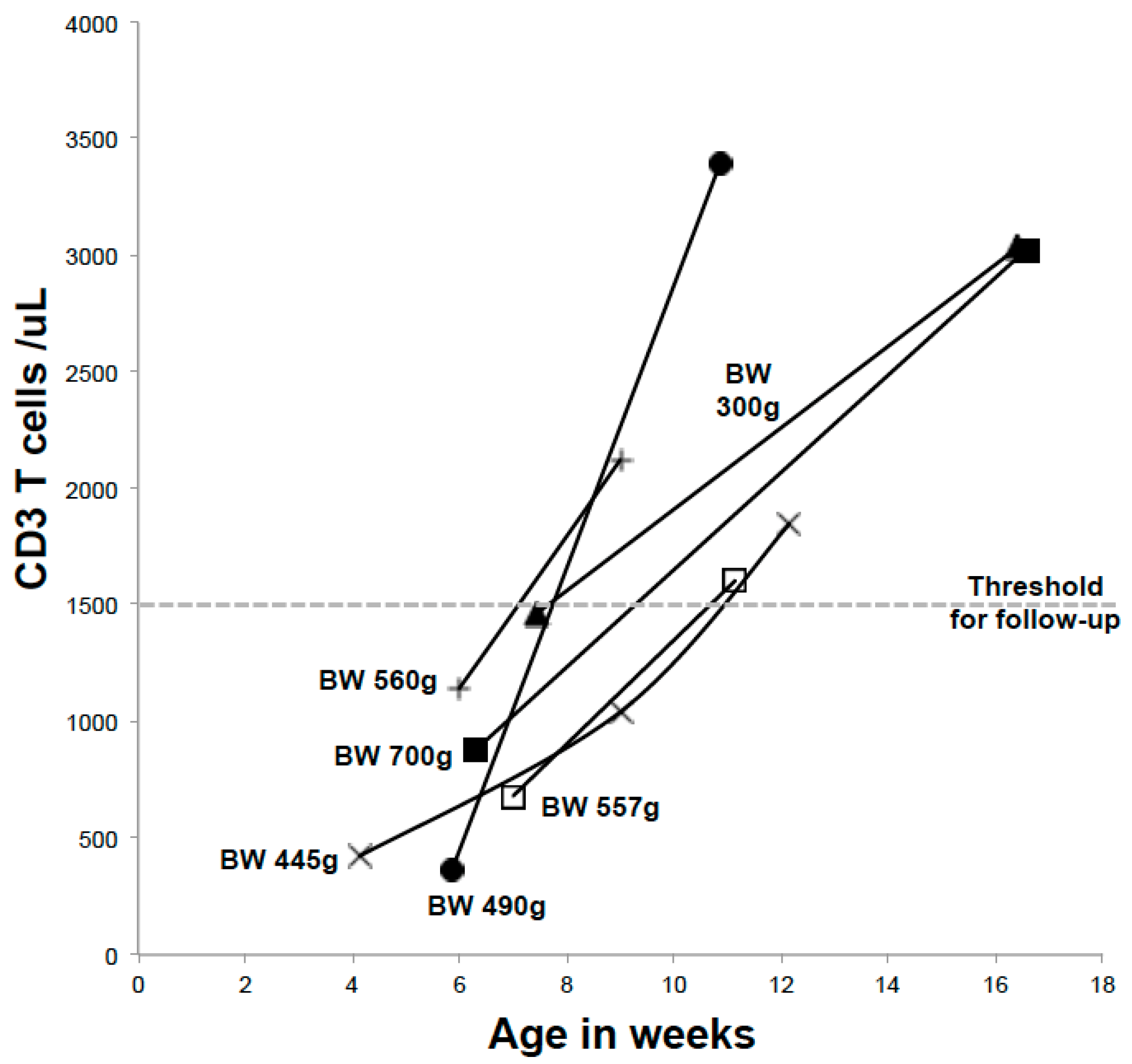
8. Preterm and Neonatal Intensive Care Infants
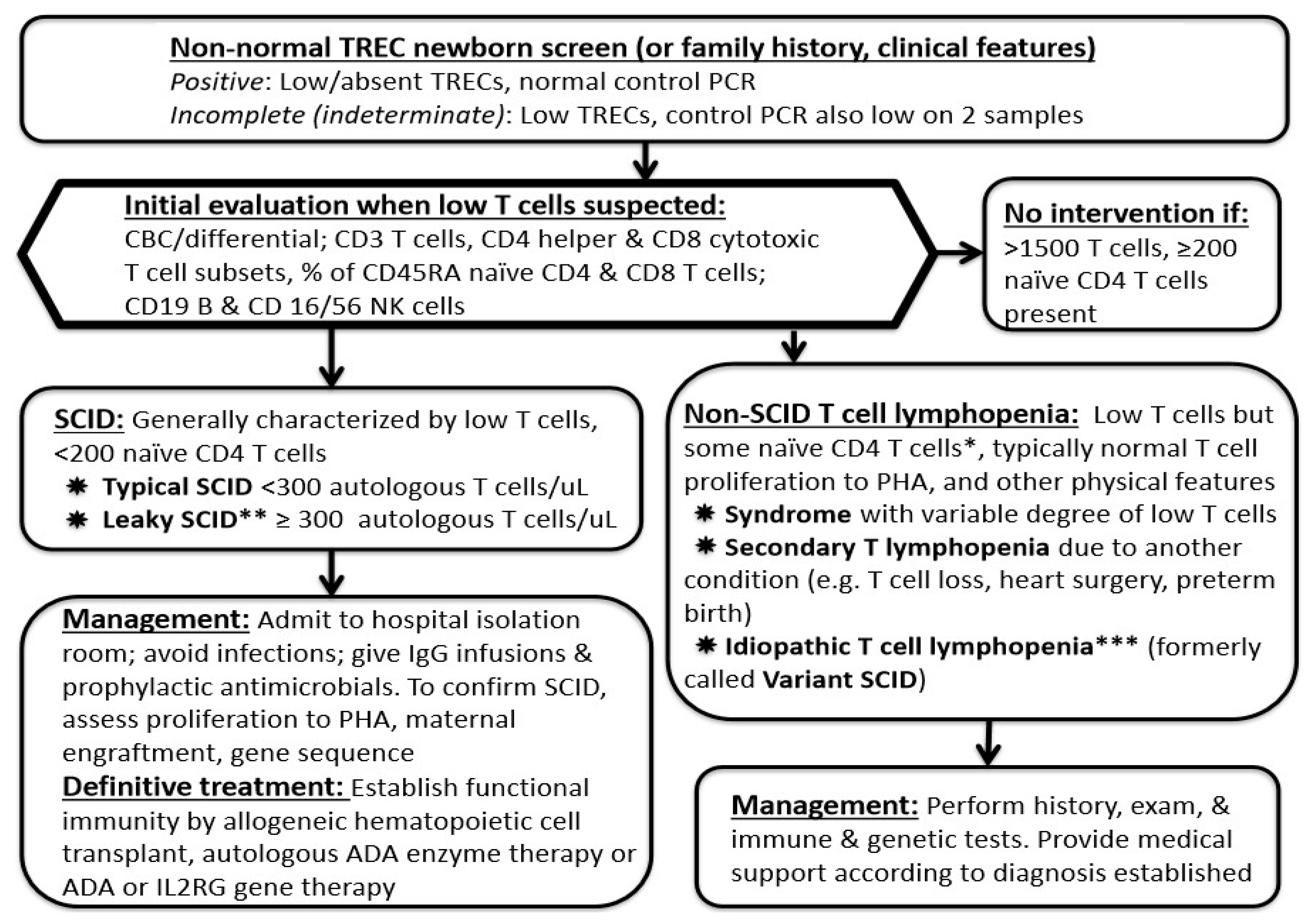
9. Early Management and Laboratory Assessment for a New Infant with Suspected SCID
10. Special Management Considerations
11. Treatment
12. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- IDF SCID Newborn Screening Campaign. Available online: http://primaryimmune.org/idf-advocacy-center/idf-scid-newborn-screening-campaign/ (accessed on 26 May 2017).
- Buckley, R.H. Molecular defects in human severe combined immunodeficiency and approaches to immune reconstitution. Annu. Rev. Immunol. 2004, 22, 625–655. [Google Scholar] [CrossRef] [PubMed]
- Ochs, H.D.; Puck, J.M. Primary Immunodeficiency Diseases: A Molecular and Genetic Approach; Oxford University Press: Oxford, UK, 2013; p. 92. [Google Scholar]
- Shearer, W.T.; Dunn, E.; Notarangelo, L.D.; Dvorak, C.C.; Puck, J.M.; Logan, B.R.; Griffith, L.M.; Kohn, D.B.; O’Reilly, R.J.; Fleisher, T.A.; et al. Establishing diagnostic criteria for severe combined immunodeficiency disease (SCID), leaky SCID, and omenn syndrome: The primary immune deficiency treatment consortium experience. J. Allergy Clin. Immunol. 2014, 133, 1092–1098. [Google Scholar] [CrossRef] [PubMed]
- Pai, S.Y.; Logan, B.R.; Griffith, L.M.; Buckley, R.H.; Parrott, R.E.; Dvorak, C.C.; Kapoor, N.; Hanson, I.C.; Filipovich, A.H.; Jyonouchi, S.; et al. Transplantation outcomes for severe combined immunodeficiency, 2000–2009. N. Engl. J. Med. 2014, 371, 434–446. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.; Puck, J.M. Development of population-based newborn screening for severe combined immunodeficiency. J. Allergy Clin. Immunol. 2005, 115, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Morinishi, Y.; Imai, K.; Nakagawa, N.; Sato, H.; Horiuchi, K.; Ohtsuka, Y.; Kaneda, Y.; Taga, T.; Hisakawa, H.; Miyaji, R.; et al. Identification of severe combined immunodeficiency by t-cell receptor excision circles quantification using neonatal guthrie cards. J. Pediatr. 2009, 155, 829–833. [Google Scholar] [CrossRef] [PubMed]
- Hitzig, W.H.; Biro, Z.; Bosch, H.; Huser, H.J. Agammaglobulinemia & alymphocytosis with atrophy of lymphatic tissue. Helv. Paediatr. Acta 1958, 13, 551–585. [Google Scholar] [PubMed]
- Giblett, E.; Anderson, J.; Cohen, F.; Pollara, B.; Meuwissen, H.J. Adenosine-deaminase deficiency in two patients with severely impaired cellular immunity. Lancet 1972, 300, 1067–1069. [Google Scholar] [CrossRef]
- Gatti, R.; Meuwissen, H.; Allen, H.; Hong, R.; Good, R.A. Immunological reconstitution of sex-linked lymphopenic immunological deficiency. Lancet 1968, 292, 1366–1369. [Google Scholar] [CrossRef]
- Noguchi, M.; Yi, H.; Rosenblatt, H.M.; Filipovich, A.H.; Adelstein, S.; Modi, W.S.; McBride, O.W.; Leonard, W.J. Interleukin-2 receptor γ chain mutation results in X-linked severe combined immunodeficiency in humans. Cell 1993, 73, 147–157. [Google Scholar] [CrossRef]
- Puck, J.M.; Deschenes, S.M.; Porter, J.C.; Dutra, A.S.; Brown, C.J.; Willard, H.F.; Henthorn, P.S. The interleukin-2 receptor γ chain maps to Xq13. 1 and is mutated in X-linked severe combined immunodeficiency, SCIDX1. Hum. Mol. Genet. 1993, 2, 1099–1104. [Google Scholar] [CrossRef] [PubMed]
- Picard, C.; Al-Herz, W.; Bousfiha, A.; Casanova, J.L.; Chatila, T.; Conley, M.E.; Rundles, C.C.; Etzioni, A.; Holland, S.M.; Klein, C.; et al. Primary immunodeficiency diseases: an update on the classification from the international union of immunological societies expert committee for primary immunodeficiency. J. Clin. Immunol. 2015, 35, 696–726. [Google Scholar] [CrossRef] [PubMed]
- Kwan, A.; Abraham, R.S.; Currier, R.; Brower, A.; Andruszewski, K.; Abbott, J.K.; Baker, M.; Ballow, M.; Bartoshesky, L.E.; Bonagura, V.R.; et al. Newborn screening for severe combined immunodeficiency in 11 screening programs in the United States. JAMA. Netw. 2014, 312, 729–738. [Google Scholar] [CrossRef] [PubMed]
- Dorsey, M.J.; Dvorak, C.C.; Cowan, M.J.; Puck, J.M. Treatment of infants identified as having severe combined immunodeficiency by means of newborn screening. J. Allergy Clin. Immunol. 2017, 139, 733–742. [Google Scholar] [CrossRef] [PubMed]
- Heimall, J.; Logan, B.R.; Cowan, M.J.; Notarangelo, L.D.; Puck, J.; Fleisher, T.; Griffith, L.M.; Kohn, D.B.; Pulsipher, M.A.; Shearer, W.; et al. Poor T cell reconstitution at 100 days after T cell-replete hematopoietic cell transplantation (HCT) for SCID is associated with later risk of death or need for 2nd transplant in the 6901 prospective study of the pidtc. Biol. Blood Marrow Transpl. 2016, 22, S101–S102. [Google Scholar] [CrossRef]
- Volpi, S.; Yamazaki, Y.; Brauer, P.M.; Rooijen, E.V.; Hayashida, A.; Slavotinek, A.; Kuehn, H.S.; Rocco, M.D.; Rivolta, C.; Bortolomai, I.; et al. EXTL3 mutations cause skeletal dysplasia, immune deficiency, and developmental delay. J. Exp. Med. 2017. [Google Scholar] [CrossRef] [PubMed]
- Hannon, W.H.; Abraham, R.S.; Kobrynski, L.; Vogt, L.; Adair, O.; Aznar, C.; Baker, M.W.; Brower, A.M.; Caggana, M.; Comeau, A.M.; et al. Newborn Blood Spot Screening for Severe Combined Immunodeficiency by Measurement of T-cell Receptor Excision Circles; Approved Guideline. Clin. Lab. Inst. Stand. 2013, 33, 1–75. [Google Scholar]
- Kwan, A.; Church, J.A.; Cowan, M.J.; Agarwal, R.; Kapoor, N.; Kohn, D.B.; Lewis, D.B.; McGhee, S.A.; Moore, T.B.; Stiehm, E.R.; et al. Newborn screening for severe combined immunodeficiency and t-cell lymphopenia in California: results of the first 2 years. J. Allergy Clin. Immunol. 2013, 132, 140–150. [Google Scholar] [CrossRef] [PubMed]
- Grunebaum, E.; Cutz, E.; Roifman, C.M. Pulmonary alveolar proteinosis in patients with adenosine deaminase deficiency. J. Allergy Clin. Immunol. 2012, 129, 1588–1593. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Conditions | % |
|---|---|
| Multisyndromes with variable T-cell deficiency | |
| DiGeorge/chromosome 22q11.2 deletion | 57 |
| Trisomy 21 | 15 |
| Ataxia telangiectasia | 3 |
| CHARGE syndrome | 2 |
| Secondary T lymphopenia | |
| Congenital cardiac anomalies | 25 |
| Other congenital anomalies | 38 |
| Vascular leakage, third spacing, hydrops | 13 |
| Neonatal leukemia | 3 |
| Maternal immunosuppressive medications | 3–5 |
| Extreme preterm birth (T cells become normal over time) | |
| Idiopathic T lymphopenia (no gene defect, few naïve cells, impaired T cell function) |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dorsey, M.; Puck, J. Newborn Screening for Severe Combined Immunodeficiency in the US: Current Status and Approach to Management. Int. J. Neonatal Screen. 2017, 3, 15. https://doi.org/10.3390/ijns3020015
Dorsey M, Puck J. Newborn Screening for Severe Combined Immunodeficiency in the US: Current Status and Approach to Management. International Journal of Neonatal Screening. 2017; 3(2):15. https://doi.org/10.3390/ijns3020015
Chicago/Turabian StyleDorsey, Morna, and Jennifer Puck. 2017. "Newborn Screening for Severe Combined Immunodeficiency in the US: Current Status and Approach to Management" International Journal of Neonatal Screening 3, no. 2: 15. https://doi.org/10.3390/ijns3020015
APA StyleDorsey, M., & Puck, J. (2017). Newborn Screening for Severe Combined Immunodeficiency in the US: Current Status and Approach to Management. International Journal of Neonatal Screening, 3(2), 15. https://doi.org/10.3390/ijns3020015