
Neonatal Screening for Primary Carnitine Deficiency: Lessons Learned from the Faroe Islands

 , and
, and
Abstract
:1. Introduction
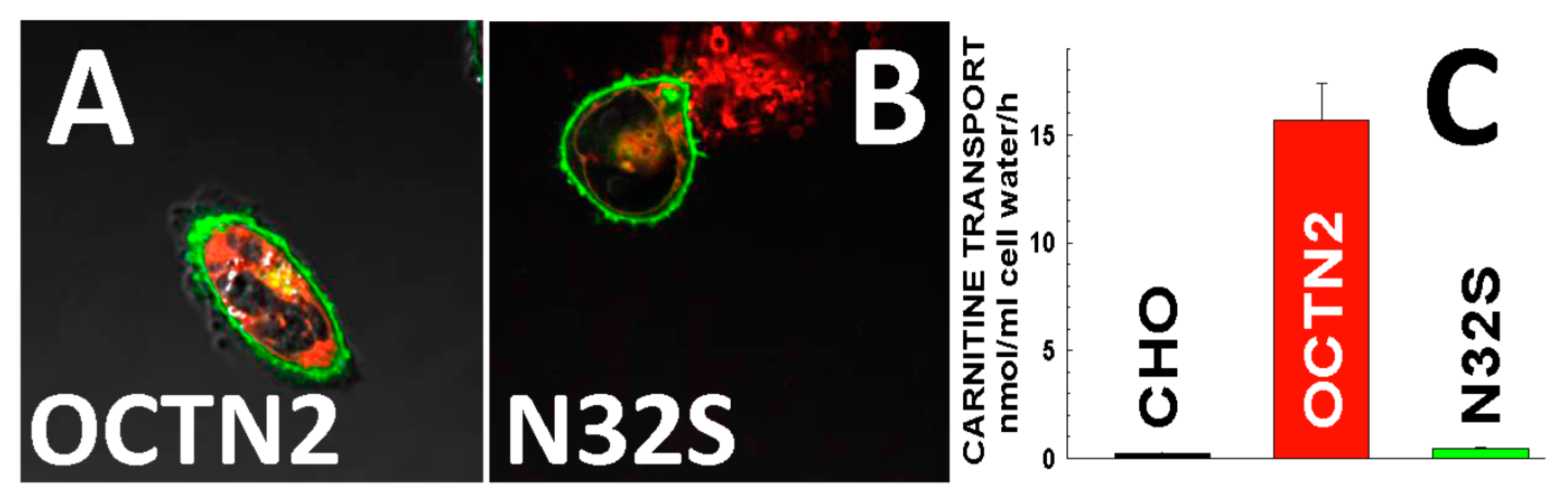
2. Molecular Bases and Prevalence of Primary Carnitine Deficiency
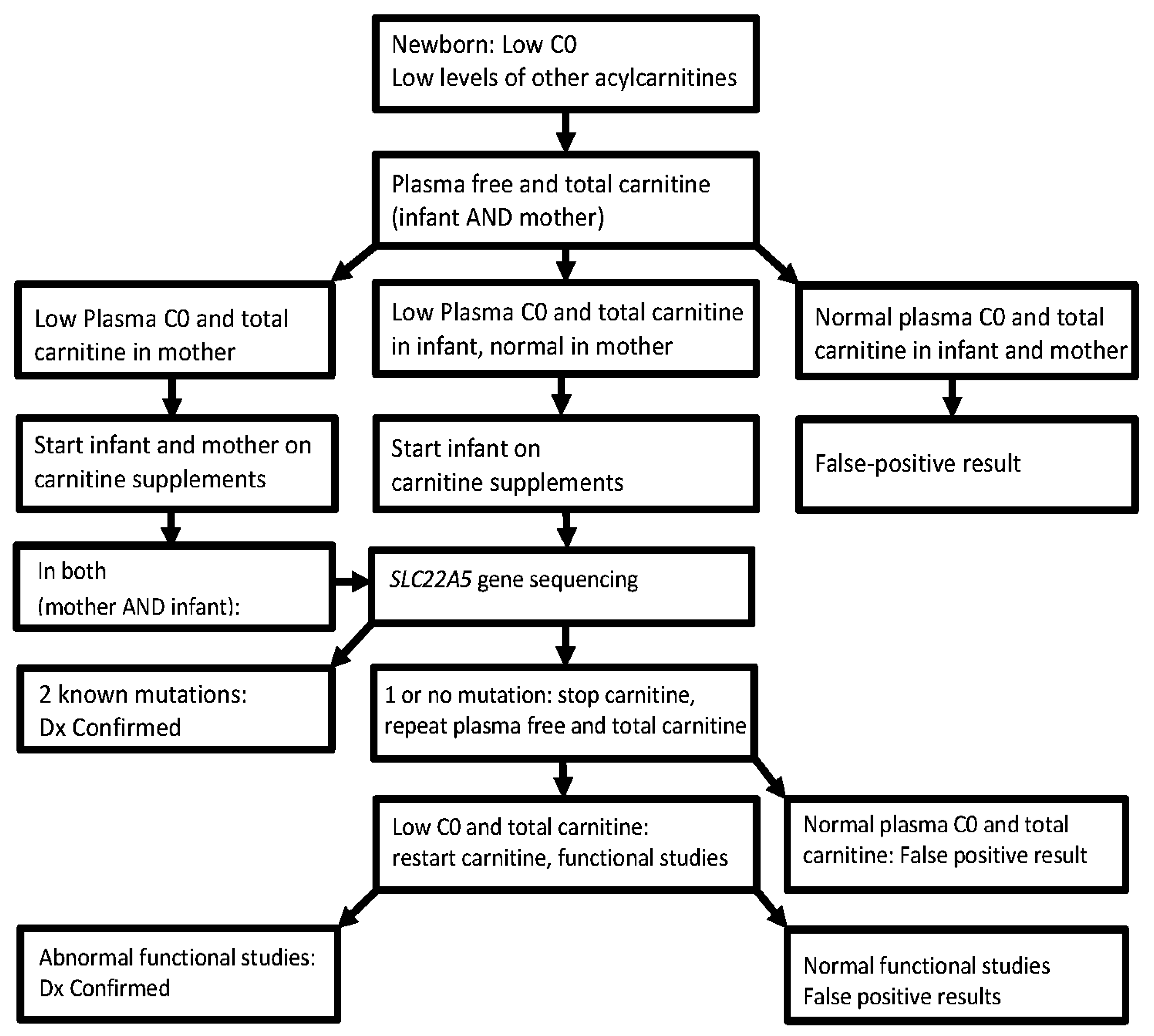
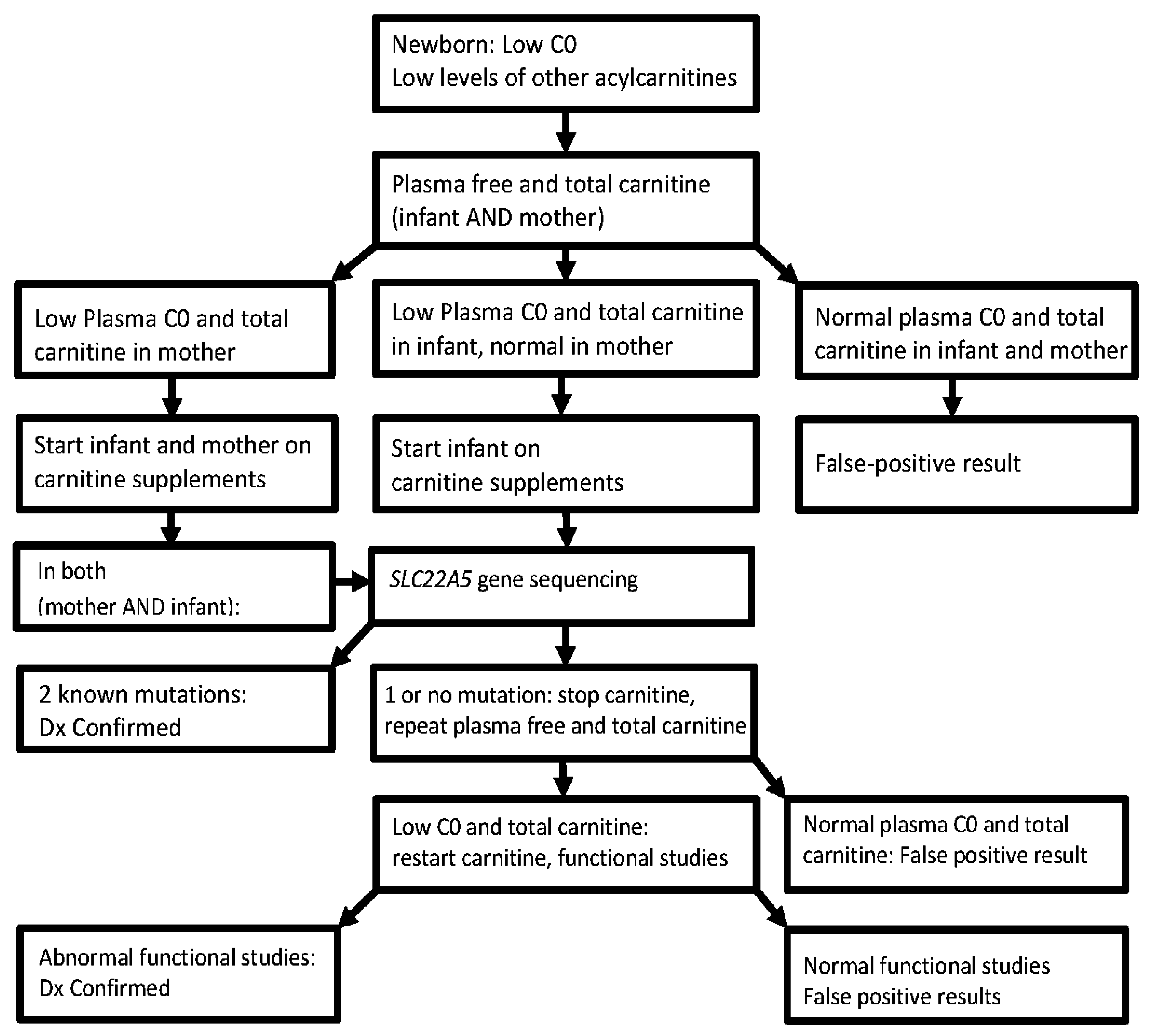
3. Diagnosis
4. Clinical Challenges/Limitations/Effectiveness of Neonatal Screening
5. Experience in the Faroe Islands
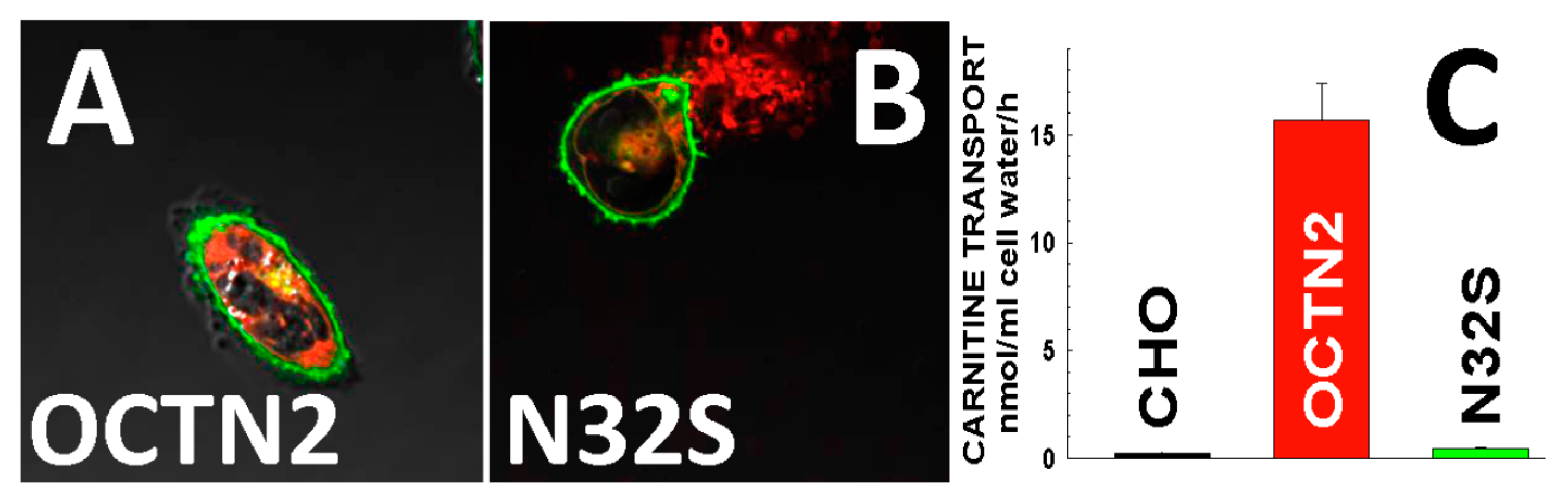
- (A)
- Expanded newborn screening for selected inborn errors of metabolism in Denmark, the Faroe Islands and Greenland was introduced in 2002, first as a pilot program and in January 2009 as a routine screening program [32]. Initial data indicated a high incidence of primary carnitine deficiency in the Faroe Islands—an archipelago of 18 islands, situated between the British Islands and Iceland [32,33], for which primary carnitine deficiency was added to the routine neonatal screening performed at the Statens Serum Institute in Copenhagen, Denmark.
- (B)
- In August 2009, following the sudden death of several young adults with undiagnosed and untreated primary carnitine deficiency [31], a nation-wide voluntary population screening-program was established in the Faroe Islands [34]. Out of a population of 49,949 people, until February 2016, 33,333 dried blood spot samples were analyzed by the Screening-Laboratory in Hannover, Germany (66.7% of the population).
- (C)
- Additionally all existing newborn screening cards of Faroese neonates born since 1986, stored at the Statens Serum Institute, were re-analyzed by tandem mass spectrometry.
- (D)
- Several cases of primary carnitine deficiency were diagnosed post mortem as well [31].
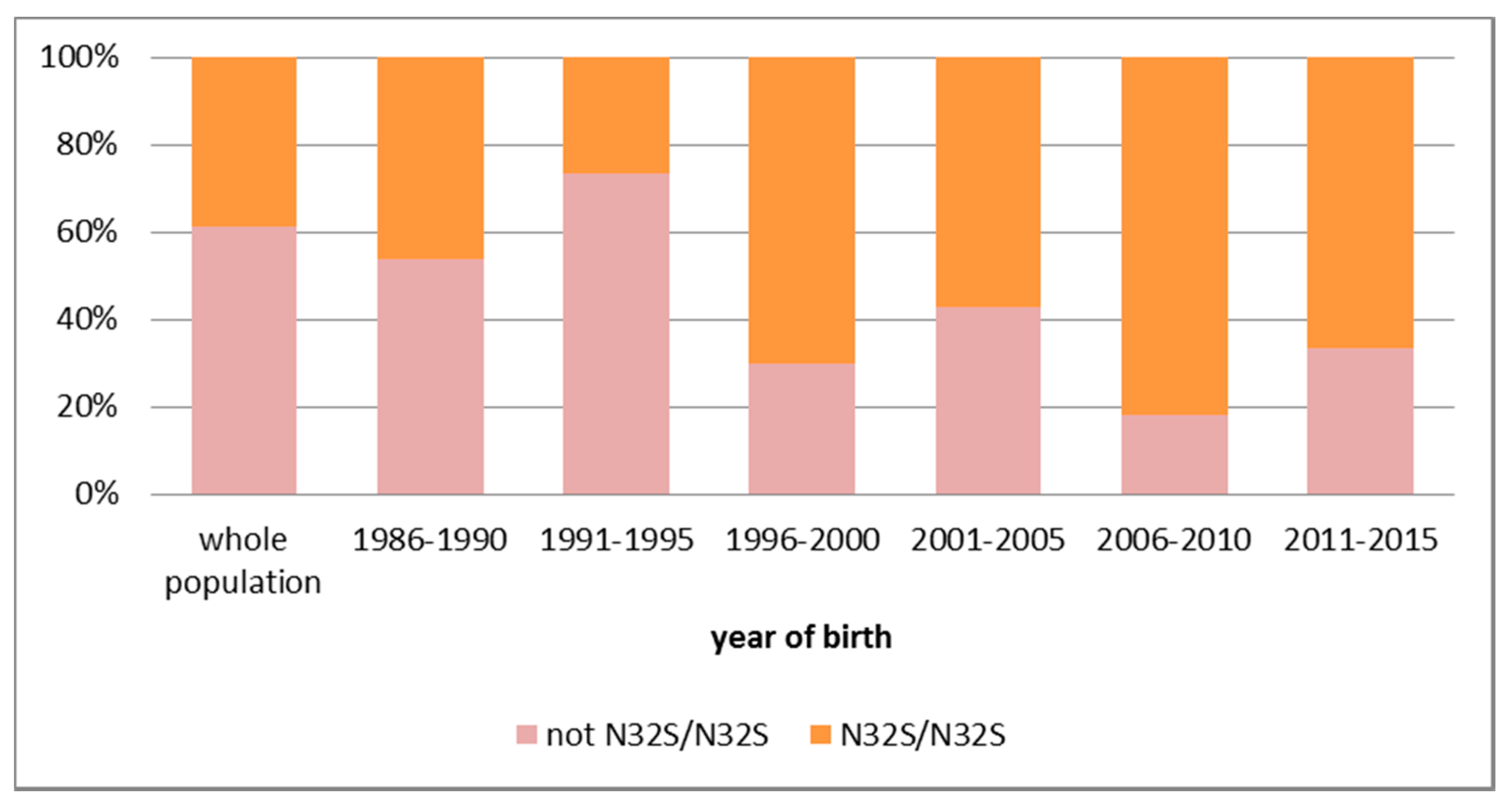
6. Molecular Bases of Primary Carnitine Deficiency in the Faroe Islands
7. Summary
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Longo, N.; Frigeni, M.; Pasquali, M. Carnitine transport and fatty acid oxidation. Biochim. Biophys. Acta 2016, 1863, 2422–2435. [Google Scholar] [CrossRef] [PubMed]
- Longo, N.; Amat di San Filippo, C.; Pasquali, M. Disorders of carnitine transport and the carnitine cycle. Am. J. Med. Genet. C Semin. Med. Genet. 2006, 142C, 77–85. [Google Scholar] [CrossRef] [PubMed]
- El-Hattab, A.W.; Scaglia, F. Disorders of carnitine biosynthesis and transport. Mol. Genet. Metab. 2015, 116, 107–112. [Google Scholar] [CrossRef] [PubMed]
- El-Hattab, A.W.; Li, F.Y.; Shen, J.; Powell, B.R.; Bawle, E.V.; Adams, D.J.; Wahl, E.; Kobori, J.A.; Graham, B.; Scaglia, F.; et al. Maternal systemic primary carnitine deficiency uncovered by newborn screening: Clinical, biochemical, and molecular aspects. Genet. Med. 2010, 12, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.C.; Tang, N.L.; Chien, Y.H.; Chen, C.A.; Lin, S.J.; Chiu, P.C.; Huang, A.C.; Hwu, W.L. Diagnoses of newborns and mothers with carnitine uptake defects through newborn screening. Mol. Genet. Metab. 2010, 100, 46–50. [Google Scholar] [CrossRef] [PubMed]
- Schimmenti, L.A.; Crombez, E.A.; Schwahn, B.C.; Heese, B.A.; Wood, T.C.; Schroer, R.J.; Bentler, K.; Cederbaum, S.; Sarafoglou, K.; McCann, M.; et al. Expanded newborn screening identifies maternal primary carnitine deficiency. Mol. Genet. Metab. 2007, 90, 441–445. [Google Scholar] [CrossRef] [PubMed]
- De Biase, I.; Champaigne, N.L.; Schroer, R.; Pollard, L.M.; Longo, N.; Wood, T. Primary carnitine deficiency presents atypically with long qt syndrome: A case report. JIMD Rep. 2012, 2, 87–90. [Google Scholar] [PubMed]
- Schoderbeck, M.; Auer, B.; Legenstein, E.; Genger, H.; Sevelda, P.; Salzer, H.; Marz, R.; Lohninger, A. Pregnancy-related changes of carnitine and acylcarnitine concentrations of plasma and erythrocytes. J. Perinat. Med. 1995, 23, 477–485. [Google Scholar] [CrossRef] [PubMed]
- Roe, C.; Ding, J. Mitochondrial fatty acid oxidation disorders. In The Metabolic and Molecular Bases of Inherited Disease, 8th ed.; Scriver, C., Beaudet, A., Sly, W., Valle, D., Eds.; McGraw-Hill: New York, NY, USA, 2001; pp. 2297–2326. [Google Scholar]
- Stanley, C.A. Carnitine deficiency disorders in children. Ann. N. Y. Acad. Sci. 2004, 1033, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Tamai, I.; Ohashi, R.; Nezu, J.; Yabuuchi, H.; Oku, A.; Shimane, M.; Sai, Y.; Tsuji, A. Molecular and functional identification of sodium ion-dependent, high affinity human carnitine transporter octn2. J. Biol. Chem. 1998, 273, 20378–20382. [Google Scholar] [CrossRef] [PubMed]
- Koizumi, A.; Nozaki, J.; Ohura, T.; Kayo, T.; Wada, Y.; Nezu, J.; Ohashi, R.; Tamai, I.; Shoji, Y.; Takada, G.; et al. Genetic epidemiology of the carnitine transporter octn2 gene in a japanese population and phenotypic characterization in japanese pedigrees with primary systemic carnitine deficiency. Hum. Mol. Genet. 1999, 8, 2247–2254. [Google Scholar] [CrossRef] [PubMed]
- Wilcken, B.; Wiley, V.; Sim, K.G.; Carpenter, K. Carnitine transporter defect diagnosed by newborn screening with electrospray tandem mass spectrometry. J. Pediatr. 2001, 138, 581–584. [Google Scholar] [CrossRef] [PubMed]
- Therrell, B.L., Jr.; Lloyd-Puryear, M.A.; Camp, K.M.; Mann, M.Y. Inborn errors of metabolism identified via newborn screening: Ten-year incidence data and costs of nutritional interventions for research agenda planning. Mol. Genet. Metab. 2014, 113, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, J.; Kober, L.; Lund, A.M.; Nielsen, O.W. Primary carnitine deficiency in the faroe islands: Health and cardiac status in 76 adult patients diagnosed by screening. J. Inherit. Metab. Dis. 2014, 37, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Taroni, F.; Garavaglia, B.; Longo, N. Functional analysis of mutations in the octn2 transporter causing primary carnitine deficiency: Lack of genotype-phenotype correlation. Hum. Mutat. 2000, 16, 401–407. [Google Scholar] [CrossRef]
- Wang, Y.; Ye, J.; Ganapathy, V.; Longo, N. Mutations in the organic cation/carnitine transporter octn2 in primary carnitine deficiency. Proc. Natl. Acad. Sci. USA 1999, 96, 2356–2360. [Google Scholar] [CrossRef] [PubMed]
- Rose, E.C.; di San Filippo, C.A.; Ndukwe Erlingsson, U.C.; Ardon, O.; Pasquali, M.; Longo, N. Genotype-phenotype correlation in primary carnitine deficiency. Hum. Mutat. 2012, 33, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Korman, S.H.; Ye, J.; Gargus, J.J.; Gutman, A.; Taroni, F.; Garavaglia, B.; Longo, N. Phenotype and genotype variation in primary carnitine deficiency. Genet. Med. 2001, 3, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Kelly, M.A.; Cowan, T.M.; Longo, N. A missense mutation in the OCTN2 gene associated with residual carnitine transport activity. Hum. Mutat. 2000, 15, 238–245. [Google Scholar] [CrossRef]
- Lamhonwah, A.M.; Olpin, S.E.; Pollitt, R.J.; Vianey-Saban, C.; Divry, P.; Guffon, N.; Besley, G.T.; Onizuka, R.; De Meirleir, L.J.; Cvitanovic-Sojat, L.; et al. Novel OCTN2 mutations: No genotype-phenotype correlations: Early carnitine therapy prevents cardiomyopathy. Am. J. Med. Genet. 2002, 111, 271–284. [Google Scholar] [CrossRef] [PubMed]
- De Sain-van der Velden, M.G.; Diekman, E.F.; Jans, J.J.; van der Ham, M.; Prinsen, B.H.; Visser, G.; Verhoeven-Duif, N.M. Differences between acylcarnitine profiles in plasma and bloodspots. Mol. Genet. Metab. 2013, 110, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Scaglia, F.; Longo, N. Primary and secondary alterations of neonatal carnitine metabolism. Semin. Perinatol. 1999, 23, 152–161. [Google Scholar] [CrossRef]
- Vijay, S.; Patterson, A.; Olpin, S.; Henderson, M.J.; Clark, S.; Day, C.; Savill, G.; Walter, J.H. Carnitine transporter defect: Diagnosis in asymptomatic adult women following analysis of acylcarnitines in their newborn infants. J. Inherit. Metab. Dis. 2006, 29, 627–630. [Google Scholar] [CrossRef] [PubMed]
- Scaglia, F.; Wang, Y.; Singh, R.H.; Dembure, P.P.; Pasquali, M.; Fernhoff, P.M.; Longo, N. Defective urinary carnitine transport in heterozygotes for primary carnitine deficiency. Genet. Med. 1998, 1, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Stanley, C.A. Carnitine disorders. Adv. Pediatr. 1995, 42, 209–242. [Google Scholar] [PubMed]
- Pochini, L.; Scalise, M.; Galluccio, M.; Indiveri, C. OCTN cation transporters in health and disease: Role as drug targets and assay development. J. Biomol. Screen. 2013, 18, 851–867. [Google Scholar] [CrossRef] [PubMed]
- Hirano, T.; Yasuda, S.; Osaka, Y.; Kobayashi, M.; Itagaki, S.; Iseki, K. Mechanism of the inhibitory effect of zwitterionic drugs (levofloxacin and grepafloxacin) on carnitine transporter (OCTN2) in caco-2 cells. Biochim. Biophys. Acta 2006, 1758, 1743–1750. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Lancaster, C.S.; Zuo, Z.; Hu, S.; Chen, Z.; Rubnitz, J.E.; Baker, S.D.; Sparreboom, A. Inhibition of OCTN2-mediated transport of carnitine by etoposide. Mol. Cancer Ther. 2012, 11, 921–929. [Google Scholar] [CrossRef] [PubMed]
- Pochini, L.; Scalise, M.; Indiveri, C. Inactivation by omeprazole of the carnitine transporter (OCTN2) reconstituted in liposomes. Chem. Biol. Interact. 2009, 179, 394–401. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, J.; Nielsen, O.W.; Lund, A.M.; Kober, L.; Djurhuus, H. Primary carnitine deficiency and pivalic acid exposure causing encephalopathy and fatal cardiac events. J. Inherit. Metab. Dis. 2013, 36, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Lund, A.M.; Hougaard, D.M.; Simonsen, H.; Andresen, B.S.; Christensen, M.; Duno, M.; Skogstrand, K.; Olsen, R.K.; Jensen, U.G.; Cohen, A.; et al. Biochemical screening of 504,049 newborns in denmark, the faroe islands and greenland--experience and development of a routine program for expanded newborn screening. Mol. Genet. Metab. 2012, 107, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Lund, A.M.; Joensen, F.; Hougaard, D.M.; Jensen, L.K.; Christensen, E.; Christensen, M.; Norgaard-Petersen, B.; Schwartz, M.; Skovby, F. Carnitine transporter and holocarboxylase synthetase deficiencies in the faroe islands. J. Inherit. Metab. Dis. 2007, 30, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, J.; Nielsen, O.W.; Janzen, N.; Duno, M.; Gislason, H.; Kober, L.; Steuerwald, U.; Lund, A.M. Carnitine levels in 26,462 individuals from the nationwide screening program for primary carnitine deficiency in the faroe islands. J. Inherit. Metab. Dis. 2014, 37, 215–222. [Google Scholar] [CrossRef] [PubMed]
- McGoey, R.R.; Marble, M. Positive newborn screen in a normal infant of a mother with asymptomatic very long-chain acyl-coa dehydrogenase deficiency. J. Pediatr. 2011, 158, 1031–1032. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, J.; Hougaard, D.M.; Sandhu, M.; Fjaellergaard, K.; Petersen, P.R.; Steuerwald, U.; Lund, A.M. Primary carnitine deficiency: Is fetal development affected and can newborn screening be improved? J. Inherit. Metab. Dis. 2017. [Google Scholar] [CrossRef]
- Rasmussen, J.; Lund, A.M.; Risom, L.; Wibrand, F.; Gislason, H.; Nielsen, O.W.; Kober, L.; Duno, M. Residual OCTN2 transporter activity, carnitine levels and symptoms correlate in patients with primary carnitine deficiency. Mol. Genet. Metab. Rep. 2014, 1, 241–248. [Google Scholar] [CrossRef] [PubMed]
- SLC22A5 gene. Available online: http://exac.broadinstitute.org/gene/ENSG00000197375 (accessed on 4 February 2017).
- De Boer, L.; Kluijtmans, L.A.; Morava, E. Primary carnitine (OCTN2) deficiency without neonatal carnitine deficiency. JIMD Rep. 2013, 10, 39–40. [Google Scholar] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Year of Birth | 1999–2000 | 2001–2005 | 2006–2010 | 2011–2015 | Overall |
|---|---|---|---|---|---|
| Cases detected by NBS [incidence of PCD] | 4 | 7 | 3 (+3) * | 4 (+2) * | 23 |
| [1:330] | [1:496] | [1:543] | [1:512] | [1:483] | |
| Additional cases (negative NBS) detected with population screening | 0 | 7 | 4 (+1) ** | 0 | 12 |
| Total incidence of PCD | 1:330 | 1:248 | 1:296 | 1:512 | 1:318 |
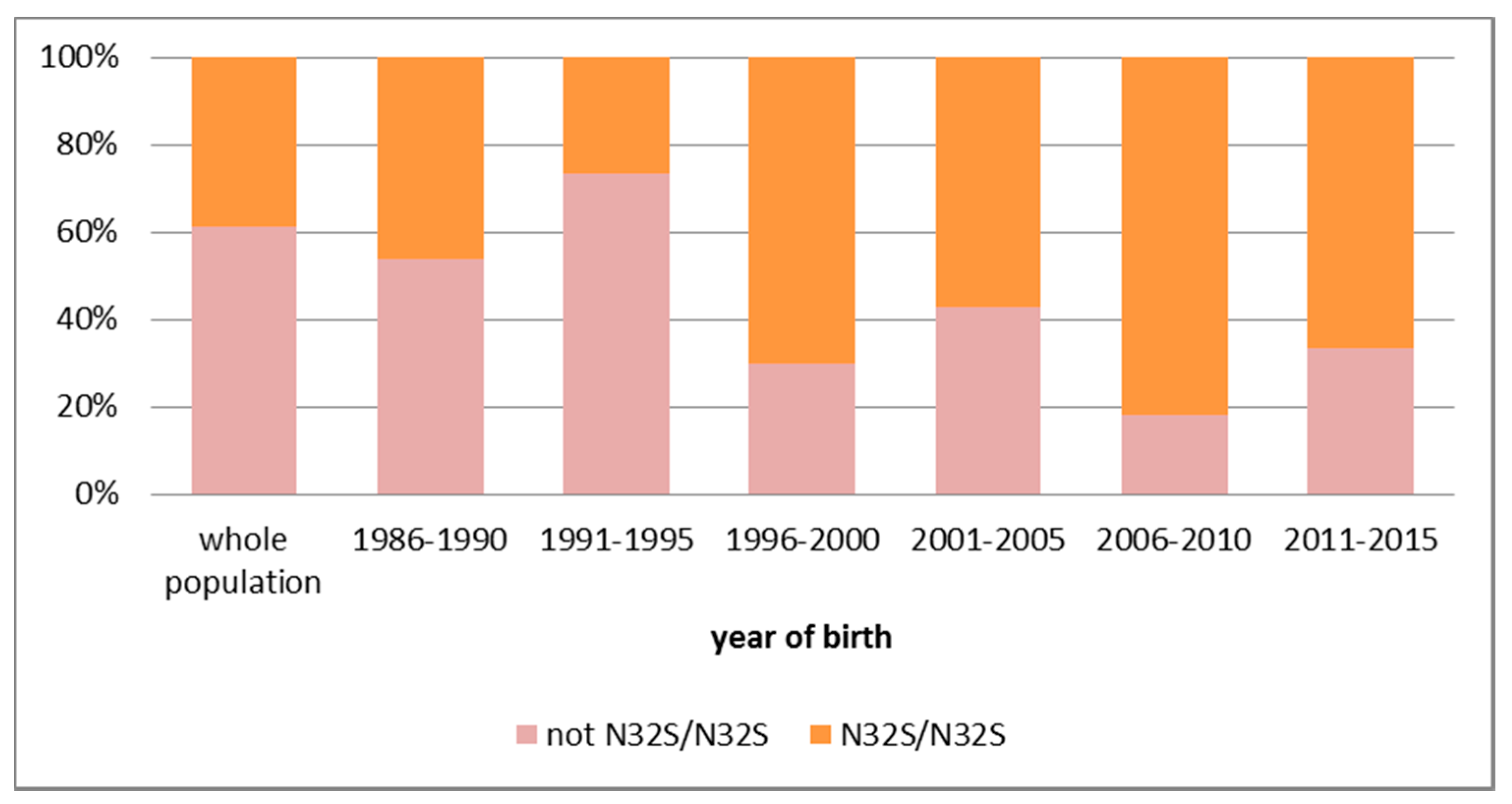
| Year of Birth | 1901–2015 | 1986–1990 | 1991–1995 | 1996–2000 | 2001–2005 | 2006–2010 | 2011–2015 |
|---|---|---|---|---|---|---|---|
| Individuals | 49,949 | 4322 | 3748 | 3283 | 3470 | 3260 | 3071 |
| Screened at >2 months (%) | 33,333 | 2954 | 2769 | 2806 | 2693 | 2038 | 713 |
| 66.7% | 68.3% | 73.9% | 85.5% | 77.6% | 62.5% | 23.2% | |
| No. of PCD-cases | 168 | 12 | 15 | 10 | 14 | 11 | 6 |
| Prevalence | 1:297 | 1:360 | 1:250 | 1:328 | 1:248 | 1:296 | 1:512 |
| Genotype | Number | Positive in Neonatal (Percent Identified) |
|---|---|---|
| c.95A>G, p.N32S/c.95A>G, p.N32S * | 20 ** | 14 (70%) |
| c.95A>G, p.N32S/c.825-52G>A (splice) | 2 | 1 (50%) |
| c.95A>G, p.N32S/RH (risk haplotype) | 6 | 2 (33%) |
| Others | 2 | 1 (50%) |
| All cases | 30 * | 18 (60%) |
© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Steuerwald, U.; Lund, A.M.; Rasmussen, J.; Janzen, N.; Hougaard, D.M.; Longo, N. Neonatal Screening for Primary Carnitine Deficiency: Lessons Learned from the Faroe Islands. Int. J. Neonatal Screen. 2017, 3, 1. https://doi.org/10.3390/ijns3010001
Steuerwald U, Lund AM, Rasmussen J, Janzen N, Hougaard DM, Longo N. Neonatal Screening for Primary Carnitine Deficiency: Lessons Learned from the Faroe Islands. International Journal of Neonatal Screening. 2017; 3(1):1. https://doi.org/10.3390/ijns3010001
Chicago/Turabian StyleSteuerwald, Ulrike, Allan M. Lund, Jan Rasmussen, Nils Janzen, David M. Hougaard, and Nicola Longo. 2017. "Neonatal Screening for Primary Carnitine Deficiency: Lessons Learned from the Faroe Islands" International Journal of Neonatal Screening 3, no. 1: 1. https://doi.org/10.3390/ijns3010001
APA StyleSteuerwald, U., Lund, A. M., Rasmussen, J., Janzen, N., Hougaard, D. M., & Longo, N. (2017). Neonatal Screening for Primary Carnitine Deficiency: Lessons Learned from the Faroe Islands. International Journal of Neonatal Screening, 3(1), 1. https://doi.org/10.3390/ijns3010001