
Rethinking Newborn Screening: A Case of GALM Deficiency

 , and
, and
Abstract
1. Introduction
2. Case Description
3. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Beutler, E.; Baluda, M.C.; Sturgeon, P.; Day, R. A new genetic abnormality resulting in galactose-1-phosphate uridyltransferase deficiency. Lancet 1965, 285, 353–354. [Google Scholar] [CrossRef]
- Isselbacher, K.J. Galactose metabolism and galactosemia. Am. J. Med. 1959, 26, 715–723. [Google Scholar] [CrossRef] [PubMed]
- Wong, L.J.; Frey, P.A. Galactose 1-phosphate uridylyltransferase. Isolation of a uridylyl-enzyme intermediate. J. Biol. Chem. 1974, 249, 2322–2324. [Google Scholar] [CrossRef] [PubMed]
- Carney, A.E.; Sanders, R.D.; Garza, K.R.; McGaha, L.A.; Bean, L.J.H.; Coffee, B.W.; Thomas, J.W.; Cutler, D.J.; Kurtkaya, N.L.; Fridovich-Keil, J.L. Origins, distribution and expression of the Duarte-2 (D2) allele of galactose-1-phosphate uridylyltransferase. Hum. Mol. Genet. 2009, 18, 1624–1632. [Google Scholar] [CrossRef] [PubMed]
- Stambolian, D.; Ai, Y.; Sidjanin, D.; Nesburn, K.; Sathe, G.; Rosenberg, M.; Bergsma, D.J. Cloning of the galactokinase cDNA and identification of mutations in two families with cataracts. Nat. Genet. 1995, 10, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Timson, D.J. Functional analysis of disease-causing mutations in human UDP-galactose 4-epimerase. FEBS J. 2005, 272, 6170–6177. [Google Scholar] [CrossRef] [PubMed]
- Wada, Y.; Kikuchi, A.; Arai-Ichinoi, N.; Sakamoto, O.; Takezawa, Y.; Iwasawa, S.; Niihori, T.; Nyuzuki, H.; Nakajima, Y.; Ogawa, E.; et al. Biallelic GALM pathogenic variants cause a novel type of galactosemia. Genet. Med. Off. J. Am. Coll. Med. Genet. 2019, 21, 1286–1294. [Google Scholar] [CrossRef]
- Timson, D.J.; Reece, R.J. Identification and characterisation of human aldose 1-epimerase. FEBS Lett. 2003, 543, 21–24. [Google Scholar] [CrossRef] [PubMed]
- Yazici, H.; Canda, E.; Altınok, Y.A.; Ucar, S.K.; Coker, M. Two siblings with galactose mutarotase deficiency: Clinical differences. JIMD Rep. 2022, 63, 25–28. [Google Scholar] [CrossRef] [PubMed]
- Carbonell, A.U.; Cho, C.H.; Tindi, J.O.; Counts, P.A.; Bates, J.C.; Erdjument-Bromage, H.; Cvejic, S.; Iaboni, A.; Kvint, I.; Rosensaft, J.; et al. Haploinsufficiency in the ANKS1B gene encoding AIDA-1 leads to a neurodevelopmental syndrome. Nat. Commun. 2019, 10, 3529. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Pintos, P.; Camba-Garea, M.J.; López-Pardo, B.M.; de Juan, J.A.C.; Bóveda, M.D.; Barbosa-Gouveia, S.; Vázquez-Mosquera, M.E.; Barros-Angueira, F.; Patiño, R.F.; Couce, M.L. Clinical and biochemical evolution after partial dietary liberalization of two cases of galactosemia due to galactose mutarotase deficiency. BMC Pediatr. 2024, 24, 620. [Google Scholar] [CrossRef]
- Mikami-Saito, Y.; Wada, Y.; Arai-Ichinoi, N.; Nakajima, Y.; Suzuki-Ajihara, S.; Murayama, K.; Tanaka, T.; Numakura, C.; Hamazaki, T.; Igarashi, N.; et al. Phenotypic and genetic spectra of galactose mutarotase deficiency: A nationwide survey conducted in Japan. Genet. Med. Off. J. Am. Coll. Med. Genet. 2024, 26, 101165. [Google Scholar] [CrossRef]
- Chen, H.-A.; Hsu, R.-H.; Chen, L.-C.; Lee, N.-C.; Chiu, P.-C.; Hwu, W.-L.; Chien, Y.-H. Twelve-year review of galactosemia newborn screening in Taiwan: Evolving methods and insights. Mol. Genet. Metab. Rep. 2024, 38, 101048. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, A.; Wada, Y.; Ohura, T.; Kure, S. The Discovery of GALM Deficiency (Type IV Galactosemia) and Newborn Screening System for Galactosemia in Japan. Int. J. Neonatal Screen. 2021, 7, 68. [Google Scholar] [CrossRef] [PubMed]
- Iwasawa, S.; Kikuchi, A.; Wada, Y.; Arai-Ichinoi, N.; Sakamoto, O.; Tamiya, G.; Kure, S. The prevalence of GALM mutations that cause galactosemia: A database of functionally evaluated variants. Mol. Genet. Metab. 2019, 126, 362–367. [Google Scholar] [CrossRef] [PubMed]
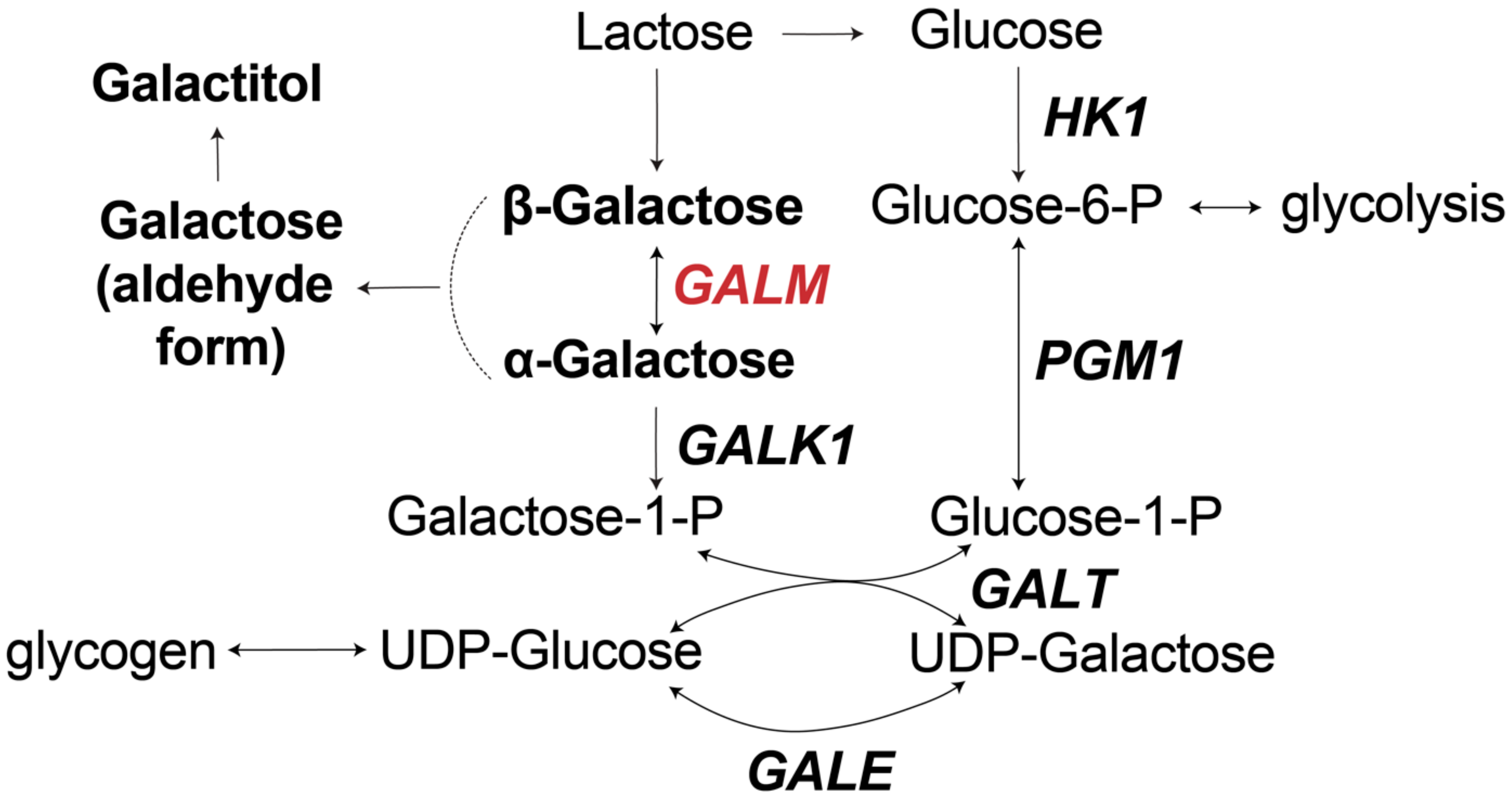
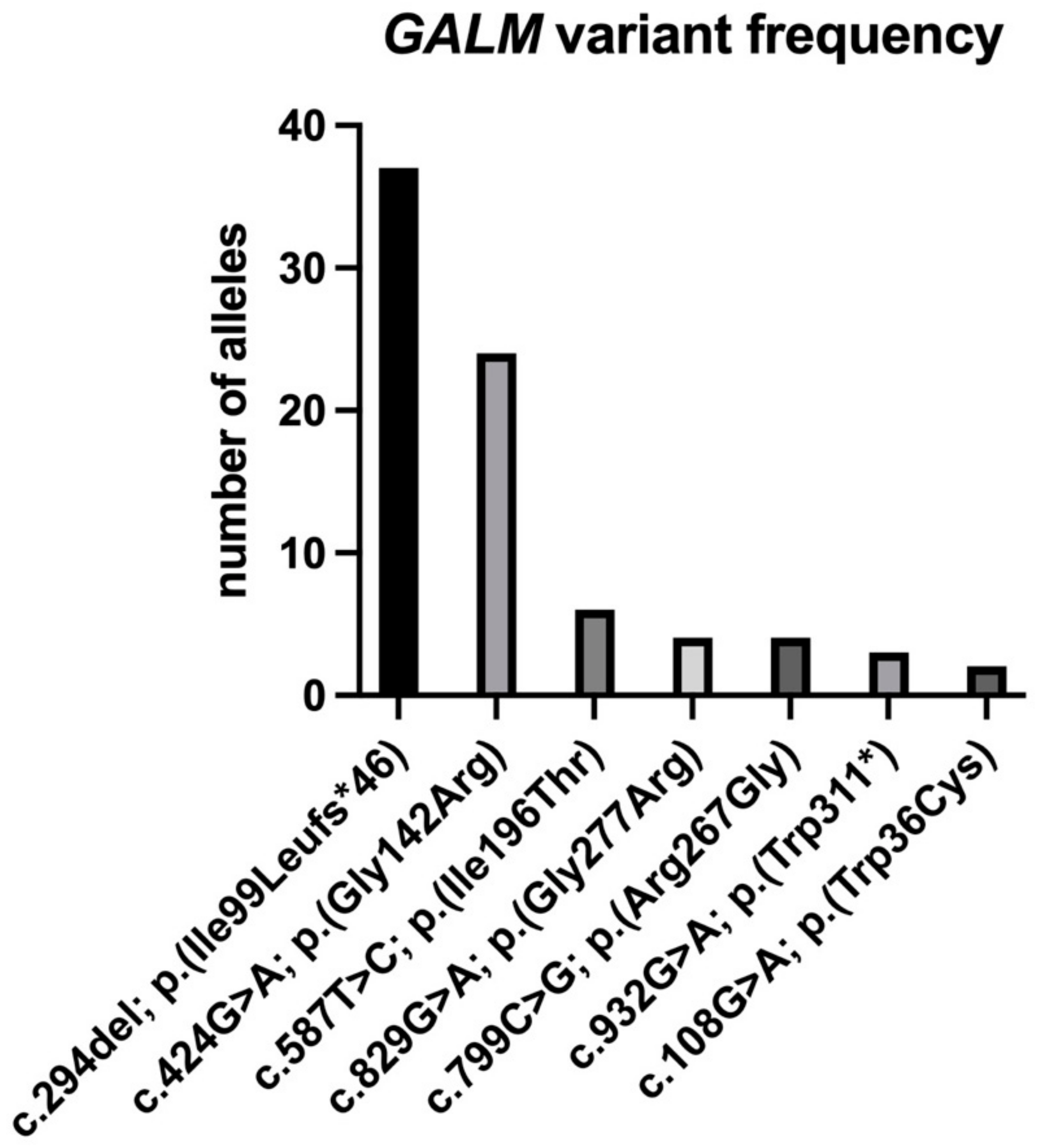

{kind=link}
{kind=link}
| Patient | Publication | NBS Total Galactose (Galactose + gal-1-p) in mg/dL * | NBS Galactose in mg/dL | NBS Gal-1-P in mg/dL # | Highest (Total) Galactose in Blood in mg/dL * (Age) | Gal-Restricted Diet (Age Started-Ended) | Clinical Presentation |
|---|---|---|---|---|---|---|---|
| P1 | This publication | Repeated measurements 842; 932; 1000 µmol/L * N< 1350 µmol/L * | x | x | 1000 µmol/L * (4 do, tgal) | 17 mo—ongoing | Nystagmus, developmental delay (probably caused by a second diagnosis of ANKS1B frameshift variant), neonatal jaundice. |
| P2 | Sánchez-Pintos et al., 2024 (pt1) [10] | 1st sample: 11 2nd sample: 21.6 N < 18 | x | 1st sample: 0.38 mmol/L # 2nd sample: 0.12 mmol/L # N < 0.7 | 21.6 (15 do, tgal) | 3–6 mo and 7–18 mo | No symptoms reported. |
| P3 | Sánchez-Pintos et al., 2024 (pt2) [10] | 1st sample: 18.9 2nd sample: 23.7 N < 18 | x | 1st sample 0.33 mmol/L # 2nd sample: 0.15 mmol/L # n < 0.7 | 23.7 (15 do, tgal) | 3–6 mo and 7–18 mo | Small for gestational age; neonatal transient tachypnea requiring respiration support; 2× febrile status epilepticus. |
| P4 | Yazici et al., 2021 (pt1) [9] | No NBS performed | No NBS performed | No NBS performed | 33.7 (3 mo, tgal) N < 10 32.2 (3 mo, gal) N < 5 | 106 do—ongoing | Mild bilateral cataracts, mildly elevated AST, ALT. Cataracts did not resolve on galactose-free diet. |
| P5 | Yazici et al., 2021 (pt2) [9] | No NBS performed | No NBS performed | No NBS performed | under non-restricted diet: 3.1 (6 yo, tgal) N < 10 1.9 (6 yo, gal) N < 5 | no | Hypermetropia |
| P6 | Wada et al., 2019 (pt1) [7] and Mikami-Saito (pt3) [11] | NR | 12.4 N < 3–6 depending on the prefecture | 8.9 N < 10–15 depending on the prefecture | 17.3 (44 do, gal) | 44 do- +/− 110 do and +/− 250 do—ongoing | Cataract at 7 mo during temporary suspension of diet, resolved at 23 mo. |
| P7 | Wada et al., 2019 (pt2) [7] and Mikami-Saito (pt15) [11] | NR | 8.7 N < 3–6 depending on the prefecture | 8.9 N < 10–15 depending on the prefecture | 41.9 (4 mo, gal) | 4 mo—ongoing | No symptoms reported |
| P8 | Wada et al., 2019 (pt3) [7] | NR | 11.9 N < 3–6 depending on the prefecture | 6.6 N < 10–15 depending on the prefecture | 19.2 (16 do, gal) | 1 mo—ongoing | Transiently elevated ALT, AST, TBA. |
| P9 | Wada et al., 2019 (pt4) [7] and Mikami-Saito (pt18) [11] | NR | 9.7 N < 3–6 depending on the prefecture | 2.4 N < 10–15 depending on the prefecture | 28.2 (5 mo, gal) | 1–20 mo | No symptoms reported |
| P10 | Wada et al., 2019 (pt5) [7] and Mikami-Saito (pt20) [11] | NR | 10.0 N < 3–6 depending on the prefecture | 10.8 N < 10–15 depending on the prefecture | during transient dietary relaxation: 29.8 (8 mo, gal) | 1.9 mo—ongoing | Neonatal jaundice, portosystemic shunt. |
| P11 | Wada et al., 2019 (pt60) [7] and Mikami-Saito (pt14) [11] | NR | 12.8 N < 3–6 depending on the prefecture | 0.3 N < 10–15 depending on the prefecture | 34 (12 do, gal) | 0.6/2 mo—ongoing | No symptoms reported. |
| P12 | Wada et al., 2019 (pt7) [7] and Mikami-Saito (pt10) [11] | NR | 15.7 N < 3–6 depending on the prefecture | 10.4 N < 10–15 depending on the prefecture | during transient dietary relaxation: 31 (6 mo, gal) | 1 mo- 11 yo after that relaxation with no high intake of dairy products | Transiently elevated TBA levels on non-restricted diet, developmental disorder. |
| P13 | Wada et al., 2019 (pt8) [7] and Mikami-Saito (pt4) [11] | NR | 11.4 N < 3–6 depending on the prefecture | 6.7 N < 10–15 depending on the prefecture | 34.1 (13 do, gal) | 3 wo—ongoing | Sustained high levels of TBA and abnormal flow signal on the abdominal ultrasound until 8 mo, but no shunt detected; mild bilateral cataracts at 10 mo. |
| P14 | Chen et al., 2024 [12] | 1st sample: 17.57 μmol/L * 2nd sample: 34.1 μmol/L N < 30 μmol/L * or repeated sample < 15 μmol/L* (this differs from the Dutch reference range) | NR | NR | 34.1 μmol/L * (9 do, tgal) (this differs from the Dutch reference range) | 15 do—ongoing | No symptoms reported. |
| P15 | Mikami-Saito et al., 2024 (pt1) [11] | NR | 8.2 N < 3–8, depending on prefecture | 7.1 N < 10–25, depending on prefecture | 18 (at onset, gal) | 1 mo—ongoing | Cataract at 1 mo, resolved at 8 months. |
| P16 | Mikami-Saito et al., 2024 (pt2) [11] | NR | 12.2 N < 3–8, depending on prefecture | 7.5 N < 10–25, depending on prefecture | 34.8 (at onset, gal) | 2 mo—ongoing | Cataract at 2 mo, liver dysfunction. |
| P17 | Mikami-Saito et al., 2024 (pt5) [11] | NR | 10 N < 3–8, depending on prefecture | NR | NR | 1 mo—ongoing | Liver dysfunction. |
| P18 | Mikami-Saito et al., 2024 (pt6) [11] | NR | 19.3 N < 3–8, depending on prefecture | 8.8 N < 10–25, depending on prefecture | NR | 2 mo—ongoing | Portosystemic shunt. |
| P19 | Mikami-Saito et al., 2024 (pt7) [11] | NR | 7 N < 3–8, depending on prefecture | 1.1 N < 10–25, depending on prefecture | NR | Unknown—2 yo | No symptoms reported. |
| P20 | Mikami-Saito et al., 2024 (pt8) [11] | NR | 6.2 N < 3–8, depending on prefecture | 14.8 N < 10–25, depending on prefecture | NR | 0.4 mo—ongoing | Hepatomegaly, liver dysfunction. |
| P21 | Mikami-Saito et al., 2024 (pt9) [11] | 14.6 N < 6–12, depending on prefecture | 6.6 N < 3–8, depending on prefecture | NR | NR | Unknown, no diet currently | Liver dysfunction, cholestasis. |
| P22 | Mikami-Saito et al., 2024 (pt11) [11] | 18.6 N < 6–12, depending on prefecture | 16.8 N < 3–8, depending on prefecture | 2.5 N < 10–25, depending on prefecture | NR | 0.1 mo—ongoing | No symptoms reported. |
| P23 | Mikami-Saito et al., 2024 (pt12) [11] | NR | 10.2 N < 3–8, depending on prefecture | 7.2 N < 10–25, depending on prefecture | NR | 0.7–31 mo | No symptoms reported. |
| P24 | Mikami-Saito et al., 2024 (pt13) [11] | NR | 7.8 N < 3-8, depending on prefecture | 5.9 N < 10-25, depending on prefecture | NR | 1–12 mo | No symptoms reported. |
| P25 | Mikami-Saito et al., 2024 (pt16) [11] | NR | 15.4 N < 3–8, depending on prefecture | 4.7 N < 10–25, depending on prefecture | NR | 1–12 mo | No symptoms reported. |
| P26 | Mikami-Saito et al., 2024 (pt17) [11] | 23.88 N < 6–12, depending on prefecture | 19.05 N < 3–8, depending on prefecture | 6.96 N < 10–25, depending on prefecture | NR | 0.1 mo—ongoing | Cholestasis. |
| P27 | Mikami-Saito et al., 2024 (pt19) [11] | 16.4 N < 6–12, depending on prefecture | 12.2 N < 3–8, depending on prefecture | 6 mg/dL N < 10–25, depending on prefecture | NR | Unknown—currently on diet | No symptoms reported. |
| P28 | Mikami-Saito et al., 2024 (pt21) [11] | 12.4 N < 6–12, depending on prefecture | 8.4 N < 3–8, depending on prefecture | 8.7 N < 10–25, depending on prefecture | NR | 3–7 mo | Liver dysfunction. |
| P29 | Mikami-Saito et al., 2024 (pt22) [11] | NR | 11.8 N < 3–8, depending on prefecture | 6.6 N < 10–25, depending on prefecture | NR | 0.5 mo—ongoing | Liver dysfunction. |
| P30 | Mikami-Saito et al., 2024 (pt23) [11] | NR | 9.9 N < 3–8, depending on prefecture | 9.3 N < 10–25, depending on prefecture | NR | 0.7 mo—ongoing | No symptoms reported. |
| P31 | Mikami-Saito et al., 2024 (pt24) [11] | 13.58 N < 6–12, depending on prefecture | 10.9 N < 3–8, depending on prefecture | NR | NR | 0.5 mo—ongoing | No symptoms reported. |
| P32 | Mikami-Saito et al., 2024 (pt25) [11] | NR | 12 N < 3–8, depending on prefecture | 4.8 N < 10–25, depending on prefecture | NR | 0.3 mo—ongoing | No symptoms reported. |
| P33 | Mikami-Saito et al., 2024 (pt26) [11] | NR | 11.5 N < 3–8, depending on prefecture | 2.4 N < 10–25, depending on prefecture | NR | 1 mo—ongoing | No symptoms reported. |
| P34 | Mikami-Saito et al., 2024 (pt27) [11] | NR | 19.4 N < 3–8, depending on prefecture | 4.2 N < 10–25, depending on prefecture | NR | 1.3 mo—ongoing | No symptoms reported. |
| P35 | Mikami-Saito et al., 2024 (pt28) [11] | NR | 12.7 N < 3–8, depending on prefecture | 0.9 N < 10–25, depending on prefecture | NR | 0.1 mo—ongoing | No symptoms reported. |
| P36 | Mikami-Saito et al., 2024 (pt29) [11] | NR | 16.22 N < 3–8, depending on prefecture | 4.1 N < 10–25, depending on prefecture | NR | 0.2 mo—ongoing | No symptoms reported. |
| P37 | Mikami-Saito et al., 2024 (pt30) [11] | NR | NR | NR | NR | 3 mo—ongoing | No symptoms reported. |
| P38 | Mikami-Saito et al., 2024 (pt31) [11] | 18.5 N < 6–12, depending on prefecture | 10.7 N < 3–8, depending on prefecture | 11.3 N < 10–25, depending on prefecture | NR | 1 mo—ongoing | No symptoms reported. |
| P39 | Mikami-Saito et al., 2024 (pt32) [11] | 15.39 N < 6–12, depending on prefecture | 9.78 N < 3–8, depending on prefecture | 8.08 N < 10–25, depending on prefecture | NR | 4–23 mo | No symptoms reported. |
| P40 | Mikami-Saito et al., 2024 (pt33) [11] | 17.9 N < 6–12, depending on prefecture | 11.9 N < 3–8, depending on prefecture | 8.6 N < 10–25, depending on prefecture | NR | 0.1 mo—ongoing | No symptoms reported. |
| P41 | Mikami-Saito et al., 2024 (pt34) [11] | 22.9 N < 6–12, depending on prefecture | 5.2 N < 3–8, depending on prefecture | 19.3 N < 10–25, depending on prefecture | NR | 0.1 mo—ongoing | No symptoms reported. |
| P42 | Mikami-Saito et al., 2024 (pt35) [11] | NR | 12.9 N < 3–8, depending on prefecture | 2.7 N < 10–25, depending on prefecture | NR | 1.6 mo—ongoing | No symptoms reported. |
| P43 | Mikami-Saito et al., 2024 (pt36) [11] | 18.1 N < 6–12, depending on prefecture | 17.6 N < 3–8, depending on prefecture | 0.8 N < 10–25, depending on prefecture | NR | 0.3 mo—ongoing | No symptoms reported. |
| P44 | Mikami-Saito et al., 2024 (pt37) [11] | 15.4 N < 6–12, depending on prefecture | 11.5 N < 3–8, depending on prefecture | 5.6 N < 10–25, depending on prefecture | NR | 0.7–22 mo | Portosystemic shunt. |
| P45 | Mikami-Saito et al., 2024 (pt38) [11] | NR | 10.8 N < 3–8, depending on prefecture | 7.9 N < 10–25, depending on prefecture | NR | 4 mo—ongoing | Liver dysfunction. |
| P46 | Mikami-Saito et al., 2024 (pt39) [11] | NR | 7.2 N < 3–8, depending on prefecture | 6.2 N < 10–25, depending on prefecture | NR | 1.6 mo—ongoing | Liver dysfunction. |
| P47 | Mikami-Saito et al., 2024 (pt40) [11] | NR | 15.34 N < 3–8, depending on prefecture | 6.36 N < 10–25, depending on prefecture | NR | 1 mo—ongoing | No symptoms reported. |
| Age of the Patient | Urinary Galactose (mmol/mol Creatinine) (ULN 326) | Urinary Galactitol (mmol/mol Creatinine) (ULN 71) |
|---|---|---|
| 15-months | 13,167 | 427 |
| 16-months | 26,078 | 929 |
| 18-months * | 56 | 126 |
| Patient | Publication | Age/Sex at Last Follow-Up | Genetic Variant 1 | Genetic Variant 2 |
|---|---|---|---|---|
| P1 | This publication | 18 mo/F | c.424G>A; p.(Gly142Arg) | c.424G>A, p.(Gly142Arg) |
| P2 | Sanches Pintos et al., 2024 (pt1) [10] | 9 yo/M | arr[hg19] 2p22.1(38,893,070 − 38,925,887) | 4arr[hg19] 2p22.1(38,916,650−38,925,887)del |
| P3 | Sanches Pintos et al., 2024 (pt2) [10] | 9 yo/M | arr[hg19] 2p22.1(38,893,070 − 38,925,887) | 4arr[hg19] 2p22.1(38,916,650−38,925,887)del |
| P4 | Yazici et al., 2021 (pt1) [9] | 3 yo/F | c.829G>A p.(Gly277Arg) | c. 829G>A, p.(Gly277Arg) |
| P5 | Yazici et al., 2021 (pt2) [9] | 10 yo/M | c.829G>A, p.(Gly277Arg) | c.829G>A, p.(Gly277Arg) |
| P6 | Wada et al., 2019 (pt1) [7] and Mikami-Saito et al., 2024 (pt3) [11] | 7 yo/M | c.244C>T p.(Arg82*) | c.294del, p.(Ile99Leufs*46) |
| P7 | Wada et al., 2019 (pt2) [7] and Mikami-Saito et al., 2024 (pt15) [11] | 10 yo/F | c.294del, p.(Ile99Leufs*46) | c.799C>G p.(Arg267Gly) |
| P8 | Wada et al., 2019 (pt3) [7] | 1 yo/M | c.294del, p.(Ile99Leufs*46) | c.294del, p.(Ile99Leufs*46) |
| P9 | Wada et al., 2019 (pt4) [7] and Mikami-Saito (pt18) [11] | 8 yo/F | c.932G>A, p.(Trp311*) | c.932G>A, p.(Trp311*) |
| P10 | Wada et al., 2019 (pt5) [7] and Mikami-Saito (pt20) [11] | 7 yo/M | c.424G>A, p.(Gly142Arg) | c.424G>A, p.(Gly142Arg) |
| P11 | Wada et al., 2019 (pt6) [7] and Mikami-Saito (pt14) [11] | 10 yo/F | c.424G>A, p.(Gly142Arg) | c.424G>A, p.(Gly142Arg) |
| P12 | Wada et al., 2019 (pt7) [7] and Mikami-Saito (pt10) [11] | 18 yo/M | c.424G>A, p.(Gly142Arg) | c.799C>G, p.(Arg267Gly) |
| P13 | Wada et al., 2019 (pt8) [7] and Mikami-Saito (pt4) [11] | 6 yo/M | c.424G>A, p.(Gly142Arg) | c.424G>A, p.(Gly142Arg) |
| P14 | Chen et al., 2024 [12] | 3 yo/M | c.325G>A, p.(Gly109Arg) | c.587T>C, p.(Ile196Thr) |
| P15 | Mikami-Saito et al., 2024 (pt1) [11] | 1 yo/M | c.256G>A, p.(Gly86Arg) | c.424G>A, p.(Gly142Arg) |
| P16 | Mikami-Saito et al., 2024 (pt2) [11] | 0 yo/M | c. 424G>A, p.(Gly142Arg) | c.587T>C, p.(Ile196Thr) |
| P17 | Mikami-Saito et al., 2024 (pt5) [11] | 34 yo/M | c.294del, p.(Ile99Leufs*46) | c.424G>A, p.(Gly142Arg) |
| P18 | Mikami-Saito et al., 2024 (pt6) [11] | 21 yo/F | c.294del, p.(Ile99Leufs*46) | c.294del, p.(Ile99Leufs*46) |
| P19 | Mikami-Saito et al., 2024 (pt7) [11] | 20 yo/F | c.294del, p.(Ile99Leufs*46) | c.294del, p.(Ile99Leufs*46) |
| P20 | Mikami-Saito et al., 2024 (pt8) [11] | 19 yo/M | c.424G>A, p.(Gly142Arg) | c.424G>A, p.(Gly142Arg) |
| P21 | Mikami-Saito et al., 2024 (pt9) [11] | 19 yo/F | c.294del, p.(Ile99Leufs*46) | c.294del, p.(Ile99Leufs*46) |
| P22 | Mikami-Saito et al., 2024 (pt11) [11] | 14 yo/F | c.108G>A, p.(Trp36Cys) | c.294del, p.(Ile99Leufs*46) |
| P23 | Mikami-Saito et al., 2024 (pt12) [11] | 13 yo/M | c.294del, p.(Ile99Leufs*46) | c.799C>G, p.(Arg267Gly) |
| P24 | Mikami-Saito et al., 2024 (pt13) [11] | 11 yo/F | c.294del, p.(Ile99Leufs*46) | c.294del, p.(Ile99Leufs*46) |
| P25 | Mikami-Saito et al., 2024 (pt16) [11] | 9 yo/M | c.294del, p.(Ile99Leufs*46) | c.294del, p.(Ile99Leufs*46) |
| P26 | Mikami-Saito et al., 2024 (pt17) [11] | 9 yo/M | c.108G>T, p.(Trp36*) | c.424G>A, p.(Gly142Arg) |
| P27 | Mikami-Saito et al., 2024 (pt19) [11] | 7 yo/M | c.108G>A, p.(Trp36Cys) | c.294del, p.(Ile99Leufs*46) |
| P28 | Mikami-Saito et al., 2024 (pt21) [11] | 7 yo/F | c.294del, p.(Ile99Leufs*46) | c.424G>A, p.(Gly142Arg) |
| P29 | Mikami-Saito et al., 2024 (pt22) [11] | 6 yo/M | c.294del, p.(Ile99Leufs*46) | c.294del, p.(Ile99Leufs*46) |
| P30 | Mikami-Saito et al., 2024 (pt23) [11] | 5 yo/M | c.294del, p.(Ile99Leufs*46) | c.424G>A, p.(Gly142Arg) |
| P31 | Mikami-Saito et al., 2024 (pt24) [11] | 5 yo/M | c.294del, p.(Ile99Leufs*46) | c.294del, p.(Ile99Leufs*46) |
| P32 | Mikami-Saito et al., 2024 (pt25) [11] | 5 yo/M | c.294del, p.(Ile99Leufs*46) | c.799C>G, p.(Arg267Gly) |
| P33 | Mikami-Saito et al., 2024 (pt26) [11] | 4 yo/F | c.294del, p.(Ile99Leufs*46) | c.878A>C p.(Lys293Thr) |
| P34 | Mikami-Saito et al., 2024 (pt27) [11] | 4 yo/M | c.294del, p.(Ile99Leufs*46) | c.294del, p.(Ile99Leufs*46) |
| P35 | Mikami-Saito et al., 2024 (pt28) [11] | 4 yo/M | c.294del, p.(Ile99Leufs*46) | c.365_392del, p.(Val122Alafs*14) |
| P36 | Mikami-Saito et al., 2024 (pt29) [11] | 4 yo/M | c.424G>A, p.(Gly142Arg) | c.587T>C, p.(Ile196Thr) |
| P37 | Mikami-Saito et al., 2024 (pt30) [11] | 4 yo/F | c.424G>A, p.(Gly142Arg) | c.845C>A p.(Thr282Lys) |
| P38 | Mikami-Saito et al., 2024 (pt31) [11] | 4 yo/F | c.294del, p.(Ile99Leufs*46) | c.424G>A, p.(Gly142Arg) |
| P39 | Mikami-Saito et al., 2024 (pt32) [11] | 4 yo/F | c.424G>A, p.(Gly142Arg) | c.587T>C, p.(Ile196Thr) |
| P40 | Mikami-Saito et al., 2024 (pt33) [11] | 3 yo/F | c.294del, p.(Ile99Leufs*46) | c.294del, p.(Ile99Leufs*46) |
| P41 | Mikami-Saito et al., 2024 (pt34) [11] | 3 yo/F | c.424G>A, p.(Gly142Arg) | c.424G>A, p.(Gly142Arg) |
| P42 | Mikami-Saito et al., 2024 (pt35) [11] | 3 yo/M | c.294del, p.(Ile99Leufs*46) | c.587T>C, p.(Ile196Thr) |
| P43 | Mikami-Saito et al., 2024 (pt36) [11] | 3 yo/M | c.587T>C, p.(Ile196Thr) | c.932G>A, p.(Trp311*) |
| P44 | Mikami-Saito et al., 2024 (pt37) [11] | 3 yo/M | c.294del, p.(Ile99Leufs*46) | c.294del, p.(Ile99Leufs*46) |
| P45 | Mikami-Saito et al., 2024 (pt38) [11] | 3 yo/F | c.221C>A, p.(Ala74Glu) | c.424G>A, p.(Gly142Arg) |
| P46 | Mikami-Saito et al., 2024 (pt39) [11] | 2 yo/F | c.294del, p.(Ile99Leufs*46) | c.424G>A, p.(Gly142Arg) |
| P47 | Mikami-Saito et al., 2024 (pt40) [11] | 1 yo/M | c.234G>T, p.(Arg78Ser) | c.294del, p.(Ile99Leufs*46) |
| Country | Target Conditions (Effected Gene) | Target (Biochemical) | Age at Screening | GALT Enzyme Activity Cut-Off | Total Galactose Cut-Off | Galactose Cut-Off | Galactose-1-Phosphate Cut-Off | Comments |
|---|---|---|---|---|---|---|---|---|
| The Netherlands | GALT | Total galactose (galactose + gal-1-p), GALT | 72–168 ho | <2.0 U/dL DBS | >1350 μmol/L DBS | X | X | Positive if both markers are outside reference range |
| GALK | Total galactose (galactose + gal-1-p) | >2.0 U/dL DBS | >2100 μmol/L DBS | X | X | Positive if both markers are outside reference range | ||
| Galicia, Spain | GALT | Galactose, gal-1-P (separately) | 48–72 h of life, second sample 15 days in special cases [11] | X | >18 mg/dL DBS | Qualitative (urine) | >0.7 mmol/L DBS | Second samples are taken in some circumstances |
| Japan | GALT, GALK | Galactose, gal-1-p, (total galactose), GALT enzyme | 96–144 ho [12] | Sometimes measured depending on prefecture | >6–12 mg/dL DBS | >3–8 mg/dL DBS | >10–25 mg/dL DBS | Different cut-offs are used by different prefectures (country regions) |
| Taiwan | GALT, GALK, GALE, (GALM) | Total galactose, referral and genetic testing for GALT, GALK, GALE, (GALM) | 48–72 ho [13] | 2nd tier measurement | >30 μmol/L or repeated sample > 15–18 μmol/L DBS | X | X | Total galactose is the primary marker |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Published by MDPI on behalf of the International Society for Neonatal Screening. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
van Konijnenburg, E.M.M.H.; Radenkovic, S.; Koop, K.; Prinsen, H.C.M.T.; de Sain-van der Velden, M. Rethinking Newborn Screening: A Case of GALM Deficiency. Int. J. Neonatal Screen. 2025, 11, 25. https://doi.org/10.3390/ijns11020025
van Konijnenburg EMMH, Radenkovic S, Koop K, Prinsen HCMT, de Sain-van der Velden M. Rethinking Newborn Screening: A Case of GALM Deficiency. International Journal of Neonatal Screening. 2025; 11(2):25. https://doi.org/10.3390/ijns11020025
Chicago/Turabian Stylevan Konijnenburg, Eva M. M. Hoytema, Silvia Radenkovic, Klaas Koop, Hubertus C. M. T. Prinsen, and Monique de Sain-van der Velden. 2025. "Rethinking Newborn Screening: A Case of GALM Deficiency" International Journal of Neonatal Screening 11, no. 2: 25. https://doi.org/10.3390/ijns11020025
APA Stylevan Konijnenburg, E. M. M. H., Radenkovic, S., Koop, K., Prinsen, H. C. M. T., & de Sain-van der Velden, M. (2025). Rethinking Newborn Screening: A Case of GALM Deficiency. International Journal of Neonatal Screening, 11(2), 25. https://doi.org/10.3390/ijns11020025