1. Introduction
Metachromatic leukodystrophy (MLD) is an ultra-rare and fatal inherited lysosomal storage disease caused by a deficiency of arylsulfatase-A (ARSA), due to mutations in the
ARSA gene. Reduced ARSA activity results in the accumulation of sulfatides in the CNS and peripheral nervous system, which leads to progressive demyelination, rapid motor and cognitive decline and premature death, particularly in early-onset MLD (late infantile [LI] and early juvenile [EJ]). The incidence of MLD in the UK has been estimated to be 1 case in 40,000–160,000 live births and the clinical spectrum of MLD is broad [
1,
2]. However, four clinical forms are commonly described on the basis of age at first symptoms’ onset, the most severe being late infantile (LI, onset of symptoms < 30 months), which typically presents between 15 and 24 months and is the most common variant, accounting for 50–60% of all MLD cases in the UK; the other three include early juvenile MLD (EJ, onset of symptoms between 30 months and <7 years), late juvenile (LJ, onset of symptoms between 7 and <17 years) and adult-onset MLD (AO, ≥17 years) [
3]. The late infantile form is rapidly progressive and characterised primarily by delays in gross and fine motor function, delays in and loss of speech, muscular hypotonia, ataxia and spasticity, which subsequently progresses to complete loss of motor and cognitive functions. The early juvenile presentation and progression of MLD is more variable, but once patients have lost the ability to walk independently, the deterioration of gross motor and cognitive function is as rapid as the infantile form [
4,
5,
6]. Earlier age at onset or presence of motor symptoms at onset is associated with a more severe and rapid disease course. For example, the median time from onset of symptoms to the inability to walk independently, characterised by being beyond the Gross Motor Classification System for MLD (GMFC-MLD) stage 1 has been reported to be 2.7 months for LI patients and approximately 7 months for EJ patients [
6]. Furthermore, the prognosis for patients without treatment is very poor—the median time from onset of symptoms to beyond GMFC-MLD 4, which can be characterised by the complete loss of any locomotion or being able to sit without support, has been reported to be only 1.12 years for LI patients and 2.7 for EJ MLD patients [
6]. The fact that the same study reported a median survival for LI patients of 8.42 years, and 68.6% of EJ patients were alive 15 years from symptom onset [
6], implies that untreated early-onset MLD patients (defined as late infantile and early juvenile MLD) spend the majority of the remainder of their lives with significant morbidity. This leads to an extremely high caregiving burden for MLD [
7] and also has a significant impact on healthcare resource use in the NHS.
Prior to the regulatory approval of atidarsagene autotemcel (arsa-cel; OTL-200), treatment of early-onset MLD in the UK consisted only of best supportive care (BSC), as no disease-modifying treatments had been approved. BSC aims to manage disease complications and maintain quality of life for as long as possible but does not target the root cause of the progressive motor and cognitive decline. Best supportive care includes and is not limited to: physical therapy to maintain mobility; muscle relaxant medications to reduce spasticity; pain management; management of skeletal deformity; respiratory physiotherapy to manage pulmonary infections; anti-convulsant drugs to control seizures; anti-psychotic medications to control psychiatric symptoms; dietary support; enteral nutrition through a feeding tube; and family and psychological counselling. However, patients still experience rapid disease progression ultimately ending in a severely debilitated state, with the median age of death reported in the literature to be 4.2 and 9.86 years of age for the most common MLD phenotype, the late infantile form [
6,
8,
9]. Haematopoietic stem cell transplantation (HSCT) is used as a treatment option for the late juvenile or adult-onset MLD phenotypes in UK clinical practice; but it is not considered a viable option for early-onset MLD by the UK clinicians with expertise in treating MLD.
Arsa-cel is an autologous CD34+ haematopoietic stem and progenitor cell (HSPC) gene therapy. HSPCs are collected from mobilised peripheral blood (mPB) and transduced ex vivo with a lentiviral vector (ARSA LVV), which inserts one or more copies of the human
ARSA complimentary deoxyribonucleic acid (cDNA) into the cell’s genome, so that genetically modified cells become capable of expressing the functional ARSA enzyme. Arsa-cel uses a lentiviral vector which allows the gene to be integrated directly into the genome of the target cell, where it can be replicated whenever the cell divides. As such, the added gene is subsequently passed on to all of the cell’s progeny. Furthermore, the self-renewal capability of HSPCs suggests that once the genetically corrected HSPCs successfully engraft in the brain, there would be a durable supply of the genetically corrected cells and their progenies. Arsa-cel received regulatory approval in the European Union in December 2020 and January 2021 in the UK, for the treatment of MLD characterised by biallelic mutations in the
ARSA gene leading to a reduction of the ARSA enzymatic activity: in children with late infantile or early juvenile forms without clinical manifestations of the disease (pre-symptomatic—PS-LI and PS-EJ), and in children with the early juvenile form with early clinical manifestations of the disease, who still have the ability to walk independently and before the onset of cognitive decline (early symptomatic—ES-EJ) [
10]. Furthermore, in February 2022 arsa-cel was recommended for reimbursement by the National Institute for Health and Care Excellence (NICE) in the UK for its licensed indication based on the provision of a patient access scheme and with the assurance that treatment be administered in a highly specialised service by a specialist multidisciplinary team [
11].
In stark contrast to the published data for BSC [
4,
5,
6], the published clinical data for arsa-cel show that with up to approximately 11 years of follow-up in 39 patients, arsa-cel provides meaningful clinical benefit to patients with early-onset MLD treated in both the pre-symptomatic and early symptomatic stages of the disease [
3,
12]. Published data show that treatment with arsa-cel resulted in sustained, clinically relevant benefits in children with early-onset MLD by preserving cognitive function and motor development in most patients, and slowing demyelination and brain atrophy [
3,
12]. Analysis of two symptomatic EJ patients included in the clinical trial who experienced rapid disease progression after gene therapy showed that levels of engraftment and pharmacodynamic effects post-gene-therapy were within the range observed in patients that responded to treatment, and no differences were observed between the two groups of patients with respect to percentage of LV+ cells, vector copy number (VCN) in the bone marrow (BM), VCN in peripheral blood mononuclear cells (PBMCs) or ARSA activity in peripheral blood and cerebral spinal fluid (CSF). However, the two ES-EJ patients (defined as treatment failures) were treated at symptomatic stages of the disease when disease progression was entering the rapid progressive phase and would not be eligible for treatment according to the eligibility definition of the populations suitable within the EU and UK indication for arsa-cel [
3,
10]. These data are important because they highlight the narrow window of time for treatment and the importance of identifying and treating early-onset MLD as soon as possible.
In the absence of newborn screening (NBS) in current clinical practice in the UK, pre-symptomatic MLD patients are only identified if an older sibling is affected, and due to the rapidly deteriorating nature of MLD, the window for treatment (before advanced nervous system damage occurs) can be limited—often only months. Without NBS, UK clinical experts in the management of MLD estimate that only about 15–20% of all early-onset MLD patients’ diagnoses will occur within the treatment window for arsa-cel (these patients will predominantly be siblings of older affected children), and worryingly, given that it is the most common phenotype, it is highly unlikely that an index case of late infantile MLD would ever be diagnosed pre-symptomatically. In fact, all the pre-symptomatic late infantile patients included in the clinical trial of arsa-cel were diagnosed early and enrolled only because of an older affected sibling, who him/herself could not be offered treatment due to advanced disease [
3]. Clinical expert opinion was corroborated by recently published real-world evidence (RWE), which examined MLD disease phenotype, presentation, eligibility and affected siblings in the year following NHS approval of arsa-cel using hospital records [
13]. The authors found that of 17 UK MLD patients who were referred for treatment, only four patients met eligibility criteria and were treated. Eleven patients failed screening, consisting of 10 symptomatic patients with late infantile disease and 1 with early juvenile disease and cognitive decline. Two further patients with later-onset subtypes did not meet the approval criteria. Three out of the four treated patients were diagnosed by screening after MLD was diagnosed in a symptomatic older sibling, and the other patient was an early-symptomatic early juvenile patient who was diagnosed within the treatment window [
13]. The authors noted that the vast majority (79%) of children referred for gene therapy were deemed ineligible due to advanced disease [
13].
The importance of early diagnosis and initiation of treatment is now increasingly recognised for a number of rare genetic conditions and inborn errors of metabolism, with the most beneficial response to treatment seen in patients prior to the onset of symptoms [
14]. For MLD specifically, this is because patients treated in the early symptomatic stage of the disease will have incurred some irreversible cell damage as a result of the accumulated sulfatides. Whilst the sulfatides are broken down by the newly restored ARSA enzyme, the damage done persists throughout the patients’ lifetime, such that the quality of life enjoyed by these patients is not perceived to be as great as that of those patients who show no manifestations of the disease. Therefore, when MLD patients are treated with arsa-cel pre-symptomatically and in advance of predicted symptom onset, arsa-cel has the potential to address the motor and cognitive aspects of disease progression and allow children to have improved length and quality of life compared to untreated patients and patients treated in the early symptomatic stages of the disease.
Due to the recent approval of a gene therapy for early-onset MLD, newborn screening using a single punch from the routine dried blood spot (DBS) sample has become a feasible option to maximise the best possible outcomes for all MLD patients, where previously only late juvenile or adult-onset MLD patients had an effective treatment option. Pilot studies using a newly developed two-tier screening algorithm for MLD are underway in multiple countries globally, including the UK. Published data from one of these de-anonymised newborn screening pilots in the US in over 27,000 newborn dried blood spots demonstrated near 100% assay specificity [
15]. To minimise the false-positive rate, the authors designed the two-tier screening algorithm such that the primary test was to quantify C16:0-sulfatide in DBS by ultraperformance liquid chromatography–tandem mass spectrometry (UPLC-MS/MS). The screening cut-off for this was established based on the results from 15 MLD newborns to achieve 100% sensitivity. The secondary test was to measure the ARSA activity in DBS from newborns with abnormal C16:0-sulfatide levels. Only newborns that displayed both abnormal C16:0-sulfatide abundance and ARSA activity were considered screen positives. Results showed that of the 27,335 samples screened, 2 high-risk cases were identified. ARSA gene sequencing identified these two high-risk subjects to be an MLD-affected patient and a heterozygote [
14]. Recently published data from a prospective study in Germany demonstrated proof of concept for the feasibility of a high-throughput method for newborn screening for MLD using the same sulfatide and ARSA enzyme-based algorithm. As of December 2023, 120,000 babies have been screened, with three cases of MLD identified [
16]. Specific to the UK, results from a pre-pilot study showed that the two-tier C16:0-S and ARSA screening strategy was successfully validated using day-5 newborn bloodspots, with high specificity for MLD; using a cut-off of 160 nmol/L for the sulfatides correctly identified all 19 positive MLD day-5 bloodspots [
17].
If treatment for MLD is administered prior to the onset of symptoms in screen-positive babies, it is imperative that the prediction of MLD subtypes in pre-symptomatic neonates is possible. European consensus-based recommendations on the clinical management of newborn screening in MLD strongly recommend predicting the age of symptom onset based on family history, genotype and ARSA enzyme activity [
18]. In neonates without affected relatives, the prediction relies primarily on genotype–phenotype correlations as published in the literature and public databases. These databases indicate whether the mutations are null (0) or residual (R) and are updated regularly. The general rule is that a 0/0 genotype is most likely to be late infantile; and for 0/R and R/R mutations, EJ MLD is suspected if it is based on known mutations which have been shown to cause EJ-MLD, e.g., the mutation of the ARSA variant c.465+1G>A coupled with the second most frequent variant c.1283C>T, p.(Pro428Leu). Homozygosity for the c.1283C>T, p.(Pro428Leu) variant is associated with late-juvenile or adult onset [
18].
There are several published studies examining the cost-effectiveness of newborn screening for other inborn errors of metabolism, but not MLD. Bessey et al. examined the cost-effectiveness of including five additional inborn errors of metabolism (glutaric aciduria type 1, homocystinuria, isovaleric acidaemia, long-chain hydroxyacyl CoA dehydrogenase deficiency and maple syrup urine disease) in the UK Newborn screening programme to provide evidence to the UK National Screening Committee [
14]. The authors found that screening for all of the conditions was more effective and cost-saving when compared to not screening for each of the conditions. As a consequence, these conditions were added to the national screening programme. In addition, a recent study by Weidlich et al. demonstrated that newborn screening for SMA was a cost-effective use of NHS resources in the UK, where health outcomes for patients with SMA were improved with NBS and less costly when compared to no screening [
19].
For NBS for MLD to be adopted nationally, there must be evidence that it is cost-effective. Therefore, this study aimed to evaluate whether it is cost-effective to add MLD to the routine UK newborn screening dried blood spot (DBS) programme using a decision analytic framework from a national health service/personal social services perspective.
4. Discussion
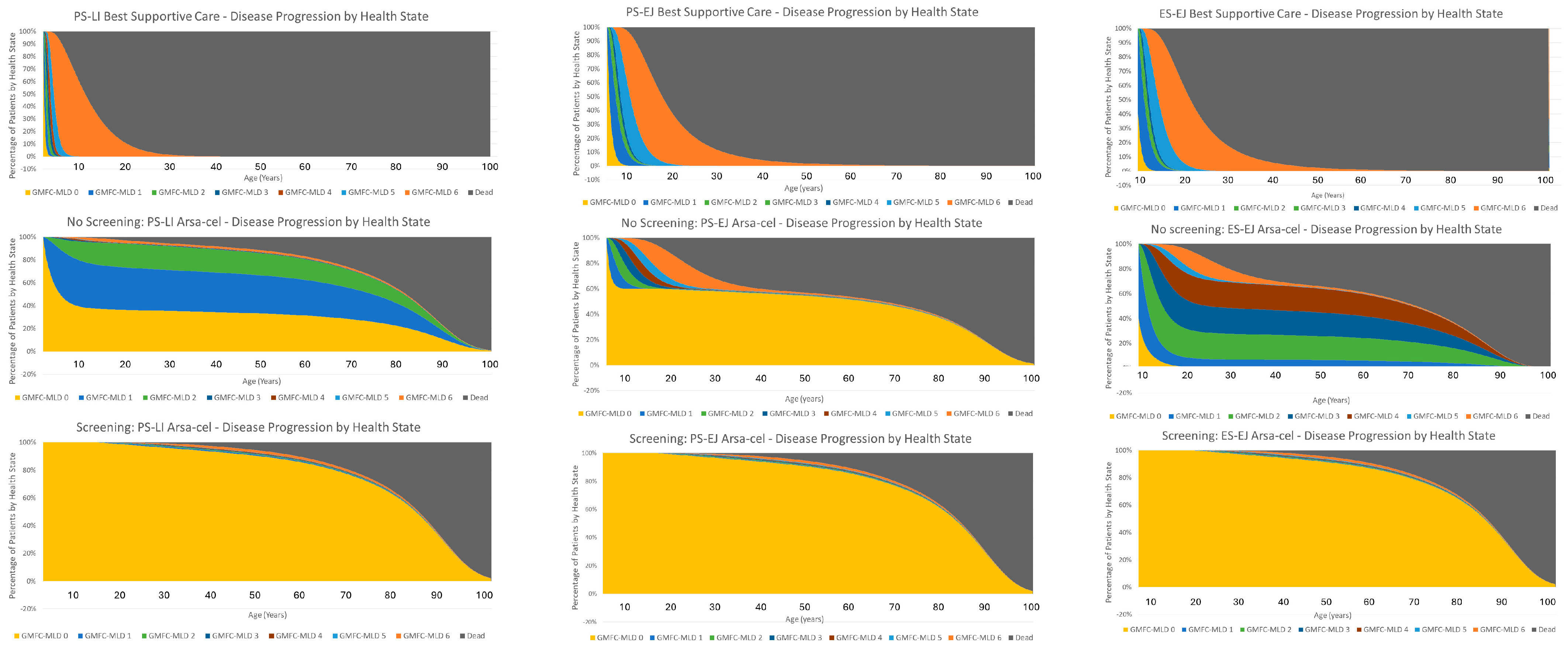
This is the first study to evaluate the cost-effectiveness of adding newborn screening for MLD to the routine dried blood spot screening programme in the UK. The decision analytic model estimated that using an incidence rate of 1/100,000 live births, screening for MLD in the UK would identify 7 newborns with MLD per year, generating an incremental total gain of 246 discounted QALYs over the newborn cohort’s lifetime compared to no screening, whereas in current clinical practice only 2.6 children are identified in time to be eligible for treatment annually. Screening for MLD will increase the total discounted lifetime costs of the newborn cohort by an estimated £8.16 million; this is almost entirely driven by the cost of treating the additional four patients identified through NBS with arsa-cel (therapy and administration costs), who in the no-screening arm would not have been identified in time to be eligible for treatment and would have experienced the symptoms of MLD before dying prematurely. Using a discount rate of 1.5%, the base-case ICER for MLD screening vs. no screening is £33,212 per QALY gained, demonstrating that adding newborn screening for MLD to the current UK routine dried blood spot screening programme in the UK is a cost-effective use of NHS resources at the willingness-to-pay threshold appropriate for the severity and impact of MLD. The main drivers for these results are two-fold: (1) Firstly, screening allows the identification and treatment of all MLD babies, the majority of which in the no-screening arm are diagnosed too late for treatment—there are therefore substantial QALY gains for these patients in terms of both survival and quality of life. (2) Secondly, for some of the patients who were treated in the no-screening arm, some were either treated close to predicted onset of symptoms or already had early symptoms. So, whilst there are comparable survival gains, the quality of life in patients who have stabilised in GMFC-MLD health states with a lower perceived quality of life leads to lower QALY gains in the no-screening arm. Conversely, in the screening arm, all early-onset MLD patients are assumed to be treated within six to eight months of life. This means that all early-onset patients would be able to receive gene therapy potentially well before any sulfatides are able to accumulate and cause any irreversible damage leading to a reduced quality of life that we see in some patients in the no-screening arm.
The structure, assumptions and limitations in the Markov model that generated the long-term cost-effectiveness data for arsa-cel are similar to the economic models reported in Fahim et al. [
26] and published health technology assessments from countries that have evaluated the cost-effectiveness of arsa-cel and in which it is reimbursed [
11,
27,
28]. This suggests that the findings from this study are likely to be generalizable to other jurisdictions, particularly in countries where the willingness-to-pay thresholds are higher than in the UK, e.g.,
$150,000/QALY gained in the US.
In the studies by Bessey and Weidlich investigating the cost-effectiveness of adding other rare neurodegenerative diseases to the DBS screening programme in the UK using similar economic model frameworks [
14,
19], the authors found that introducing screening for these conditions improved health outcomes and was less costly than no screening (i.e., screening dominated no screening). Whilst screening for MLD significantly improves health outcomes, it is not less costly compared with no screening. The difference between the findings of our study and those of other published studies looking at the cost-effectiveness of screening for a genetic condition is primarily due to the proportion of patients in each decision arm that receive treatment. In the studies by Bessey and Weidlich, all patients in both decision arms received treatment (and therefore incurred the cost); it was the timing of treatment in relation to pre- or post-disease onset that differed between the decision arms. Therefore, differences in costs and QALYs were attributable to the achievable health gains and cost savings due to lack of disease progression in the screening arm and not due to treatment costs. For MLD, screening results in significant QALY gains over no screening; however, due to the strict eligibility criteria required for treatment with arsa-cel, very few patients in the no-screening arm actually received treatment (and therefore incurred any treatment costs). Therefore, whilst implementing screening for MLD is cost-effective, it is not cost-saving.
Recently published real-world evidence for the number of annual cases of MLD referred for potential treatment in the UK [
13] showed that the number of referrals for gene therapy in a year exceeded the reported incidence rate of 1/40,000 [
1], and far exceeds the average incidence that has been assumed for the base case in this study (1/100,000). It is likely that the higher number of patients in that study reflects the prevalent pool of MLD rather than the incident pool, given that many patients were symptomatic on referral and were of varying ages ranging from 17 months to 9 years. Increasing the number of MLD patients going through each arm of the decision model to these numbers results in incremental QALY gains similar to those reported in Horgan et al. Given that a higher incidence rate of MLD improves the ICER for screening vs. no screening, it can be inferred that the results from this study are a conservative estimate of the cost-effectiveness of newborn screening for MLD.
There are some limitations associated with this decision analytic model. Firstly, for the late juvenile and adult-onset (LJ/AO) MLD input parameters, there are no published data for the long-term costs and benefits of HSCT treatment for MLD that could be used to inform the model; consequently, these parameters were informed by the HSCT cost and outcomes for an analogous disease. Real-life outcomes for these patients could be better or worse than assumed in the model; however, given that the percentage of LJ/AO incident patients is very small, even large changes in the costs and QALYs estimated for this group have minimal impact on the overall ICER for screening vs. no screening. In addition, because the data have been derived from the NICE appraisal of Strimvelis for ADA-SCID [
23], the long-term costs of HSCT have been applied assuming that LJ/AO MLD patients will undergo HSCT as infants, the costs of which may differ from those for a teen or adult population. This is contrary to European consensus-based recommendations which suggest a deferred approach to treatment until subclinical evidence of disease onset is present. Finally, for the screening arm for the LJ/AO QALY inputs, an arbitrary disutility of −1.0 QALY has been applied to reflect the potential disutility of knowing from birth about having MLD that will develop in later life, as there was no published evidence to estimate this potential disutility. Removing this disutility improves the ICER for the LJ/AO subtype by £6242/QALY gained but only improves the overall ICER by £105, demonstrating that this potential disutility has minimal impact on the overall cost-effectiveness of screening. Furthermore, it could be argued that knowing about MLD earlier means that these late-onset patients could have earlier access to upcoming innovative treatments that they may not have if they were only diagnosed after having symptoms. More importantly, it gives patients and their families the option for treatment, which for the majority of MLD patients in current clinical practice is not possible.
Whilst the impact of key input parameters in the decision tree have been tested in scenario analyses, particularly the probabilities at the chance nodes that were based on expert opinion, an improvement to the model structure would be to embed the Markov model in the decision tree rather than just use the results from the model as input parameters, so that probabilistic sensitivity analyses (PSA) could be performed for the long-term costs and outcomes. This would allow each input parameter to be varied within its specific distribution simultaneously to ascertain what proportion of the 10,000 iterations results in an ICER that falls below the £50,000/QALY-gained threshold, therefore establishing the probability of being cost-effective whilst capturing uncertainty in the input parameters. However, the results from this study provide a reliable indication of the cost-effectiveness profile for a screening programme for MLD and a probabilistic analysis, and more information is unlikely to yield findings that would contravene what has been observed here.
Another area for enhancement would be to broaden the perspective to a societal scope. The current study has been performed for the healthcare perspective, of most interest and direct relevance to the NHS. There are considerable additional potential benefits associated with treatment which have been captured in other accounts, e.g., productivity gains [
7,
26,
29,
30]. These will serve to improve the ICERs illustrated here.
In conclusion, from a clinical point of view it is unequivocal that NBS for MLD has the potential to reduce the current high proportion of ineligible patients by ensuring rapid diagnosis at birth and timely treatment before symptom onset, while without NBS, the unfortunate and stark reality is that the majority of patients will be ineligible for treatment and will succumb to their disease due to a late diagnosis. Treating patients at an early age provides life chances, e.g., education and work-productivity benefits which are important considerations within the context of health technology appraisals. Whilst the feasibility of the MLD screening algorithm and the clinical rationale for screening for MLD have been evidenced in a number of publications, this study is the first to demonstrate that newborn screening for MLD is also a cost-effective use of NHS resources based on the outlined assumptions. These findings strongly support the inclusion of MLD in the official NBS program in the UK.

 ,
, 

{kind=link}
{kind=link}
{kind=link}