
Newborn Screening for Inborn Errors of Metabolism by Next-Generation Sequencing Combined with Tandem Mass Spectrometry
 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Recruitment and Sample Collection
2.2. Panel of Screened Disorders
2.3. Expended NBSs by MS/MS
2.4. NBS by NGS
2.5. Diagnosis
2.6. Statistical Analysis
3. Results
3.1. Performance of NBS by NGS Combined with MS/MS
3.2. Assessment of NGS as a First-Tier Screening Test
3.3. Assessment of MS-MS as a First-Tier Screening Test
3.4. Biochemical and Molecular Characteristics of the Diagnosed Patients
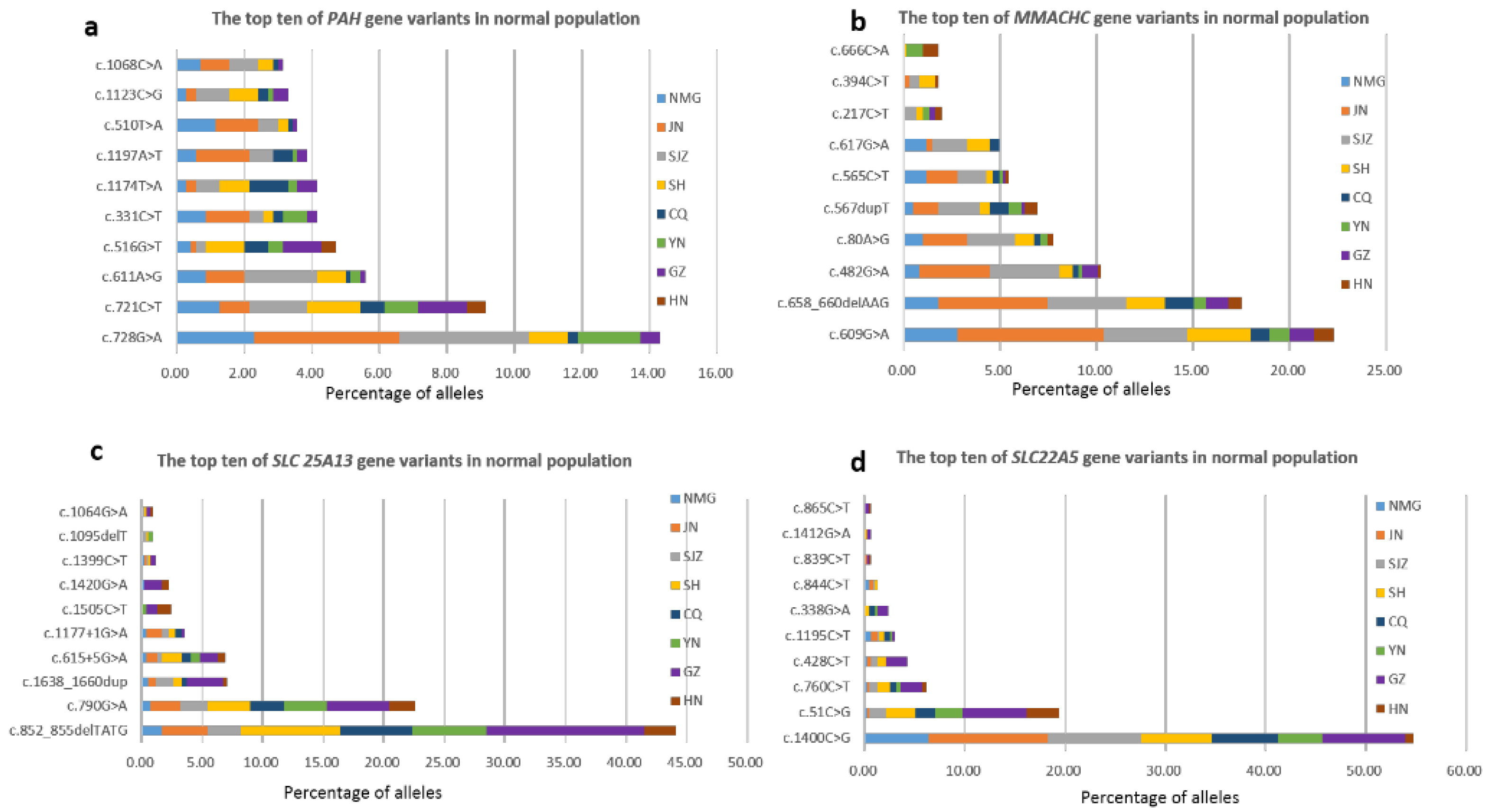
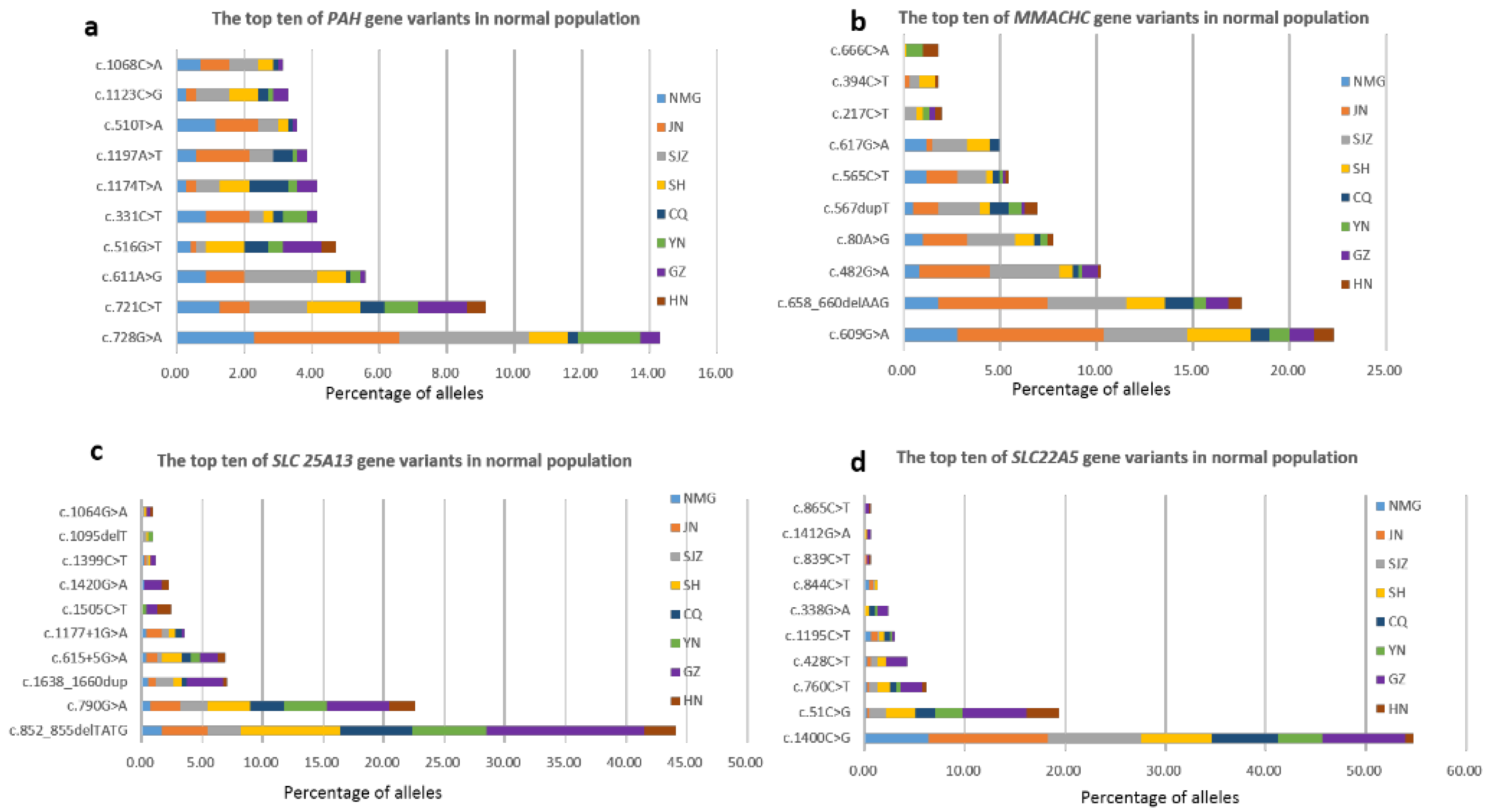
3.5. Carrier Frequencies and Geographical Distribution of Targeted Genes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| MMA | Methylmalonic academia |
| cblC-MMA | Methylmalonic academia, Cobalamin C defect |
| cblA-MMA | Methylmalonic acidemia, Cobalamin A defect |
| PCD | Primary carnitine deficiency |
| PKU | Phenylketonuria |
| SCADD | Short chain acyl-CoA dehydrogenase deficiency |
| IBD | Isobutyryl-CoA dehydrogenase deficiency |
| NICCD | Citrin deficiency |
| MCAD | Medium-chain acyl-CoA dehydrogenase deficiency |
| CPTII | Carnitine palmitoyltransferase II deficiency |
| MSUD | Maple syrup urine disease |
| MSUD (type II) | Maple syrup urine disease, type II |
| MADD | Multiple acyl-CoA dehydrogenase deficiency |
| OTCD | Ornithine transcarbamylase deficiency |
| SH | Shanghai Xinhua Hospital |
| GZ | Guangzhou Women and Children’s Medical Center |
| JN | Jinan Maternal and Child Health Care Hospital |
| SJZ | Shijiazhuang Maternal and Child Health Care Hospital |
| CQ | Chongqing Maternal and Child Health Care Hospital |
| YN | First People’s Hospital of Yunnan Province |
| NMG | Inner Mongolia Maternal and Child Health Care Hospital |
| HN | Hainan Women and Children’s Medical Center |
| P | Pathogenic |
| LP | Likely pathogenic |
| VUS | Variant of uncertain significance or unclassified |
| Het | Heterozygous |
| Hom | Homozygous |
| AR | Autosomal recessive |
| NBS | Newborn screening |
| NGS | Next-generation sequencing |
| IEMs | Inborn errors of metabolism |
| MS/MS | Tandem mass spectrometry |
| DBS | Dried blood spots |
| PPV | Positive predictive value |
References
- Loeber, J.G.; Platis, D.; Zetterstrom, R.H.; Almashanu, S.; Boemer, F.; Bonham, J.R.; Borde, P.; Brincat, I.; Cheillan, D.; Dekkers, E.; et al. Neonatal Screening in Europe Revisited: An ISNS Perspective on the Current State and Developments Since 2010. Int. J. Neonatal Screen. 2021, 7, 15. [Google Scholar] [CrossRef]
- Messina, M.; Meli, C.; Raudino, F.; Pittala, A.; Arena, A.; Barone, R.; Giuffrida, F.; Iacobacci, R.; Muccilli, V.; Sorge, G.; et al. Expanded Newborn Screening Using Tandem Mass Spectrometry: Seven Years of Experience in Eastern Sicily. Int. J. Neonatal Screen. 2018, 4, 12. [Google Scholar] [CrossRef]
- Tang, C.; Liu, S.; Wu, M.; Lin, S.; Lin, Y.; Su, L.; Zhang, J.; Feng, Y.; Huang, Y. Clinical and molecular characteristics of carnitine-acylcarnitine translocase deficiency: Experience with six patients in Guangdong China. Clin. Chim. Acta 2019, 495, 476–480. [Google Scholar] [CrossRef]
- Zhao, Z.; Chen, C.; Sun, X.; Zhou, D.; Huang, X.; Dong, H. Newborn screening for inherited metabolic diseases using tandem mass spectrometry in China: Outcome and cost-utility analysis. J. Med. Screen. 2022, 29, 12–20. [Google Scholar] [CrossRef]
- Tarini, B.A.; Christakis, D.A.; Welch, H.G. State newborn screening in the tandem mass spectrometry era: More tests, more false-positive results. Pediatrics 2006, 118, 448–456. [Google Scholar] [CrossRef]
- Schnabel, E.; Kölker, S.; Gleich, F.; Feyh, P.; Hörster, F.; Haas, D.; Fang-Hoffmann, J.; Morath, M.; Gramer, G.; Röschinger, W.; et al. Combined Newborn Screening Allows Comprehensive Identification also of Attenuated Phenotypes for Methylmalonic Acidurias and Homocystinuria. Nutrients 2023, 15, 3355. [Google Scholar] [CrossRef]
- Younesi, S.; Yazdani, B.; Taheri Amin, M.M.; Saadati, P.; Jamali, S.; Modarresi, M.H.; Savad, S.; Amidi, S.; Razavi, H.; Ghafouri-Fard, S. Incorporation of second-tier tests and secondary biomarkers to improve positive predictive value (PPV) rate in newborn metabolic screening program. J. Clin. Lab. Anal. 2022, 36, e24471. [Google Scholar] [CrossRef]
- Hall, P.L.; Marquardt, G.; McHugh, D.M.; Currier, R.J.; Tang, H.; Stoway, S.D.; Rinaldo, P. Postanalytical tools improve performance of newborn screening by tandem mass spectrometry. Genet. Med. 2014, 16, 889–895. [Google Scholar] [CrossRef]
- Peng, G.; Tang, Y.; Cowan, T.M.; Enns, G.M.; Zhao, H.; Scharfe, C. Reducing False-Positive Results in Newborn Screening Using Machine Learning. Int. J. Neonatal Screen. 2020, 6, 16. [Google Scholar] [CrossRef]
- Ombrone, D.; Giocaliere, E.; Forni, G.; Malvagia, S.; la Marca, G. Expanded newborn screening by mass spectrometry: New tests, future perspectives. Mass. Spectrom. Rev. 2016, 35, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Mordaunt, D.; Cox, D.; Fuller, M. Metabolomics to Improve the Diagnostic Efficiency of Inborn Errors of Metabolism. Int. J. Mol. Sci. 2020, 21, 1195. [Google Scholar] [CrossRef]
- Hays, T.; Wapner, R.J. Genetic testing for unexplained perinatal disorders. Curr. Opin. Pediatr. 2021, 33, 195–202. [Google Scholar] [CrossRef]
- Boemer, F.; Fasquelle, C.; d’Otreppe, S.; Josse, C.; Dideberg, V.; Segers, K.; Guissard, V.; Capraro, V.; Debray, F.G.; Bours, V. A next-generation newborn screening pilot study: NGS on dried blood spots detects causal mutations in patients with inherited metabolic diseases. Sci. Rep. 2017, 7, 17641. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, J.B.; Lescai, F.; Grove, J.; Baekvad-Hansen, M.; Christiansen, M.; Hagen, C.M.; Maller, J.; Stevens, C.; Li, S.; Li, Q.; et al. High-Quality Exome Sequencing of Whole-Genome Amplified Neonatal Dried Blood Spot DNA. PLoS ONE 2016, 11, e0153253. [Google Scholar] [CrossRef]
- Ko, J.M.; Park, K.S.; Kang, Y.; Nam, S.H.; Kim, Y.; Park, I.; Chae, H.W.; Lee, S.M.; Lee, K.A.; Kim, J.W. A New Integrated Newborn Screening Workflow Can Provide a Shortcut to Differential Diagnosis and Confirmation of Inherited Metabolic Diseases. Yonsei Med. J. 2018, 59, 652–661. [Google Scholar] [CrossRef] [PubMed]
- Park, K.J.; Park, S.; Lee, E.; Park, J.H.; Park, J.H.; Park, H.D.; Lee, S.Y.; Kim, J.W. A Population-Based Genomic Study of Inherited Metabolic Diseases Detected Through Newborn Screening. Ann. Lab. Med. 2016, 36, 561–572. [Google Scholar] [CrossRef]
- Huang, X.; Wu, D.; Zhu, L.; Wang, W.; Yang, R.; Yang, J.; He, Q.; Zhu, B.; You, Y.; Xiao, R.; et al. Application of a next-generation sequencing (NGS) panel in newborn screening efficiently identifies inborn disorders of neonates. Orphanet J. Rare Dis. 2022, 17, 66. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, A.N.; Gallagher, R.C.; Wang, Y.; Currier, R.J.; Amatuni, G.; Bassaganyas, L.; Chen, F.; Kundu, K.; Kvale, M.; Mooney, S.D.; et al. The role of exome sequencing in newborn screening for inborn errors of metabolism. Nat. Med. 2020, 26, 1392–1397. [Google Scholar] [CrossRef]
- Chen, T.; Fan, C.; Huang, Y.; Feng, J.; Zhang, Y.; Miao, J.; Wang, X.; Li, Y.; Huang, C.; Jin, W.; et al. Genomic Sequencing as a First-Tier Screening Test and Outcomes of Newborn Screening. JAMA Netw. Open 2023, 6, e2331162. [Google Scholar] [CrossRef]
- Tong, F.; Wang, J.; Xiao, R.; Wu, B.B.; Zou, C.C.; Wu, D.W.; Wang, H.; Zou, H.; Han, L.S.; Yang, L.; et al. Application of next generation sequencing in the screening of monogenic diseases in China, 2021: A consensus among Chinese newborn screening experts. World J. Pediatr. 2022, 18, 235–242. [Google Scholar] [CrossRef]
- Shi, X.T.; Cai, J.; Wang, Y.Y.; Tu, W.J.; Wang, W.P.; Gong, L.M.; Wang, D.W.; Ye, Y.T.; Fang, S.G.; Jing, P.W. Newborn screening for inborn errors of metabolism in mainland china: 30 years of experience. JIMD Rep. 2012, 6, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Tamamori, A.; Fujimoto, A.; Okano, Y.; Kobayashi, K.; Saheki, T.; Tagami, Y.; Takei, H.; Shigematsu, Y.; Hata, I.; Ozaki, H.; et al. Effects of citrin deficiency in the perinatal period: Feasibility of newborn mass screening for citrin deficiency. Pediatr. Res. 2004, 56, 608–614. [Google Scholar] [CrossRef] [PubMed]
- Chengfang, T.; Sichi, L.; Yi, F.; Huifen, M.; Haiping, L.; Jinwen, F.; Lixin, Y.; Guoqing, W.; Li, L.; Yonglan, H. Newborn screening program and blood amino acid profiling in early neonates with citrin deficiency. Zhonghua Er Ke Za Zhi 2019, 57, 797–801. [Google Scholar]
- Almannai, M.; Marom, R.; Divin, K.; Scaglia, F.; Sutton, V.R.; Craigen, W.J.; Lee, B.; Burrage, L.C.; Graham, B.H. Milder Clinical and Biochemical Phenotypes Associated with the c.482G>A (p.Arg161Gln) Pathogenic Variant in Cobalamin C Disease: Implications for Management and Screening. Mol. Genet. Metab. 2017, 122, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Dong, H.; Liu, Y.; He, R.; Song, J.; Jin, Y.; Li, M.; Liu, Y.; Liu, X.; Yan, H.; et al. Late-onset cblC defciency around puberty: A retrospective study of the clinical characteristics, diagnosis, and treatment. Orphanet J. Rare Dis. 2022, 17, 330. [Google Scholar] [CrossRef]
- Liu, J.; Peng, Y.; Zhou, N.; Liu, X.; Meng, Q.; Xu, H.; Zhao, S. Combined methylmalonic acidemia and homocysteinemia presenting predominantly with late-onset diffuse lung disease: A case series of four patients. Orphanet J. Rare Dis. 2017, 12, 58. [Google Scholar] [CrossRef]
- Liu, X.; Xiao, H.; Yao, Y.; Wang, S.; Zhang, H.; Zhong, X.; Yang, Y.; Ding, J.; Wang, F. Prominent renal complications associated with MMACHC pathogenic variant c.80A > G in Chinese children with cobalamin C deficiency. Front. Pediatr. 2022, 10, 1057594. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Li, D.; Cai, F.; Zhang, X.; Xu, X.; Liu, X.; Zhang, C.; Wang, D.; Lin, S.; Zhang, Y.; et al. Mutation spectrum of MMACHC in Chinese pediatric patients with cobalamin C disease: A case series and literature review. Eur. J. Med. Genet. 2019, 62, 103713. [Google Scholar] [CrossRef]
- Liu, M.Y.; Yang, Y.L.; Chang, Y.C.; Chiang, S.H.; Lin, S.P.; Han, L.S.; Qi, Y.; Hsiao, K.J.; Liu, T.T. Mutation spectrum of MMACHC in Chinese patients with combined methylmalonic aciduria and homocystinuria. J. Hum. Genet. 2010, 55, 621–626. [Google Scholar] [CrossRef]
- Tao, J.; Li, N.; Jia, H.; Liu, Z.; Li, X.; Song, J.; Deng, Y.; Jin, X.; Zhu, J. Correlation between genotype and the tetrahydrobiopterin-responsive phenotype in Chinese patients with phenylketonuria. Pediatr. Res. 2015, 78, 691–699. [Google Scholar] [CrossRef]
- Li, N.; He, C.; Li, J.; Tao, J.; Liu, Z.; Zhang, C.; Yuan, Y.; Jiang, H.; Zhu, J.; Deng, Y.; et al. Analysis of the genotype-phenotype correlation in patients with phenylketonuria in mainland China. Sci. Rep. 2018, 8, 11251. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Jia, L.; Gong, M.; Ma, C.; Feng, J. Tandem Mass Spectrometry Screening and Gene Mutation Analysis of Neonatal Hyperprolinemia. J. Int. Reprod. Health/Fam. Plan. 2022, 41, 18–21. [Google Scholar] [CrossRef]
- Tuchman, M.; Morizono, H.; Rajagopal, B.S.; Plante, R.J.; Allewell, N.M. Identification of ‘private’ mutations in patients with ornithine transcarbamylase deficiency. J. Inher. Metab. Dis. 1997, 20, 525–527. [Google Scholar] [CrossRef] [PubMed]
- Martín-Hernández, E.; Aldámiz-Echevarría, L.; Castejón-Ponce, E.; Pedrón-Giner, C.; Couce, M.L.; Serrano-Nieto, J.; Pintos-Morell, G.; Bélanger-Quintana, A.; Martínez-Pardo, M.; García-Silva, M.T.; et al. Urea cycle disorders in Spain: An observational, cross-sectional and multicentric study of 104 cases. Orphanet J. Rare Dis. 2014, 9, 187. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Han, F.; Qiu, W.; Zhang, H.; Ye, J.; Liang, L.; Wang, Y.; Ji, W.; Zhan, X.; Gu, X.; et al. Clinical and molecular characteristics of 69 Chinese patients with ornithine transcarbamylase deficiency. Orphanet J. Rare Dis. 2020, 15, 340. [Google Scholar] [CrossRef] [PubMed]
- Caldovic, L.; Abdikarim, I.; Narain, S.; Tuchman, M.; Morizono, H. Genotype-Phenotype Correlations in Ornithine Transcarbamylase Deficiency: A Mutation Update. J. Genet. Genom. 2015, 42, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Donati, M.A.; Guerrini, R.; Guéant, J.L.; Morrone, A. PRDX1 gene-related epi-cblC disease is a common type of inborn error of cobalamin metabolism with mono- or bi-allelic MMACHC epimutations. Clin. Epigenet. 2021, 13, 137. [Google Scholar] [CrossRef]
- Graffelman, J.; Weir, B.S. The transitivity of the Hardy–Weinberg law. Forensic Sci. Int. Genet. 2022, 58, 102680. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Cai, H.; Li, H.; Ji, Z.; Gu, M. Quantification of Differential Metabolites in Dried Blood Spots Using Second-Tier Testing for SCADD/IBDD Disorders Based on Large-Scale Newborn Screening in a Chinese Population. Front. Pediatr. 2021, 9, 757424. [Google Scholar] [CrossRef]
- Zhang, R.; Qiang, R.; Song, C.; Ma, X.; Zhang, Y.; Li, F.; Wang, R.; Yu, W.; Feng, M.; Yang, L.; et al. Spectrum analysis of inborn errors of metabolism for expanded newborn screening in a northwestern Chinese population. Sci. Rep. 2021, 11, 2699. [Google Scholar] [CrossRef]
- Tan, J.; Chen, D.; Chang, R.; Pan, L.; Yang, J.; Yuan, D.; Huang, L.; Yan, T.; Ning, H.; Wei, J.; et al. Tandem Mass Spectrometry Screening for Inborn Errors of Metabolism in Newborns and High-Risk Infants in Southern China: Disease Spectrum and Genetic Characteristics in a Chinese Population. Front. Genet. 2021, 12, 631688. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Zheng, Q.; Zheng, T.; Zheng, Z.; Lin, W.; Fu, Q. Expanded newborn screening for inherited metabolic disorders and genetic characteristics in a southern Chinese population. Clin. Chim. Acta 2019, 494, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Xin, F.; Bobo, X.; Qiang, Z.; Shang, Y.; Guoxing, G.; Qi, Y.; Jingsi, L.; Jin, W.; Chuan, L.; Shaoke, C.; et al. Analysis of four carnitine-acylcarnitine translocase deficiency cases caused by homozygous mutation of SLC25A20 c.199-10T> G. Zhonghua Er Ke Za Zhi 2018, 56, 545–549. [Google Scholar]
- Wang, X.; Guan, X.W.; Wang, Y.Y.; Zhang, Z.L.; Li, Y.H.; Yang, P.Y.; Sun, Y.; Jiang, T. Current attitudes and preconceptions on newborn genetic screening in the Chinese reproductive-aged population. Orphanet J. Rare Dis. 2022, 17, 322. [Google Scholar] [CrossRef]

{kind=link}
| Disease Name | Gene | Diagnosed Cases | Incidence Rate | Carrier Frequencies |
|---|---|---|---|---|
| cblC-MMA | MMACHC | 5 | 1/5920 | 1/52 |
| PCD | SLC22A5 | 5 | 1/5920 | 1/63 |
| PKU | PAH | 3 | 1/9867 | 1/42 |
| SCADD | ACADS | 2 | 1/14801 | 1/127 |
| IBD | ACAD8 | 2 | 1/14801 | 1/211 |
| NICCD | SLC25A13 | 1 | 1/29,601 | 1/55 |
| MCAD | ACADM | 1 | 1/29,601 | 1/251 |
| CPTII | CPT2 | 1 | 1/29,601 | 1/580 |
| cblA-MMA | MMAA | 1 | 1/29,601 | 1/1139 |
| MSUD (type II) | DBT | 1 | 1/29,601 | 1/1480 |
| MADD | ETFA 1 | 1 | 1/29,601 | / |
| Total | 23 | 1/1287 |
| MS/MS Screening | Genetic Screening | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Units | Neonates | Positive Cases | Number of Recalls (Recall Rate) | Confirmed Cases | PPV | False Negative Cases | Confirmed Disease | Positive Cases | Number of Recalls (Recall Rate) | Confirmed Cases | PPV | False Negative Cases | Confirmed Disease |
| SH | 4888 | 47 | 41 (87.2%) | 1 | 2.44 | 0 | MMA | 1 | 1 (100%) | 1 | 100 | 0 | MMA |
| GZ | 4813 | 50 | 47 (94%) | 4 | 8.51 | 1 | MMA, PCD (2), MADD | 5 | 5 (100%) | 4 | 80 | 1 | MMA, NICCD, PCD (2) |
| JN | 4797 | 149 | 138 (92.6%) | 6 | 4.35 | 0 | MMA (2), IBD, CPTII, PCD, MCAD | 6 | 6 (100%) | 4 | 66.67 | 2 | MMA (2), PCD, MCAD |
| SJZ | 4899 | 67 | 65 (97.0%) | 2 | 3.08 | 0 | PKU, MSUD | 1 | 1 (100%) | 1 | 100 | 1 | PKU |
| CQ | 2988 | 30 | 22 (73.3%) | 2 | 9.09 | 0 | PKU, PCD | 3 | 3 (100%) | 1 | 33.33 | 1 | PKU |
| YN | 3006 | 43 | 40 (93.0%) | 2 | 5.00 | 1 | SCADD, IBD | 3 | 3 (100%) | 3 | 100 | 0 | MMA, SCADD, IBD |
| NMG | 3233 | 113 | 36 (31.86%) | 2 | 5.56 | 0 | PKU, MMA | 3 | 3 (100%) | 2 | 66.67 | 0 | PKU, MMA |
| HN | 977 | 8 | 8 (100%) | 2 | 25 | 0 | PCD, SCADD | 2 | 2 (100%) | 1 | 50 | 1 | PCD |
| Total | 29601 | 507 | 397 (78.3%) | 21 | 5.29 | 2 | 9 (kinds) | 24 | 24 (100%) | 17 | 70.83 | 6 | 7 (kinds) |
| Diagnosed Cases No. | Genetic/MS-MS Screening | Primary Screening Results (Cutoff) | Recall Review Results (Cutoff) | Genes | Exon | Nucleotide Change | Protein Change | ACMG | Zygosity | Disorders | Mode of Inheritance |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Category | |||||||||||
| P1 | +/+ | C3 15.19 (5) | 9.01 (5.5) | MMAA | EX2 | c.365T>C | p.Leu122Pro | LP | Hom | cblA-MMA | AR |
| P2′ | +/+ | C3 4.4 (4.5), | C3 4.37 (4.5), | MMACHC | EX1 | c.80A>G | p.Gln27Arg | LP | Het | cblC-MMA | AR |
| C3/C2 0.356 (0.2) | C3/C2 0.344 (0.2) | MMACHC | EX4E | c.482G>A | p.Arg161Gln | LP | Het | AR | |||
| P3 | +/+ | C3 3.53 (5), Met8.36 (9), | C3 0.92 (5), Met7.17 (9), | MMACHC | EX1 | c.80A>G | p.Gln27Arg | LP | Het | cblC-MMA | AR |
| C3/C2 0.18 (0.2) | C3/C2 0.25 (0.2) | MMACHC | EX4E | c.609G>A | p.Trp203 * | LP | Het | AR | |||
| P4 | +/+ | C3 2.23 (4.5), | C3 3.64 (4.5), | MMACHC | EX1 | c.80A>G | p.Gln27Arg | LP | Het | cblC-MMA | AR |
| C3/C2 0.235 (0.2) | C3/C2 0.44 (0.2) | MMACHC | EX4E | c.609G>A | p.Trp203 * | LP | Het | AR | |||
| P5 1 | +/- | C3 1.99 (3.59) | C3 2.16 (4.5) | MMACHC | EX4E | c.482G>A | p.Arg161Gln | LP | Het | cblC-MMA | AR |
| C3/C2 0.14 (0.2) | C3/C2 0.11 (0.22) | MMACHC | EX4E | c.565C>T | p.Arg189Cys | LP | Het | AR | |||
| P6 | +/+ | C3 9.74 (4.8), C3/C2 0.57 (0.23) | C3 7.83 (4.8), C3/C2 0.96 (0.23) | MMACHC | EX4E | c.658_660delAAG | p.Lys220del | P | Hom | cblC-MMA | AR |
| P7 | +/+ | Phe 276 (116), | Phe 393 (116), | PAH | EX3 | c.331C>T | (p.Arg111 *) | P | Het | PKU | AR |
| Phe/Tyr 3.02 (1.5) | Phe/Tyr 4.57 (1.5) | PAH | IVS12 | c.1315+6T>A | LP | Het | AR | ||||
| P8 | +/+ | Phe 606 (120), | Phe 10.89 (mg/dL) | PAH | EX5 | c.482T>C | p.Phe161Ser | LP | Het | PKU | AR |
| Phe/Tyr 14.11 (1.5) | PAH | EX11 | c.1197A>T | p.V399V | P | Het | AR | ||||
| P9 | +/+ | Phe 2.45 (mg/dL) (2.1) | Phe 166 (120), | PAH | EX6 | c.688G>A | p.Val230Ile | LP | Het | PKU | AR |
| Phe/Tyr 1.88 (1.2) | PAH | EX12 | c.1238G>C | p.Arg413Pro | P | Het | AR | ||||
| P10 | +/+ | C0 6.78 (10) | C0 10.09 (10) | SLC22A5 | EX1 | c.51C>G | p.Phe17Leu | LP | Hom | PCD | AR |
| P11 | +/+ | C0 7.46 (10) | C0 4.64 (10) | SLC22A5 | EX2 | c.428C>T | p.Pro143Leu | P | Het | PCD | AR |
| SLC22A5 | EX8 | c.1400C>G | p.Ser467Cys | P | Het | AR | |||||
| P12 | +/+ | C0 8.82 (10) | C0 4.81 (10) | SLC22A5 | EX4 | c.760C>T | p.Arg254 * | P | Het | PCD | AR |
| SLC22A5 | EX8 | c.1400C>G | p.Ser467Cys | P | Het | AR | |||||
| P13 | +/+ | C0 5.16 (9) | C0 7.76 (9) | SLC22A5 | EX8 | c.1400C>G | p.Ser467Cys | P | Hom | PCD | AR |
| P14 | +/+ | C4 2.8 (0.46) C4/C3 3.79 (0.4) | C4 1.98 (0.46) C4/C3 6.04 (0.4) | ACADS | EX9 | c.1031A>G | p.Glu344Gly | P | Hom | SCADD | AR |
| P15 | +/+ | C4 2.08 (0.46) | C4 1.99 (0.46) | ACAD8 | EX3 | c.289G>A | p.Gly97Arg | LP | Het | IBD | AR |
| C4/C3 4.1 (0.4) | C4/C3 2.02 (0.4) | ACAD8 | EX4 | c.413delA | p.Asn138Metfs * 36 | P | Het | AR | |||
| P16 | +/+ | C8 3.64 (0.13), | C8 2.16 (0.13), | ACADM | IVS10 | c.946-1G>C | LP | Het | MCAD | AR | |
| C8/C1015.8 (1.4) | C8/C10 12 (1.4) | ACADM | EX11 | c.1085G>A | p.Gly362Glu | LP | Het | AR | |||
| P17 | +/- | Cit 20.13 (30) | Cit 449.85 (35) | SLC25A13 | EX16 | c.1638_1660dup | p.Ala554Glyfs * 17 | P | Het | NICCD | AR |
| SLC25A13 | EX9 | c.852_855delTATG | p.Met285Profs * 2 | P | Het | AR | |||||
| P18 | -/+ | Leu 1088 (270), | Leu 4182.3 (270), | DBT | EX2 | c.75_76delAT | p.Cys26Trpfs * 2 | P | Het | MSUD | AR |
| Val 602 (269) | Val 998.6 (269) | DBT | EX11E | c.1359_1360delAG | p.Arg453Serfs * 3 | VUS | Het | AR | |||
| P19 | -/+ | C4 1.19 (0.5) | C4 1.23 (0.5) | ACAD8 | EX4 | c.473A>G | p,Tyr158Cys | VUS | Het | IBD | AR |
| ACAD8 | EX10 | c.1165C>T | p.Arg389Trp | VUS | Het | AR | |||||
| P20 | -/+ | C4 0.9 (0.45) | C4 2.11 (0.45) | ACADS | EX3 | c.322G>A | p.Gly108Ser | LP | Het | SCADD | AR |
| ACADS | EX6 | c.779G>T | p.Gly260Val | VUS | Het | AR | |||||
| P21 | -/+ | C0 6.65 (9) | C0 8.96 (9) | SLC22A5 | EX8 | c.1400C>G | p.Ser467Cys | P | Het | PCD | AR |
| SLC22A5 | EX3 | c.621G>T | p.Gln207His | VUS | AR | ||||||
| P22 | -/+ | C12 1.18 (0.3), C14 2.52 (0.4), C16 25.1 (4.27), | C12 0.9 (0.33), C14 0.57 (0.4), | CPT2 | EX1 | c.125C>T | p.Thr42IIe | VUS | Het | CPTII | AR |
| C18 5.82 (1.8), C18:1 8.94 (3) | C18:1 4.24 (3) | CPT2 | EX4 | c.1613delA | p.Tyr538Serfs * 5 | VUS | Het | AR | |||
| P23 | -/+ | C4 2.19 (0.8), C5 3.11 (0.35), C6 1.14 (0.12), | C4 5.83 (0.8), C5 6.2 (0.35), C6 3.15 (0.12), | ETFA 1 | EX5 | c.369G>A | p.R122K | P | Het | MADD | AR |
| C8 1.76 (0.16) | C8 5.2(0.16) | ETFA 1 | EX8 | c.659delC | p.S220Lfs * 6 | P | Het | AR |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, C.; Li, L.; Chen, T.; Li, Y.; Zhu, B.; Zhang, Y.; Yin, Y.; Liu, X.; Huang, C.; Miao, J.; et al. Newborn Screening for Inborn Errors of Metabolism by Next-Generation Sequencing Combined with Tandem Mass Spectrometry. Int. J. Neonatal Screen. 2024, 10, 28. https://doi.org/10.3390/ijns10020028
Tang C, Li L, Chen T, Li Y, Zhu B, Zhang Y, Yin Y, Liu X, Huang C, Miao J, et al. Newborn Screening for Inborn Errors of Metabolism by Next-Generation Sequencing Combined with Tandem Mass Spectrometry. International Journal of Neonatal Screening. 2024; 10(2):28. https://doi.org/10.3390/ijns10020028
Chicago/Turabian StyleTang, Chengfang, Lixin Li, Ting Chen, Yulin Li, Bo Zhu, Yinhong Zhang, Yifan Yin, Xiulian Liu, Cidan Huang, Jingkun Miao, and et al. 2024. "Newborn Screening for Inborn Errors of Metabolism by Next-Generation Sequencing Combined with Tandem Mass Spectrometry" International Journal of Neonatal Screening 10, no. 2: 28. https://doi.org/10.3390/ijns10020028
APA StyleTang, C., Li, L., Chen, T., Li, Y., Zhu, B., Zhang, Y., Yin, Y., Liu, X., Huang, C., Miao, J., Zhu, B., Wang, X., Zou, H., Han, L., Feng, J., & Huang, Y. (2024). Newborn Screening for Inborn Errors of Metabolism by Next-Generation Sequencing Combined with Tandem Mass Spectrometry. International Journal of Neonatal Screening, 10(2), 28. https://doi.org/10.3390/ijns10020028