
Expanded Newborn Screening for Inborn Errors of Metabolism in Hong Kong: Results and Outcome of a 7 Year Journey

 , , , , and
, , , , and
Abstract
1. Background
2. Materials
2.1. Subjects, Eligibility, and Coverage
2.2. Data Collection
2.3. Sample Collection
2.4. Panel of Conditions Screened
3. Methods
3.1. First-Tier Biochemical Tests
3.2. Second-Tier Biochemical Tests
3.3. Second-Tier Genetic Screening Tests
3.4. Reporting
3.5. Data Analysis
4. Results
4.1. Baseline Characteristics and Analysis
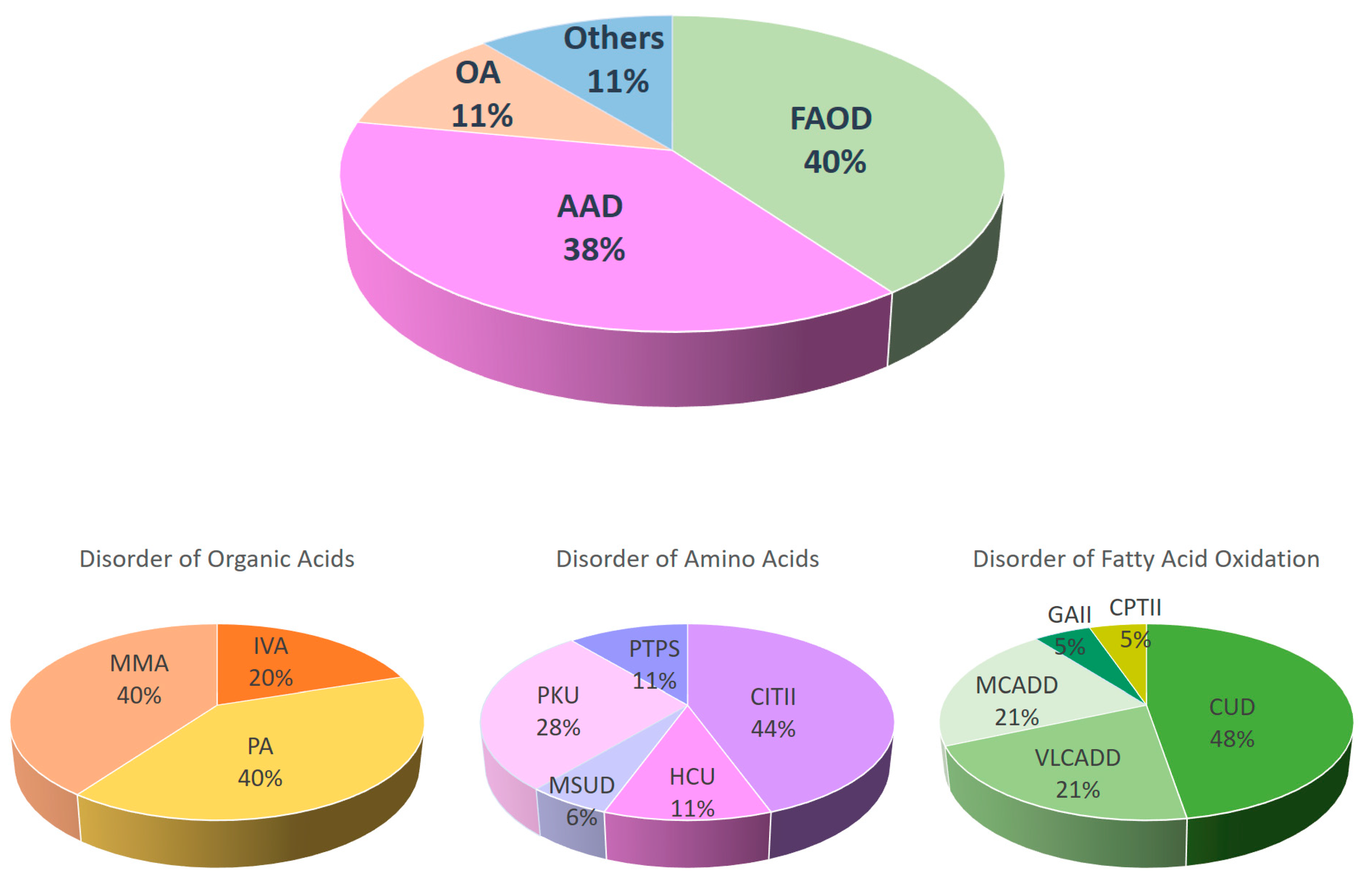
4.2. Disease Spectrum of IMD
4.3. Outcome of the Newborns Screened by NBSIEM
4.4. False Positives and Specificity
4.5. False Negatives and Sensitivity
5. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Guthrie, R.; Susi, A. A simple phenylalanine method for detecting phenylketonuria in large populations of newborn infants. Pediatrics 1963, 32, 338–343. [Google Scholar] [CrossRef]
- Mak, C.M. Newborn Screening: Past, Present and the Future; Topical Update; The Hong Kong College of Pathologist: Hong Kong, China, 2016; Volume 11. [Google Scholar]
- Hau, E.W.L. Evaluation of the 18-month “Pilot Study of Newborn Screening for Inborn Errors of Metabolism” in Hong Kong: The Task Force on the Pilot Study of Newborn Screening for Inborn Errors of Metabolism. HK J. Paediatr. 2020, 25, 16–22. [Google Scholar]
- Lee, H.C.; Mak, C.M.; Lam, C.W.; Yuen, Y.P.; Chan, A.O.K.; Shek, C.C.; Siu, T.-S.; Lai, C.-K.; Ching, C.-K.; Siu, W.-K.; et al. Analysis of inborn errors of metabolism: Disease spectrum for expanded newborn screening in Hong Kong. Chin. Med. J. 2011, 124, 983–989. [Google Scholar]
- Lacey, J.M.; Minutti, C.Z.; Magera, M.J.; Tauscher, A.L.; Casetta, B.; McCann, M.; Lymp, J.; Hanhn, S.H.; Rinaldo, P.; Matern, D. Improved specificity of newborn screening for congenital adrenal hyperplasia by second tier steroid profiling using tandem mass spectrometry. Clin. Chem. 2004, 50, 621–625. [Google Scholar] [CrossRef] [PubMed]
- Yeung, M.C.; Chan, T.C.; Mak, C.M. Clinical Utility of Second-tier Testing in Newborn Screening for Congenital Adrenal Hyperplasia: The Hong Kong Experience. HK J. Paediatr. 2020, 25, 3–7. [Google Scholar]
- Turgeon, C.T.; Magera, M.J.; Cuthbert, C.D.; Loken, P.R.; Gavrilov, D.K.; Tortorelli, S.; Raymond, K.M.; Oglesbee, D.; Rinaldo, P.; Matern, D. Determination of total homocysteine, methylmalonic acid, and 2-methylcitric acid in dried blood spots by tandem mass spectrometry. Clin. Chem. 2010, 56, 1686–1695. [Google Scholar] [CrossRef]
- Tsang, K.Y.; Chan, T.C.; Yeung, M.C.; Wong, T.K.; Lau, W.T.; Mak, C.M. Validation of amplicon-based next generation sequencing panel for second-tier test in newborn screening for inborn errors of metabolism. J. Lab. Med. 2021, 45, 267–274. [Google Scholar] [CrossRef]
- Deng, K.; Zhu, J.; Yu, E.; Xiang, L.; Yuan, X.; Yao, Y.; Li, X.; Liu, H. Incidence of inborn errors of metabolism detected by tandem mass spectrometry in China: A census of over seven million newborns between 2016 and 2017. J. Med. Screen. 2021, 28, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Yunus, Z.; Rahman, S.A.; Choy, Y.S.; Keng, W.T.; Ngu, L.H. Pilot study of newborn screening of inborn error of metabolism using tandem mass spectrometry in Malaysia: Outcome and challenges. J. Pediatr. Endocrinol. Metab. 2016, 29, 1031–1039. [Google Scholar] [CrossRef]
- Martin-Rivada, A.; Perez, L.P.; Ruiz-Sala, P.; Navarrete, R.; Conejero, A.C.; Fraile, P.Q.; Lopez, A.M.; Belanger-Quintana, A.; Martin-Hernandez, E.; Bellusci, M.; et al. Diagnosis of inborn errors of metabolism within the expanded newborn screening in the Madrid region. JIMD Rep. 2022, 63, 146–161. [Google Scholar] [CrossRef]
- Mohamed, S.; Elsheikh, W.; Al-Aqeel, A.I.; Alhashem, A.M.; Alodaib, A.; Alahaideb, L.; Almashary, M.; Alharbi, F.; AlMalawi, H.; Ammari, A.; et al. Incidence of newborn screening disorders among 56632 infants in Central Saudi Arabia. Saudi Med. J. 2020, 41, 703–708. [Google Scholar] [CrossRef]
- Sorensen, L.; von Dobeln, U.; Ahlman, H.; Ohlsson, A.; Engvall, M.; Naess, K.; Backman-Johansson, C.; Nordqvist, Y.; Wedell, A.; Zetterstrom, R.H. Expanded Screening of One Million Swedish Babies with R4S and CLIR for Post Analytical Evaluation of Data. Int. J. Neonatal Screen. 2020, 6, 42. [Google Scholar] [CrossRef]
- Lam, S.T.; Cheng, M.L. Neonatal screening in Hong Kong and Macau. Southeast Asian J. Trop. Med. Public Health 2003, 34 (Suppl. 3), 73–75. [Google Scholar]
- Census and Statistics Departments. The Government of the Hong Kong Special Administrative Region Website. Available online: https://www.censtatd.gov.hk/en/web_table.html?id=115-01012# (accessed on 13 December 2023).
- IEMbase. Inborn Errors of Metabolism Knowledgebase Website. Available online: http://www.iembase.org/index.asp (accessed on 26 January 2024).
- Lim, J.S.; Tan, E.S.; John, C.M.; Poh, S.; Yeo, S.J.; Ang, J.S.M.; Adakalaisamy, P.; Rozalli, R.A.; Hart, C.; Tan, E.T.H.; et al. Inborn Error of Metabolism (IEM) screening in Singapore by electrospray ionization-tandem mass spectrometry (ESI/MS/MS): An 8-year journey from pilot to current program. Mol. Genet. Metab. 2014, 113, 53–61. [Google Scholar] [CrossRef]
- Schulze, A.; Lindner, M.; Kohlmuller, D.; Olgemoller, K.; Mayatepek, E.; Hoffmann, G.G. Expanded Newborn Screening for Inborn Errors of Metabolism by Electrospray Ionization-Tandem Mass Spectrometry: Results, Outcome and Implications. Paediatrics 2003, 11, 1399–1406. [Google Scholar] [CrossRef]
- Men, S.; Liu, S.; Zheng, Q.; Yang, S.; Mao, H.; Wang, Z.; Gu, Y.; Tang, X.; Wang, L. Incidence and genetic variants of inborn errors of metabolism identified through newborn screening: A 7-year study in eastern coastal areas of China. Mol. Genet. Genom. Med. 2023, 11, e2152. [Google Scholar] [CrossRef] [PubMed]
- Tangeraas, T.; Saeves, I.; Klingeberg, C.; Jorgensen, J.; Kristensen, E.; Gunnarsdottir, G.; Hansen, E.V.; Strand, J.; Lundman, E.; Ferdinandusse, S.; et al. Performance of Expanded Newborn Screening in Norway Supported by Post Analytical Bioinformatics Tools and Rapid Second Tier DNA Analyses. Int. J. Neonatal Screen. 2020, 6, 51. [Google Scholar] [CrossRef]
- Shibata, N.; Hasegawa, Y.; Yamada, K.; Kobayashi, H.; Purevsuren, J.; Yang, Y.; Dung, V.C.; Khanh, N.; Verma, I.C.; Bijarnia-Mahay, S.; et al. Diversity in the incidence and spectrum of organic acidemias, fatty acid oxidation disorders, and amino acid disorders in Asian countries: Selective vs newborn screening. Mol. Genet. Metab. Rep. 2018, 16, 5–10. [Google Scholar] [CrossRef] [PubMed]
- New Zealand Government, Ministry of Health. National Screening Unit Website. Available online: https://www.nsu.govt.nz/pregnancy-newborn-screening/newborn-metabolic-screening-programme-heel-prick-test/about-test (accessed on 15 December 2023).
- Grunert, S.C.; Mullerleile, S.; de Silva, L.; Barth, M.; Walter, M.; Walter, K.; Meissner, T.; Lindner, M.; Enzenauer, R.; Santer, R.; et al. Propionic acidemia: Neonatal versus selective metabolic screening. J. Inherit. Metab. Dis. 2012, 35, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Heringer, J.; Valayannopoulou, V.; Lund, A.M.; Wijburg, F.A.; Freisinger, P.; Baric, I.; Baumgartner, M.R.; Burgard, P.; Burlina, A.B.; Chapman, K.A.; et al. Impact of age at onset and newborn screening on outcome in organic acidurias. J. Inherit. Metab. Dis. 2016, 39, 341–353. [Google Scholar] [CrossRef] [PubMed]
- State of Michigan. Department of Health and Human Services Website. Available online: https://www.michigan.gov/-/media/Project/Websites/mdhhs/Folder3/Folder60/Folder2/Folder160/Folder1/Folder260/Letter_Announcing_Removal_of_SCAD_and_IBD.pdf?rev=fcba2eef56bd44859c35e552a8b7213c (accessed on 15 December 2023).
- Ruoppolo, M.; Malvagia, S.; Boenzi, S.; Carducci, C.; Dionisi-Vici, C.; Teofoli, F.; Burlina, A.; Angeloni, A.; Aronica, T.; Bordugo, A.; et al. Expanded Newborn Screening in Italy Using Tandem Mass Spectrometry: Two Years of National Experience. Int. J. Neonatal Screen. 2022, 8, 47. [Google Scholar] [CrossRef]
- Tang, N.L.S.; Hwu, W.L.; Chan, R.T.; Law, L.K.; Fung, L.M.; Zhang, W.M. A Founder Mutation (R254X) of SLC22A5 (OCTN2) in Chinese Primary Carnitine Deficiency Patients. Hum. Mutat. 2002, 20, 232. [Google Scholar] [CrossRef]
- Ji, X.; Ge, Y.; Ni, Q.; Xu, S.; Xiong, Z.; Yang, L.; Hu, L.; Cao, Y.; Lu, Y.; Wei, Q.; et al. Primary carnitine deficiency: Estimation of prevalence in Chinese population and insights into newborn screening. Front. Genet. 2023, 14, 1304458. [Google Scholar] [CrossRef]
- Chen, H.-A.; Hsu, R.-H.; Chen, Y.-H.; Hsu, L.-W.; Chiang, S.-C.; Lee, N.-C.; Hwu, W.-L.; Chiu, P.-C.; Chien, Y.-H. Improved diagnosis of citrin deficiency by newborn screening using a molecular second-tier test. Mol. Genet. Metab. 2022, 136, 330–336. [Google Scholar] [CrossRef]
- Kido, J.; Häberle, J.; Tanaka, T.; Nagao, M.; Wada, Y.; Numakura, C.; Bo, R.; Nyuzuki, H.; Dateki, S.; Maruyama, S.; et al. Improved sensitivity and specificity for citrin deficiency using selected amino acids and acylcarnitines in the newborn screening. J. Inherit. Metab. Dis. 2023, 1–10. [Google Scholar] [CrossRef]
- Tabata, A.; Sheng, J.-S.; Ushikai, M.; Song, Y.-Z.; Gao, H.-Z.; Lu, Y.-B.; Okumura, F.; Iijima, M.; Mutoh, K.; Kishida, S.; et al. Identification of 13 novel mutations including a retrotransposal insertion in SLC25A13 gene and frequency of 30 mutations found in patients with citrin deficiency. J. Hum. Genet. 2008, 53, 534–545. [Google Scholar] [CrossRef]
- Yamaguchi-Kabata, Y.; Yasuda, J.; Uruno, A.; Shimokawa, K.; Koshiba, S.; Suzuki, Y.; Fuse, N.; Kawame, H.; Tadaka, S.; Nagasaki, M.; et al. Estimating carrier frequencies of newborn screening disorders using a whole-genome reference panel of 3552 Japanese individuals. Hum. Genet. 2019, 138, 389–409. [Google Scholar] [CrossRef] [PubMed]
- Chong, S.C.; Lo, P.; Chow, C.W.; Yuen, L.; Chu, W.C.W.; Leung, T.Y.; Hui, J.; Scaglia, F. Molecular and clinical characterization of citrin deficiency in a cohort of Chinese patients in Hong Kong. Mol. Genet. Metab. Rep. 2018, 17, 3–8. [Google Scholar] [CrossRef]
- Okano, Y.; Ohura, T.; Sakamoto, O.; Inui, A. Current treatment for citrin deficiency during NICCD and adaptation/compensation stages: Strategy to prevent CTLN2. Mol. Genet. Metab. 2019, 127, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.H.; Gong, J.Y.; Wang, J.S. Citrin Deficiency presenting as acute liver failure in an eight-month-old infant. World J. Gastroenterol. 2015, 21, 7331–7334. [Google Scholar] [CrossRef] [PubMed]
- Estrella, J.; Wilcken, B.; Carpenter, K.; Bhattacharya, K.; Tchan, M.; Wiley, V. Expanded newborn screening in New South Wales: Missed cases. J. Inherit. Metab. Dis. 2014, 37, 881–887. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.Y.; Wong, L.M.; Belaramani, K.M. Newborn Screening Pitfalls: A Missed Case of Salt-losing Type of Congenital Adrenal Hyperplasia. HK J. Paediatr. 2020, 25, 49–52. [Google Scholar]
- Pearce, M.; Demartino, L.; McMahon, R.; Hamel, R.; Maloney, B.; Stansfield, D.M.; McGrath, E.C.; Occhionero, A.; Gearhart, A.; Caggana, M.; et al. Newborn screening for congenital adrenal hyperplasia in New York State. Mol. Genet. Metab. Rep. 2016, 7, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Votava, F.; Török, D.; Kovács, J.; Moslinger, D.; Baumgartner-Parzer, S.M.; Sólyom, J.; Pribilincová, Z.; Battelino, T.; Lebl, J.; Frisch, H.; et al. Estimation of the false-negative rate in newborn screening for congenital adrenal hyperplasia. Eur. J. Endocrinol. 2005, 152, 869–874. [Google Scholar] [CrossRef]
- Held, P.K.; Shapira, S.K.; Hinton, C.F.; Jones, E.; Hannon, W.H.; Ojodu, J. Congenital adrenal hyperplasia cases identified by newborn screening in one- and two-screen states. Mol. Genet. Metab. 2015, 116, 133–138. [Google Scholar] [CrossRef]
- Sarafoglou, K.; Banks, K.; Kyllo, J.; Pittock, S.; Thomas, W. Cases of congenital adrenal hyperplasia missed by newborn screening in Minnesota. J. Am. Med. Assoc. 2012, 307, 2371–2374. [Google Scholar] [CrossRef]
- Lund, A.; Wibrand, F.; Skogstrand, K.; Cohen, A.; Christensen, M.; Japelt, R.B.; Duno, M.; Skovby, F.; Norgaard-Pedersen, B.; Gregersen, N.; et al. Danish expanded newborn screening is a successful preventive public health programme. Dan. Med. J. 2020, 67, A06190341. [Google Scholar]
- Chien, Y.-H.; Hwu, W.-L. The modern face of newborn screening. Pediatr. Neonatol. 2023, 64, S22–S29. [Google Scholar] [CrossRef]
- Belaramani, K.M.; Fung, C.W.; Kwok, A.M.K.; Lee, S.Y.R.; Yau, E.K.C.; Luk, H.M.; Mak, C.M.; Yeung, M.C.; Ngan, O.M.Y. Public and Healthcare Provider Receptivity toward the Retention for Dried Blood Spot Cards and Their Usage for Extended Genetic Testing in Hong Kong. Int. J. Neonatal Screen. 2023, 9, 45. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Disorders of Organic Acid Metabolism (8 Conditions) | Primary Marker/Ratio | Second-Tier Testing |
|---|---|---|
| Glutaric acidemia type I | ↑ C5DC | Genetic (September 2021 onwards) |
| Isovaleric acidemia | ↑ C5, ↑ C5/C2, ↑ C5/C3 | |
| Methylmalonic acidemia | ↑ C3, ↑C3/C2 | Biochemical: MMA, MCA, tHCY by LC-MS/MS (January 2021 onwards) |
| Methylmalonic acidemia and homocystinaemia (cobalamin C deficiency) (October 2019 onwards) | ||
| Propionic acidemia | ||
| Multiple carboxylase deficiency | ↑ C5OH, ↑ C5OH/C8 | Genetic (September 2021 onwards) |
| Beta-ketothiolase deficiency | ↑ C5:1, ↑ C5OH, ↑C5OH/C8 | Genetic (September 2021 onwards) |
| 3-hydroxy-3-methylglutaryl-coA lyase deficiency | ↑ C5OH, ↑ C6DC, ↑C5OH/C8 | Genetic (September 2021 onwards) |
| Disorders of Amino Acid Metabolism (9 conditions) | ||
| Argininemia (October 2019 onwards) | ↑ Arg | Genetic (October 2022 onwards) |
| Argininosuccinic acidemia | ↑Cit | |
| Citrullinemia type I | ||
| Citrullinemia type II | Genetic (September 2021 onwards) | |
| Phenylketonuria | ↑ Phe, ↑ Phe/Tyr Ratio | |
| 6-pyruvoyl-tetrahydropterin synthase deficiency | ||
| Homocystinuria | ↑ Met, ↑ Met/Phe | Biochemical: tHCY by LC-MS/MS (January 2021 onwards) |
| Maple syrup urine disease | ↑ Xle, ↑Valine | |
| Tyrosinemia type I | ↑ Tyr, ↑Succinylacetone | |
| Disorders of Fatty Acid Oxidation Metabolism (6 conditions) | ||
| Carnitine-acylcarnitine translocase deficiency | ↑C16, C18, ↑C18:1, ↑C18:2, ↓C0/(C16+C18) ↑ (C16+C18:1)/C2 | |
| Carnitine palmitoyltransferase II deficiency | ||
| Carnitine uptake deficiency | ↓ C0 | Genetic (September 2021 onwards) |
| Glutaric acidemia Type II | ↑ C4-C18 | |
| Medium chain acyl-coA dehydrogenase deficiency | ↑ C6, ↑ C8, ↑C10, ↑ C10:1, ↑C8/C10 | |
| Very long-chain acyl-coA dehydrogenase deficiency | ↑ C14, ↑ C14:1, ↑ C14:2, ↑ C14:1/C16, ↑C14:1/C2, ↑C14:1/C12:1 | |
| Other IMD (3 conditions) | ||
| Biotinidase deficiency (April 2016 onwards) | ↓ BTD activity | Genetic (October 2022 onwards) |
| Classic galactosemia (April 2016 onwards) | ↓ GALT activity | |
| Congenital adrenal hyperplasia (April 2016 onwards) | ↑ 17OHP by GSP | Biochemical: 17OHP, D4A and cortisol by LC-MS/MS. Positive if elevated (17OHP +D4A)/cortisol ratio with elevated 17OHP (April 2016 onwards) |
| Newborn Screened 125,688 | True Positive | Incidence | False Positive | False Negative | Positive Predictive Value |
|---|---|---|---|---|---|
| Specificity (%) | Sensitivity (%) | ||||
| Total | 47 | 1 in 2674 | 248 * (99.8) | 16 (74.6) | 15.9% |
| Disorders of Organic Acids | 5 | 1 in 25,138 | 32 (99.97) | 0 (100) | 13.5% |
| Isovaleric acidemia | 1 | 1 in 125,688 | 3 (99.99) | 0 (100) | 25% |
| Methylmalonic acidemia (methylmalonyl-coA mutase deficiency) | 2 | 1 in 62,844 | 29 (99.98) | 0 (100) | 12.1% # |
| Propionic acidemia | 2 | 1 in 62,844 | 0 (100) | ||
| Disorders of Amino Acid | 18 | 1 in 6983 | 70 (99.94) | 13 (58.1) | 20.5% |
| Citrullinemia type II | 8 | 1 in 15,711 # | 49 (99.96) | 13 (38.1) | 14.3% # |
| Maple syrup urine disease | 1 | 1 in 125,688 | 1 (99.99) | 0 (100) | 50% |
| Classic phenylketonuria | 5 | 1 in 25,137 | 15 (99.98) | 0 (100) | 22.7% |
| 6-pyruvoyl-tetrahydropterin synthase deficiency | 2 | 1 in 62,844 | 0 (100) | ||
| Homocystinuria | 2 | 1 in 62,844 | 5 (99.99) | 0 (100) | 28.6% |
| Disorders of Fatty Acid Oxidation | 19 | 1 in 6615 | 58 (99.93) | 0 (100) | 24.7% |
| Carnitine palmitoyltransferase II deficiency | 1 | 1 in 125,688 | 10 (99.99) | 0 (100) | 9.1% |
| Carnitine uptake deficiency | 9 | 1 in 13,965 | 42 (99.96) | 0 (100) | 16.6% |
| Glutaric acidemia type II | 1 | 1 in 125,688 | 1 (99.98) | 0 (100) | 3.6% |
| Medium chain acyl-coA dehydrogenase deficiency | 4 | 1 in 31,422 | 3 (99.99) | 0 (100) | 57.1% |
| Very long-chain acyl-coA dehydrogenase deficiency | 4 | 1 in 31,422 | 2 (99.99) | 0 (100) | 57.1% |
| Other IMD | 5 | 1 in 25,138 | 35 (99.97) | 3 (62.5) | 12.5% |
| Biotidinase deficiency | 2 | 1 in 62,844 | 20 (99.98) | 0 (100) | 9% |
| Congenital adrenal hyperplasia | 3 | 1 in 41,896 # | 15 (99.99) | 3 (50) | 16.7% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Belaramani, K.M.; Chan, T.C.H.; Hau, E.W.L.; Yeung, M.C.W.; Kwok, A.M.K.; Lo, I.F.M.; Law, T.H.F.; Wu, H.; Wong, S.S.N.; Lam, S.W.; et al. Expanded Newborn Screening for Inborn Errors of Metabolism in Hong Kong: Results and Outcome of a 7 Year Journey. Int. J. Neonatal Screen. 2024, 10, 23. https://doi.org/10.3390/ijns10010023
Belaramani KM, Chan TCH, Hau EWL, Yeung MCW, Kwok AMK, Lo IFM, Law THF, Wu H, Wong SSN, Lam SW, et al. Expanded Newborn Screening for Inborn Errors of Metabolism in Hong Kong: Results and Outcome of a 7 Year Journey. International Journal of Neonatal Screening. 2024; 10(1):23. https://doi.org/10.3390/ijns10010023
Chicago/Turabian StyleBelaramani, Kiran Moti, Toby Chun Hei Chan, Edgar Wai Lok Hau, Matthew Chun Wing Yeung, Anne Mei Kwun Kwok, Ivan Fai Man Lo, Terry Hiu Fung Law, Helen Wu, Sheila Suet Na Wong, Shirley Wai Lam, and et al. 2024. "Expanded Newborn Screening for Inborn Errors of Metabolism in Hong Kong: Results and Outcome of a 7 Year Journey" International Journal of Neonatal Screening 10, no. 1: 23. https://doi.org/10.3390/ijns10010023
APA StyleBelaramani, K. M., Chan, T. C. H., Hau, E. W. L., Yeung, M. C. W., Kwok, A. M. K., Lo, I. F. M., Law, T. H. F., Wu, H., Wong, S. S. N., Lam, S. W., Ha, G. H. Y., Lau, T. P. Y., Wong, T. K., Or, V. W. C., Wong, R. M. S., Ming, W. L., Chow, J. C. K., Yau, E. K. C., Fu, A., ... Fung, C. W. (2024). Expanded Newborn Screening for Inborn Errors of Metabolism in Hong Kong: Results and Outcome of a 7 Year Journey. International Journal of Neonatal Screening, 10(1), 23. https://doi.org/10.3390/ijns10010023