
Leading Risk Factors for Congenital Deafness in the Context of Universal Neonatal Screening: Our Observations in a Four-Year Retrospective Study

 and
and
Abstract
1. Introduction
2. Materials and Methods
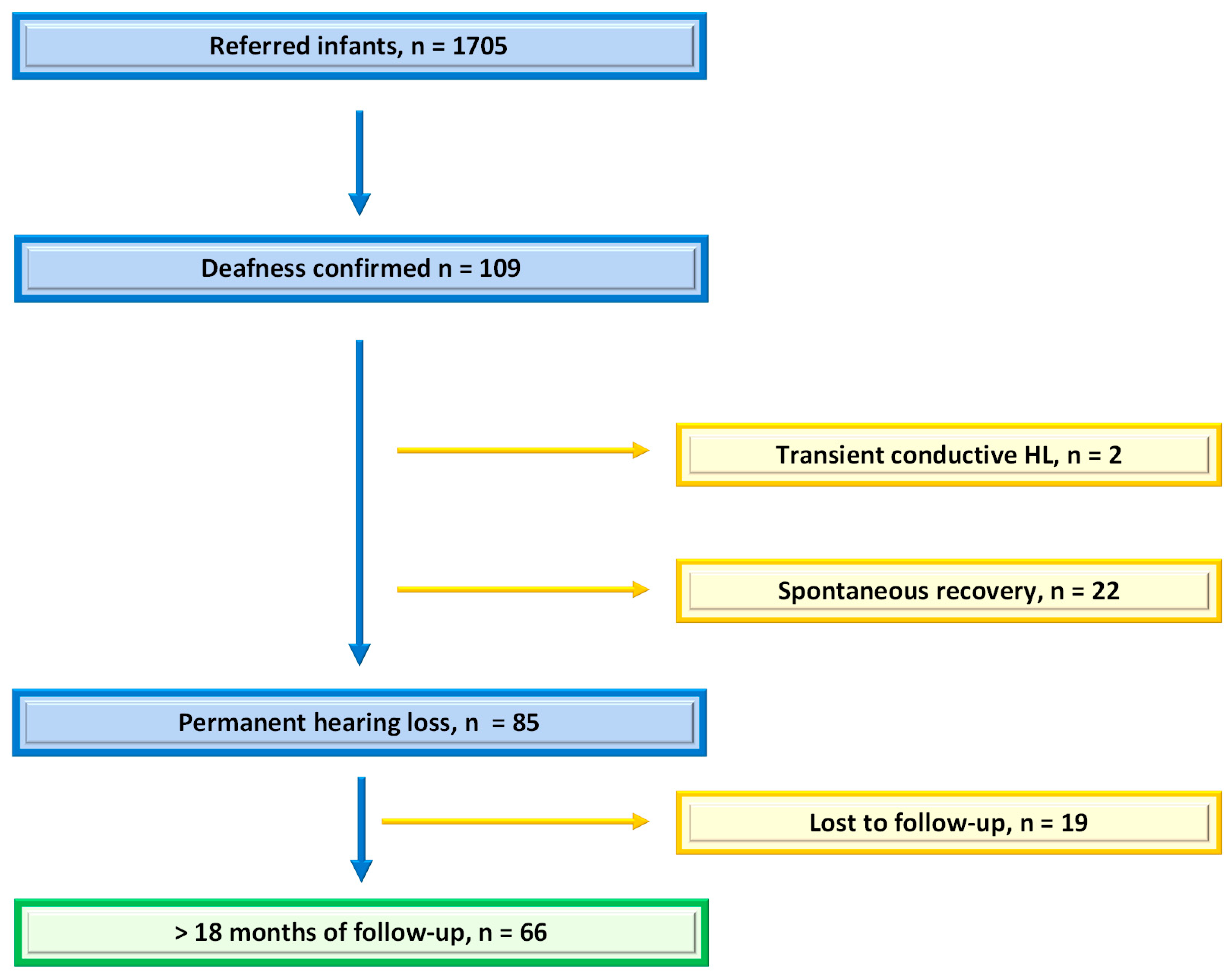
3. Results
3.1. Cohort Characteristics
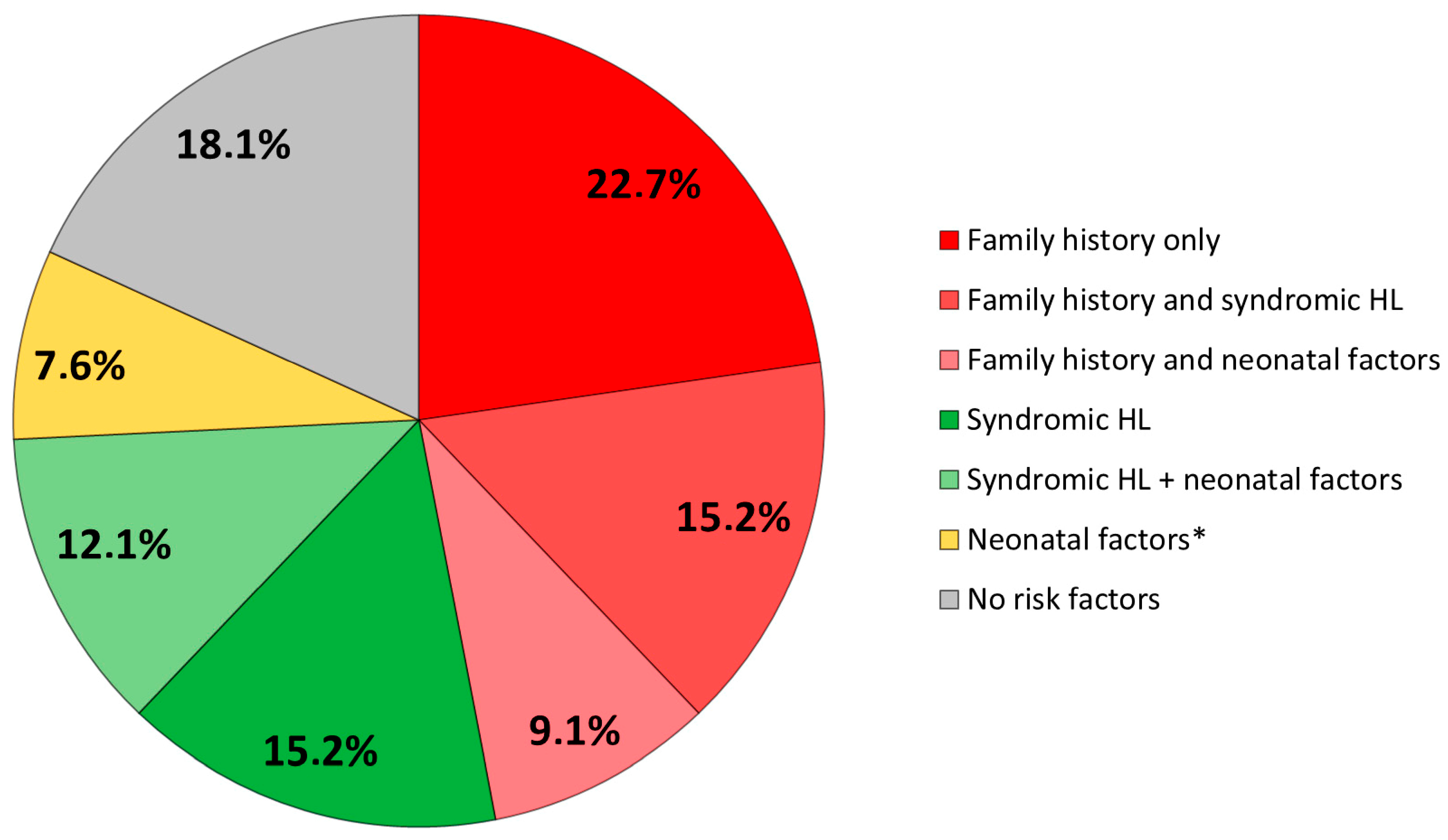
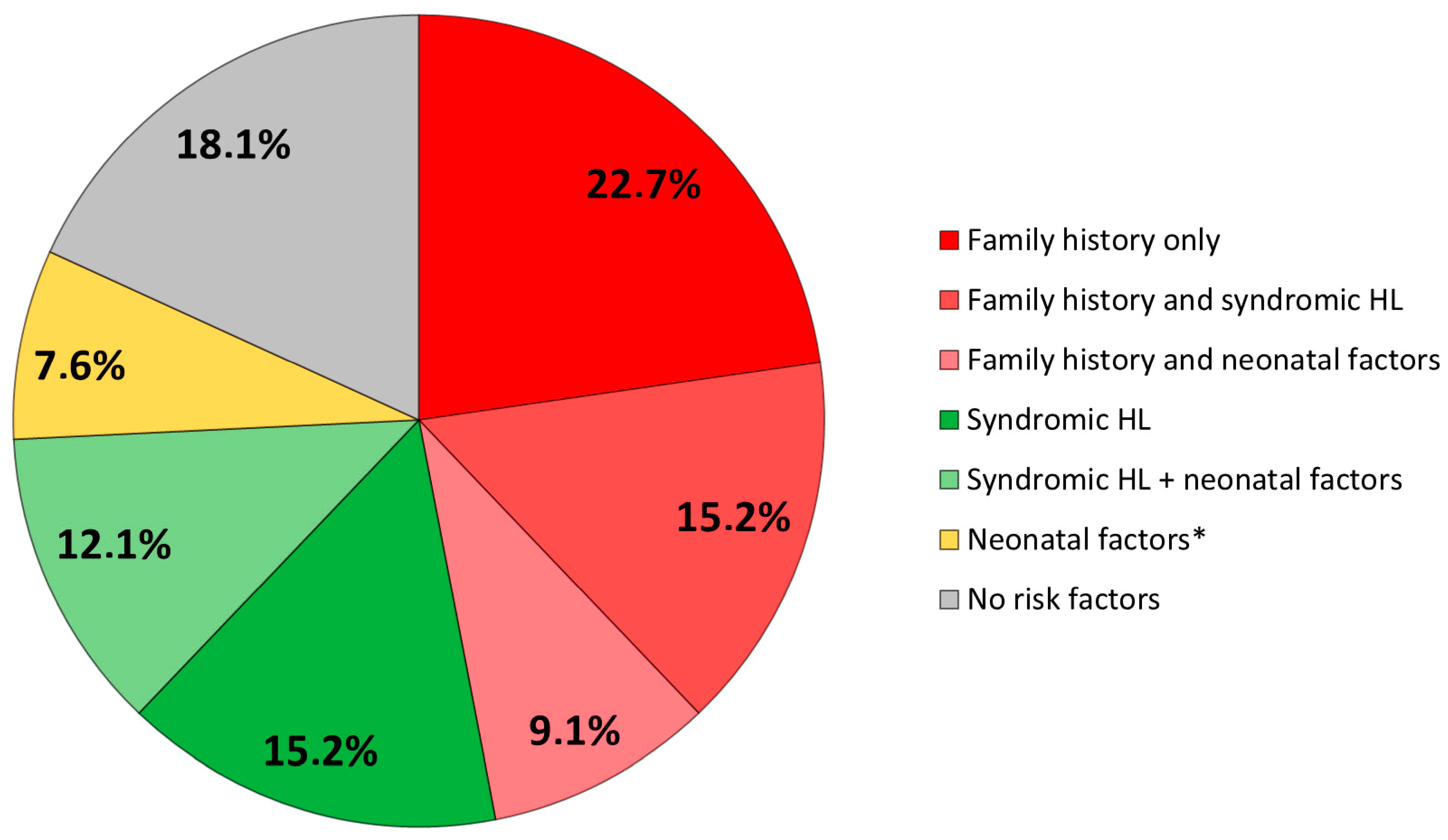
3.2. Risk Factors
3.3. Imaging Findings
3.4. Genetic Causes
4. Discussion
- Zhou et al. (2022) shared the idea that the epidemiology of the usual risk factors studied has evolved, at least in developed countries, due to the “improvement in the quality of medical care” [3]. Four main factors were observed in their cohort as follows: craniofacial anomalies, NICU admission, family history, and advanced maternal age (AMA). However, their study dealt with fewer infants (n = 25), and surprisingly, the frequency of congenital conductive deafness was greater than sensorineural deafness (15 versus 8), making a comparison with our work difficult.
- It must also be taken into account that neonatal screening has been set up in developed countries with a suitable healthcare system, but it is hardly affordable in poor countries. Data from the recent literature should help physicians in such countries to select infants with a higher risk of congenital hearing loss and to focus their limited means on this specific population.
- The limitations of current devices must also be taken into account as they have some difficulty in screening mild deafness. TEOAE devices provide the detection of outer hair cell function when a hearing threshold does not exceed 35 dBHL, whereas aABR devices validate responses at a threshold ranging from 25 to 40 dBHL [7].
- Moreover, as both aABR and OAE were not systematically performed together, we could have underestimated cases of auditory neuropathy spectrum disorders.
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A. Main Malformations in Syndromic Deafness

{kind=link}
{kind=link}
| Associated Abnormalities | n (%) |
|---|---|
| Craniofacial | 14 (21.2) |
| Microtia | 5 |
| Fistula—pits | 5 |
| Hemifacial microsomia | 2 |
| Palate cleft | 2 |
| Brain | 8 (12.1) |
| Epilepsy (west) | 1 |
| Microcephaly | 1 |
| Sacrococcygeal defects | 1 |
| Brain atresia | 1 |
| Anoxo-ischemic brain | 1 |
| Cerebellar cyst | 1 |
| Axial hypotonia—white matter anomaly | 1 |
| Intraparenchymal calcification | 1 |
| Eye | 4 (6) |
| Coloboma | 1 |
| Iris heterochromia | 3 |
| Heart | 4 (6) |
| Ischemic cardiomyopathy | 1 |
| Atrioventricular canal | 1 |
| Aortic hypoplasia | 1 |
| Tetralogy of Fallot | 1 |
| Kidney | 3 |
| Hypoplasia | 1 |
| Renal fusion | 1 |
| Renal insufficiency | 1 |
| Limbs | 3 (4.5) |
| Spastic equinus deformity | |
| Polydactyly | |
| Talus valgus | |
| Digestive tracts | 3 (4.5) |
| Oesophageal atresia | 2 |
| Digestive tract stenosis | 1 |
| Vertebra | 2 (3) |
| Sacrococcygeal anomalies | 1 |
| Hemivertebral hypoplasia | 1 |
| n (%) | |
|---|---|
| Molecular diagnosis | 8 (12) |
| GJB2 | 3 |
| SLC26A4 | 1 |
| CHD23 | 1 |
| Chromosome 6 duplication | 1 |
| TECTA | 1 |
| ANKRD11 (KGB syndrome) | 1 |
| B4GALT7 | 1 |
| OTUD6B | 1 |
| Syndromic deafness | 4 (6) |
| Waardenburg syndrome | 3 |
| CHARGE syndrome | 1 |
| Unidentified syndromic deafness | 20 |
References
- Mytton, J.; Mackenzie, I. Observed and expected prevalence of permanent childhood hearing impairment in Oldham. J. Public Health 2005, 27, 298–302. [Google Scholar] [CrossRef] [PubMed]
- Kvestad, E.; Lie, K.K.; Eskild, A.; Engdahl, B. Sensorineural hearing loss in children: The association with Apgar score. A registry-based study of 392,371 children in Norway. Int. J. Pediatr. Otorhinolaryngol. 2014, 78, 1940–1944. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Wang, L.; Jin, F.; Guo, Y.; Zhou, Y.; Zhang, X.; Zhang, Y.; Ni, X.; Li, W.; Liu, H. The prevalence and risk factors for congenital hearing loss in neonates: A birth cohort study based on CHALLENGE study. Int. J. Pediatr. Otorhinolaryngol. 2022, 162, 111308. [Google Scholar] [CrossRef] [PubMed]
- Anastasio, A.R.T.; Yamamoto, A.Y.; Massuda, E.T.; Manfredi, A.K.S.; Cavalcante, J.M.S.; Lopes, B.C.P.; Aragon, D.C.; Boppana, S.; Fowler, K.B.; Britt, W.J.; et al. Comprehensive evaluation of risk factors for neonatal hearing loss in a large Brazilian cohort. J. Perinatol. 2021, 41, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Antoni, M.; Rouillon, I.; Denoyelle, F.; Garabédian, E.-N.; Loundon, N. Newborn hearing screening: Prevalence and medical and paramedical treatment of bilateral hearing loss in a neonatal series in the Île-de-France region of France. Eur. Ann. Otorhinolaryngol. Head Neck Dis. 2016, 133, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Fischer, C.; Lieu, J. Unilateral hearing loss is associated with a negative effect on language scores in adolescents. Int. J. Pediatr. Otorhinolaryngol. 2014, 78, 1611–1617. [Google Scholar] [CrossRef] [PubMed]
- JCIH. Year 2019 Position Statement: Principles and Guidelines for Early Hearing Detection and Intervention Programs. J. Early Hear. Detect. Interv. 2019, 4, 1–44. [Google Scholar] [CrossRef]
- Kral, A.; Kronenberger, W.G.; Pisoni, D.B.; O’Donoghue, G.M. Neurocognitive factors in sensory restoration of early deafness: A connectome model. Lancet Neurol. 2016, 15, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Shearer, A.E.; Hildebrand, M.S.; Schaefer, A.M.; Smith, R.J. Genetic Hearing Loss Overview. In GeneReviews®; Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993. Available online: http://www.ncbi.nlm.nih.gov/books/NBK1434/ (accessed on 28 August 2023).
- Rance, G.; Starr, A. Pathophysiological mechanisms and functional hearing consequences of auditory neuropathy. Brain 2015, 138, 3141–3158. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.-B.; Shi, H.-B.; Wang, J.; Ding, D.-L.; Yu, D.-Z.; Chen, Z.-N.; Li, C.-Y.; Zhang, W.-T.; Yin, S.-K. Bilirubin induces auditory neuropathy in neonatal guinea pigs via auditory nerve fiber damage. J. Neurosci. Res. 2012, 90, 2201–2213. [Google Scholar] [CrossRef] [PubMed]
- Verstappen, G.; Foulon, I.; Van den Houte, K.; Heuninck, E.; Van Overmeire, B.; Gordts, F.; Topsakal, V. Analysis of congenital hearing loss after neonatal hearing screening. Front. Pediatr. 2023, 11, 1153123. [Google Scholar] [CrossRef] [PubMed]
- Fitzgibbons, E.J.; Driscoll, C.; Myers, J.; Nicholls, K.; Beswick, R. Predicting hearing loss from 10 years of universal newborn hearing screening results and risk factors. Int. J. Audiol. 2021, 60, 1030–1038. [Google Scholar] [CrossRef] [PubMed]

| n (%) | |
|---|---|
| Number of infants | 66 (100) |
| Male | 35 (53) |
| Female | 31 (47) |
| Delivery | |
| Normal delivery | 40 (60) |
| Cesarean | 14 (21) |
| Unspecified | 12 (19) |
| Mean age of diagnosis (SD) | 5.7 mo (3.7) |
| Mean birth weight (SD) | 3061.9 g (789) |
| Unknown | 2 |
| Unilateral deafness | 14 (21) |
| Severity of deafness at diagnosis | |
| Mild | 0 |
| Moderate | 5 |
| Severe | 4 |
| Profound | 5 |
| Bilateral deafness | 52 (79) |
| Severity of deafness at diagnosis | |
| Mild | 3 |
| Asymmmetrical mild/moderate | 2 |
| Moderate | 14 |
| Asymmmetrical moderate/profound | 4 |
| Severe | 8 |
| Profound | 21 |
| Screening tests in maternity/NICU | |
| TEOAE only | 22 (33) |
| aABR only | 28 (42) |
| Both TEOAE and aABR | 11 (17) |
| Unknown | 5 (8) |
| CONGENITAL DEAFNESS RISK FACTORS | n (%) |
|---|---|
| Family history of deafness | 31 (47) |
| 1st degree | 28 |
| 2nd degree | 3 |
| Syndromic forms | 27 (41) |
| Prematurity and/or intrauterine growth retardation | 13 (19.7) |
| NICU hospitalization | 12 (18) |
| Noninvasive ventilation > 5 days | 2 |
| Invasive ventilation > 5 days | 2 |
| Cardiopulmonary arrest | 4 |
| Mean duration of hospitalization (days) | 32 (1–180) |
| Jaundice | 8 (12) |
| Jaundice + phototherapy | 6 |
| Benign jaundice − no treatment | 1 |
| Hyberbilirubinemia > 350 umoL/mL and/or needing exsanguino-transfusion | 1 |
| CMV maternofetal infection | 3 (4.5) |
| Other maternofetal infections | - |
| Perinatal ototoxic treatment | 2 (3) |
| <5 days | 1 |
| >5 days | 1 |
| Bacterial meningitis | 1 (1.5) |
| FHd | Sd | Prematurity | IGR | NICUa | Ventilation | CMVmi | HB | BM | Ode | |
|---|---|---|---|---|---|---|---|---|---|---|
| FH | 1 | |||||||||
| SD | 0.42 | 1 | ||||||||
| Prematurity | 0.242 | 0.234 | 1 | |||||||
| IGR | 0.412 | 0.433 | 0.804 ** | 1 | ||||||
| NICUa | 0.354 | 0.512 | 0.874 ** | 0.782 | 1 | |||||
| Ventilation | 0.204 | 0.199 | 0.948 | 0.755 * | 0.889 ** | 1 | ||||
| cCMV | −0.175 | 0.411 | −0.267 | 0.005 | −0.01 | −0.268 | 1 | |||
| HB | 0.387 | 0.055 | 0.872 ** | 0.700 * | 0.735 * | 0.767 ** | −0.391 | 1 | ||
| BM | −0.273 | −0.275 | −0.511 | −0.484 | −0.517 | −0.509 | −0.232 | −0.482 | 1 | |
| Ode | 0.325 | 0.021 | 0.298 | 0.173 | 0.373 | 0.461 | −0.453 | 0.299 | −0.481 | 1 |
| n | |
|---|---|
| Middle/inner ear malformations | 15 |
| CV anomalies | 5 |
| CV + CND | 4 |
| Cochlear nerve deficiency | 2 |
| Enlarged vestibular aqueduct | 1 |
| EVA + CV malformations | 1 |
| Gusher syndrome | 1 |
| Ossicles dysplasia | 1 |
| Normal imaging | 23 |
| Unknown | 28 |
| TOTAL | 66 |
| Risk factors among children with ear malformations | |
| Familial history | 4 |
| syndromic hearing loss | 3 |
| Family history and syndromic hearing loss | 2 |
| Syndromic hearing loss and perinatal factors | 3 |
| perinatal factors | 2 |
| No risk factors | 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paul, A.; Bense, F.; Boithias Guerot, C.; De La Rubia, S.; Lebeaux, C.; Papon, J.-F. Leading Risk Factors for Congenital Deafness in the Context of Universal Neonatal Screening: Our Observations in a Four-Year Retrospective Study. Int. J. Neonatal Screen. 2024, 10, 11. https://doi.org/10.3390/ijns10010011
Paul A, Bense F, Boithias Guerot C, De La Rubia S, Lebeaux C, Papon J-F. Leading Risk Factors for Congenital Deafness in the Context of Universal Neonatal Screening: Our Observations in a Four-Year Retrospective Study. International Journal of Neonatal Screening. 2024; 10(1):11. https://doi.org/10.3390/ijns10010011
Chicago/Turabian StylePaul, Antoine, Fanny Bense, Claire Boithias Guerot, Sofia De La Rubia, Cécile Lebeaux, and Jean-François Papon. 2024. "Leading Risk Factors for Congenital Deafness in the Context of Universal Neonatal Screening: Our Observations in a Four-Year Retrospective Study" International Journal of Neonatal Screening 10, no. 1: 11. https://doi.org/10.3390/ijns10010011
APA StylePaul, A., Bense, F., Boithias Guerot, C., De La Rubia, S., Lebeaux, C., & Papon, J.-F. (2024). Leading Risk Factors for Congenital Deafness in the Context of Universal Neonatal Screening: Our Observations in a Four-Year Retrospective Study. International Journal of Neonatal Screening, 10(1), 11. https://doi.org/10.3390/ijns10010011