
Newborn Screening for Primary Immunodeficiencies: Focus on Severe Combined Immunodeficiency (SCID) and Other Severe T-Cell Lymphopenias

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
: Primary immunodeficiencies (PID) are congenital disorders of immune competence, which are mainly characterized by a pathological susceptibility to infection. More than 240 PID disease entities have been defined so far, accounting for a broad spectrum of clinical symptoms and severity. Severe PID are increasingly becoming appreciated as a relevant health problem, and diagnostic procedures and screening profiles to allow earliest possible diagnosis on a population scale have already been developed in the USA and few European countries. The most severe PID are characterized by significant mortality in the first years of life, as well as serious morbidity with irreversible organ damage. This applies in particular to PID that are defined by the absence or functional anergy of T-lymphocytes (severe combined immunodeficiency; SCID) or B-lymphocytes (e.g., X-linked agammaglobulinemia; XLA). A strategy to improve the outcome of severe PID by prompt diagnosis and immediate adequate treatment is screening newborns for the presence of T and B cells.1. Introduction
Mass screening of newborn infants started in the early 1960s in the EU with a method for detecting phenylketonuria. Once this procedure was established, other disorders were considered for newborn screening (NBS), prompting the creation of guidelines to help in deciding which diseases were suitable for population-based screening. Different concepts for NBS evaluation exist, yet most EU countries require compliance of the proposed diseases with the expanded Wilson-Jungner framework [1,2,3].
Primary immunodeficiency diseases (PID) represent a heterogeneous group of inborn defects of the immune system that result in recurrent and severe infectious complications. Most PID can be characterized by a functional impairment of the tightly regulated capacity of leukocytes to clear infections, yet some disorders are due to limited differentiation of cells of the lymphoid lineage or increased lymphocyte apoptosis. Newborns that cannot produce T cells themselves are characterized by a severe T-cell lymphopenia, associated in many cases with the clinical diagnosis of “severe combined immunodeficiencies” (SCID).
Newborns with SCID appear healthy at birth and remain clinically “silent” during the first months of life until maternal antibodies which confer a certain degree of protection against infection vanish and the inability to develop T cells cannot be compensated anymore by maternal immunity. SCID is lethal during the first two years of life, mandating curative haematopoietic stem cell transplantation (HSCT) or gene therapy. In addition, for adenosine deaminase (ADA) SCID, enzyme replacement therapy (ERT) also offers a treatment alternative. These therapies work best if initiated before overwhelming infectious complications occur.
The clinical severity and the associated economic burden of a delayed diagnosis, as well as the potentially fatal outcome if not treated promptly, emphasize the importance of early identification of the affected newborns. Although the condition is rare, precise estimates of its frequency based on large population studies are lacking. Early data from pilot screening programs in the USA suggest that one in 30,000–50,000 infants may be born with SCID. Although incidence rates are not strictly part of the extended Wilson-Jungner evaluation framework, SCID may well be detected at frequencies similar to disorders of fatty acid or amino acid metabolism that are screened for in numerous EU country NBS programs.
The benefit of a neonatal screening for SCID is best exemplified by the extremely effective treatment for SCID, with approximately 95% survival following HSCT when transplantation is performed before 3.5 months of age. However, if SCID is not screened for at birth, patients will suffer from recurrent infections and survival is severely compromised. Furthermore, following onset of infectious periods, the pharmacological preparation for stem cell transplantation has to be adjusted and becomes more difficult, often resulting in less successful engraftment. Thus, early intervention not only reduces the disease-related mortality and morbidity but is also curative, in contrast to the dietary therapeutic approaches that have to be taken in the metabolic diseases currently screened for.
2. Severe Combined Immunodeficiency and Severe T-Cell Lymphopenia—A Life-Threatening Group of Disorders
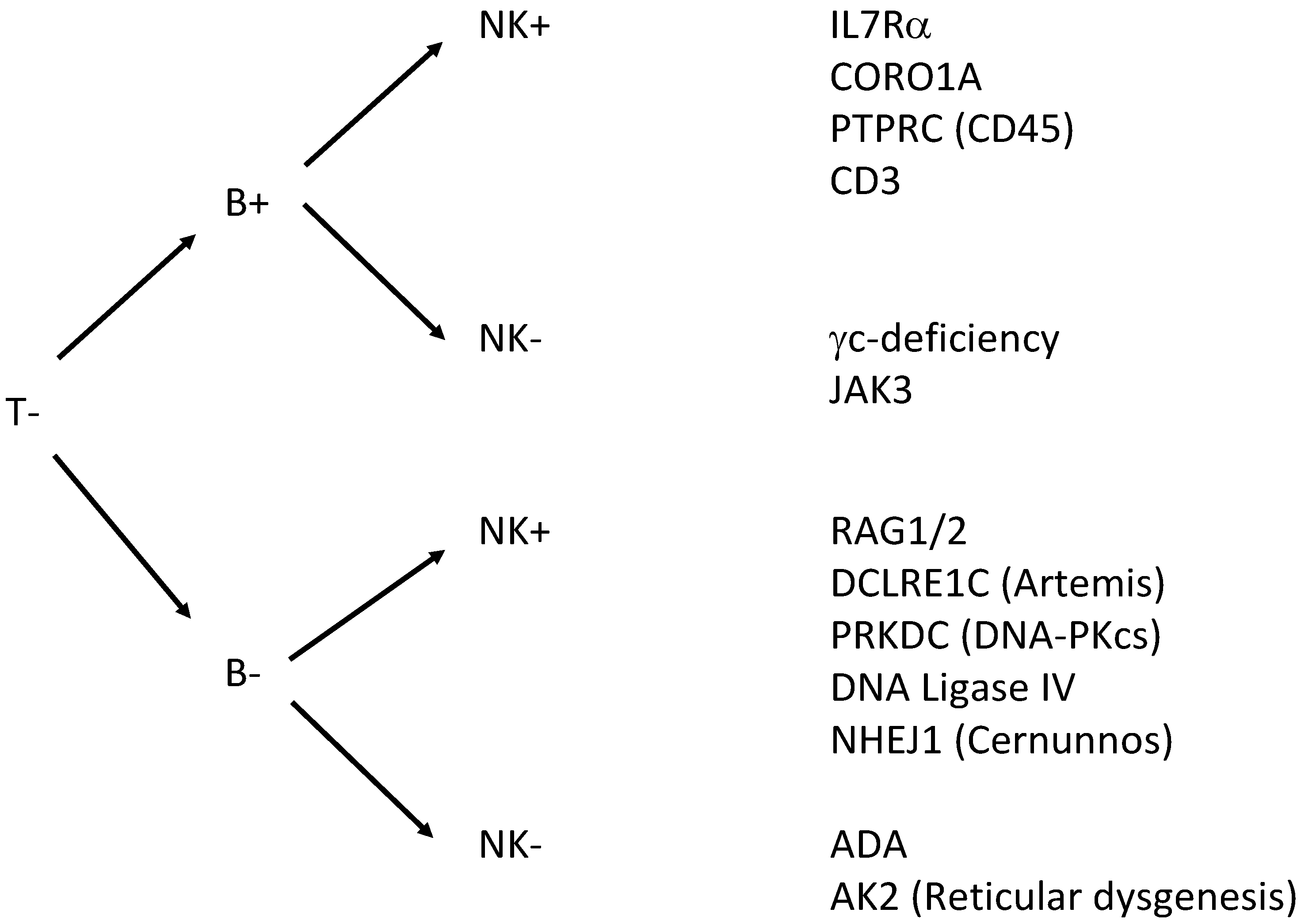
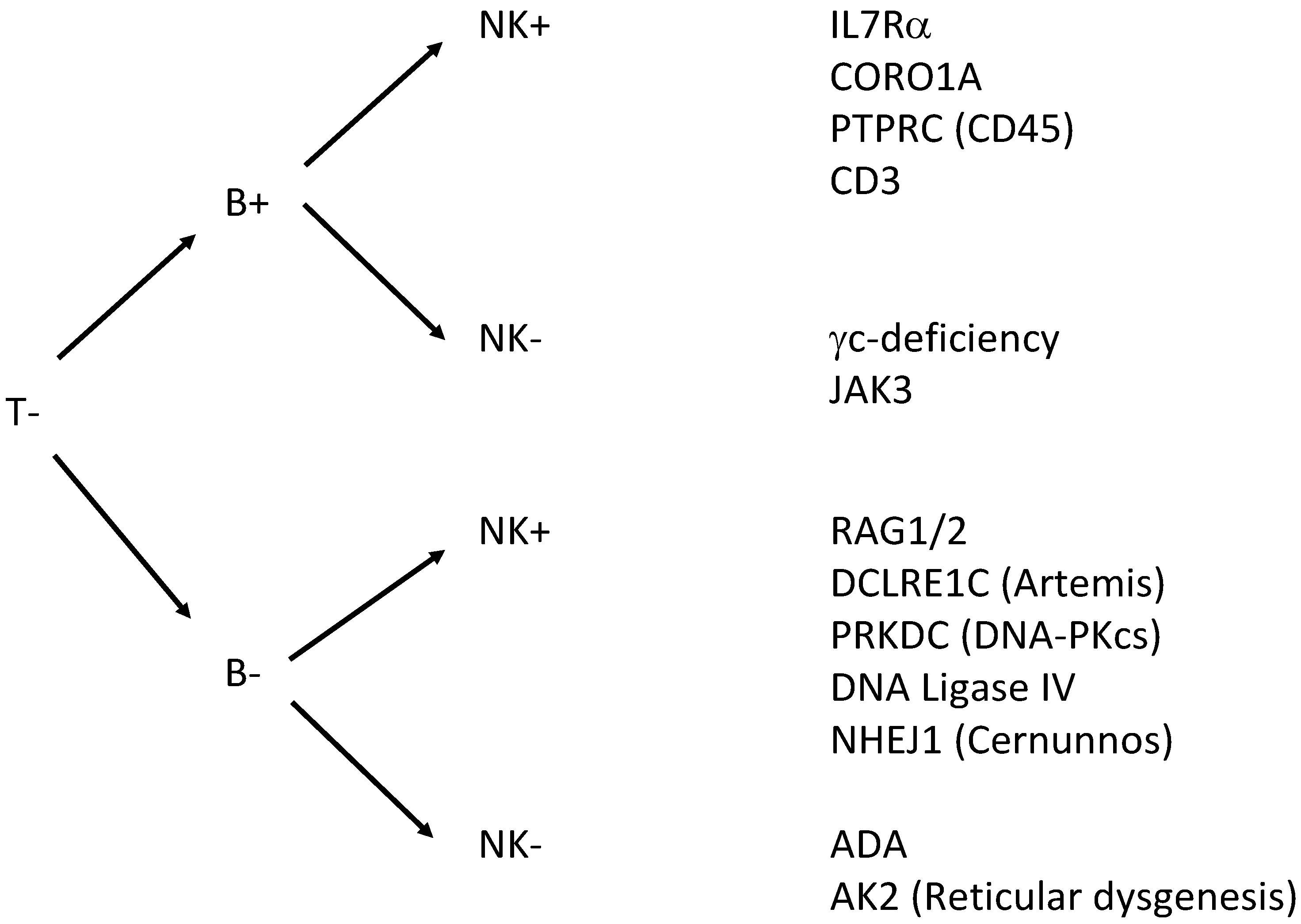
Severe combined immunodeficiency (SCID) is a group of life-threatening immune disorders arising from a variety of genetic defects that lead to the absence of lymphocyte development and function. Nearly all patients with SCID have absent T-cells, and are further grouped by absence or presence of B-cells and NK-cells (Figure 1). The diagnosis of SCID is a pediatric emergency, as affected children have extreme susceptibility to bacterial, viral, fungal, and opportunistic infections, which are fatal in the first 1–2 years of life without curative treatment [4,5]. In most cases, children with SCID appear well at birth and present with recurrent severe infections and failure to thrive at 3–6 months as passively transferred protective maternal immunoglobulins are diminishing [6,7,8]. Severe T-cell deficiency syndromes may also present early in infancy with autoimmune-like phenomena and susceptibility to infection. These disorders may be caused by hypomorphic mutations in known SCID-causing genes.
When diagnosis is delayed, a significant number of children die before a definitive curative treatment such as haematopoietic stem cell transplantation (HSCT) or gene therapy (GT, currently available for ADA-SCID and X-SCID) can be undertaken. Early diagnosis allows timely start of preventive measures, including prophylactic antibiotics, antifungal or antiviral treatment and immunoglobulin replacement therapy, as well as avoidance of live vaccines. In addition, early searches for HLA-matched haematopoietic stem cell (HSC) donors are enabled [9]. This is important, as HSCT in the first 3.5 months of life is associated with up to 95% chance of survival, irrespective of donor type or type of SCID [10,11,12], compared with 76% survival for later HSCT [13].
3. Newborn Screening for SCID and Severe T-Cell Lymphopenia: TREC Assay
There has been a long-standing search for a suitable biomarker for normal T-cell development and for methods that allow NBS for SCID. Newborn screening for SCID was initially proposed in the 1990s by Puck and Buckley, suggesting that a complete (differential) blood count might help diagnose the disease based on a reduced absolute lymphocyte count in blood. As this method relied on fresh blood samples, the efforts for sample logistics and handling prevented its large-scale implementation. In addition, T−B+NK+ forms of SCID with elevated numbers of B- or NK-cells might yield false-negative test results. ELISA-like immunoassays for Interleukin-7 have also been proposed for SCID screening, yet technical issues of inferior sample stability and tedious protein purification have prevented their broader application [14]. Similarly, bead-capture assays for the T-cell receptor-CD3 complex and CD45 have been developed, but false negative results in SCID patients with maternal engraftment or expansion of oligoclonal T cells was encountered, thereby disqualifying this technique from implementation [15].
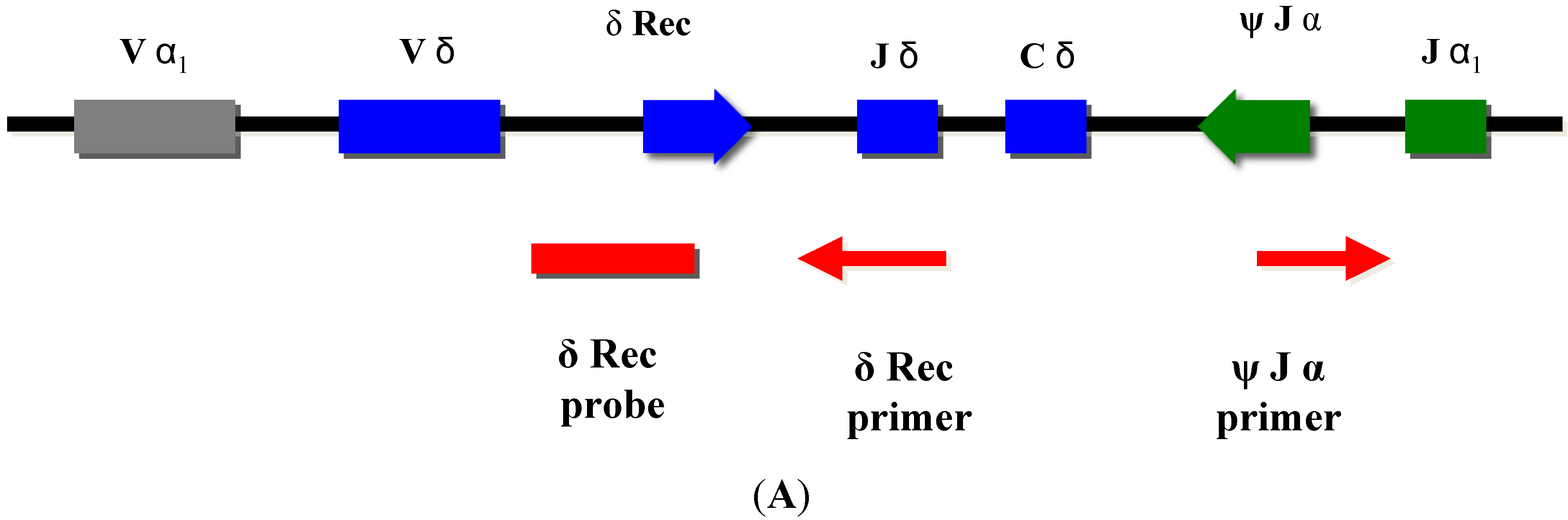
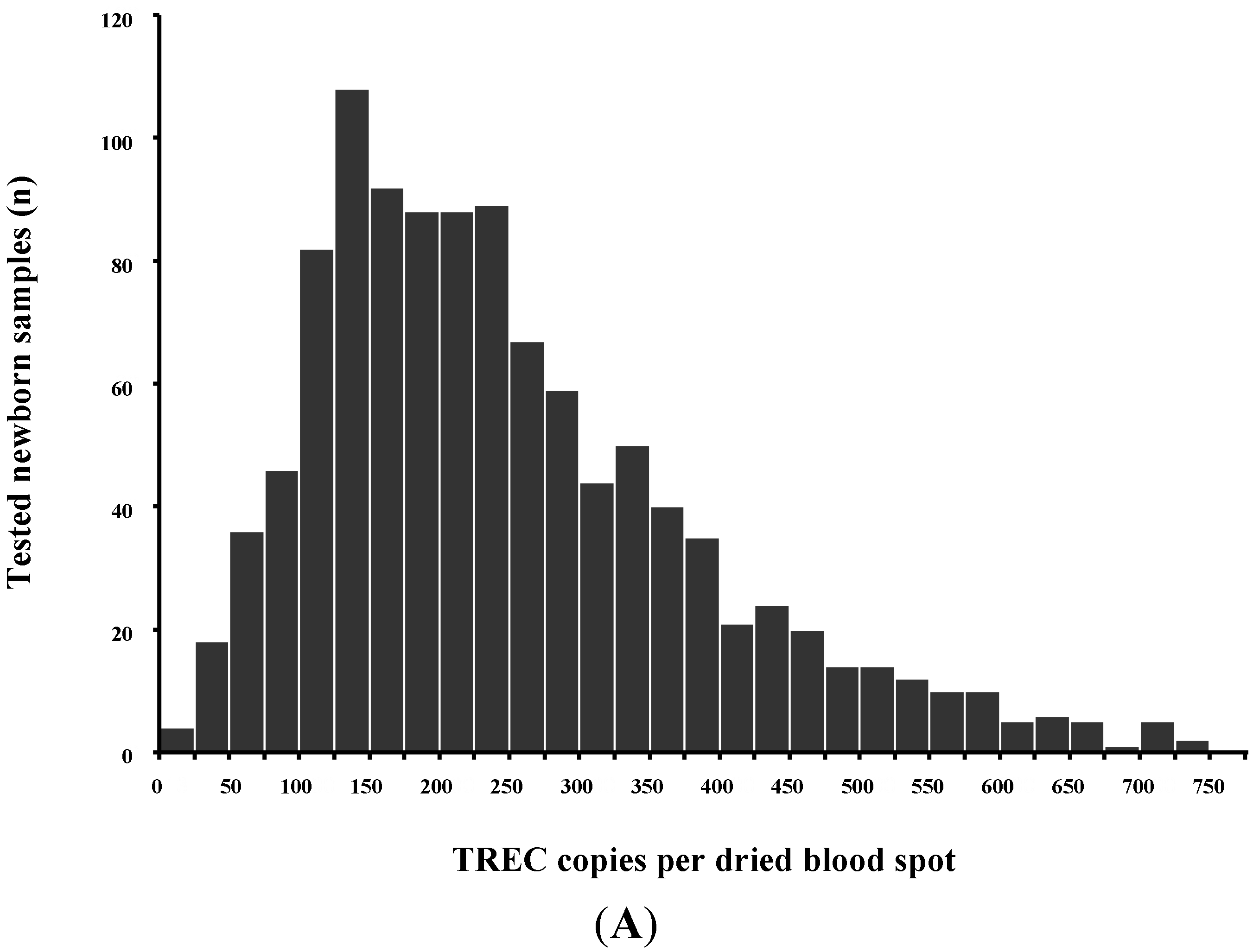
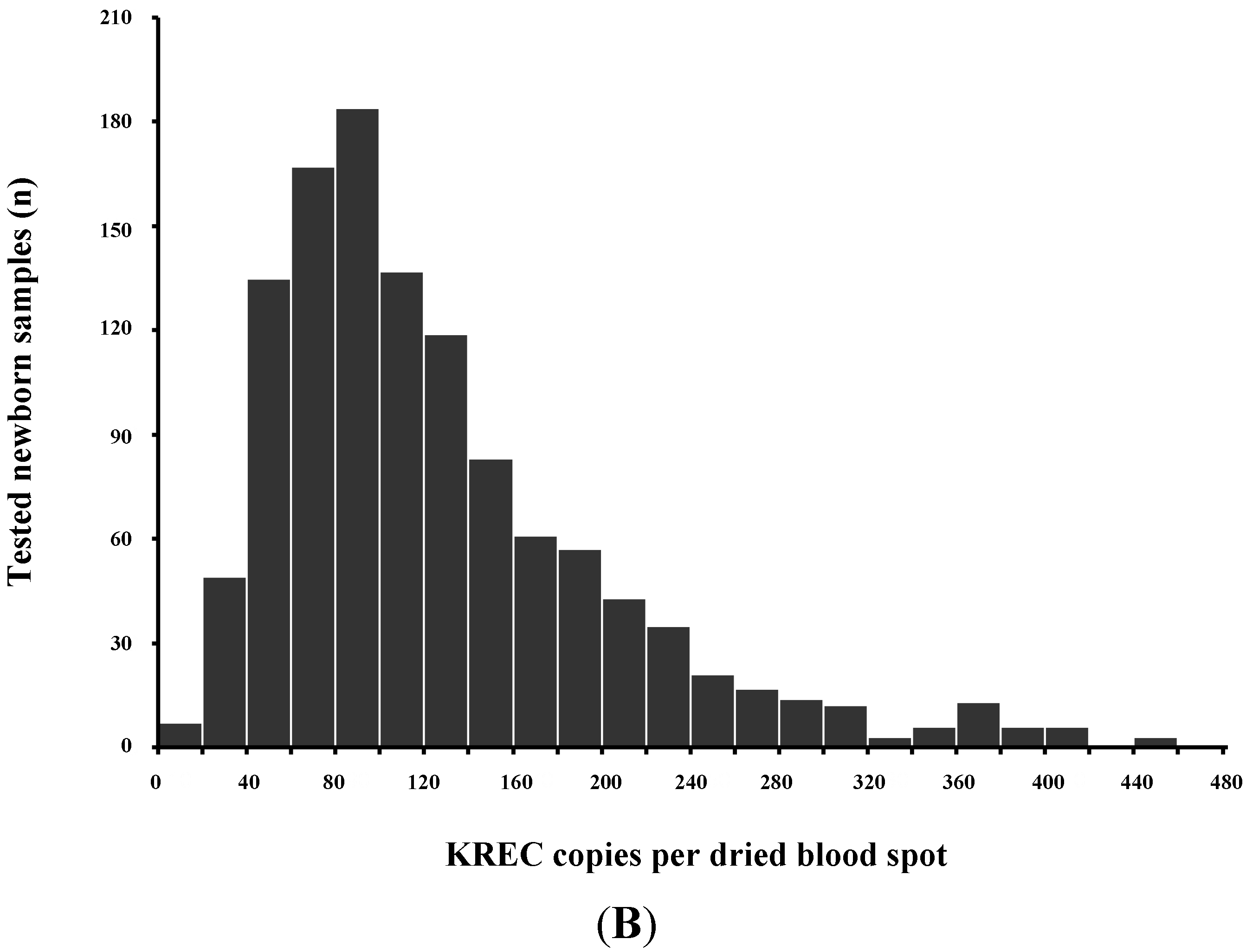
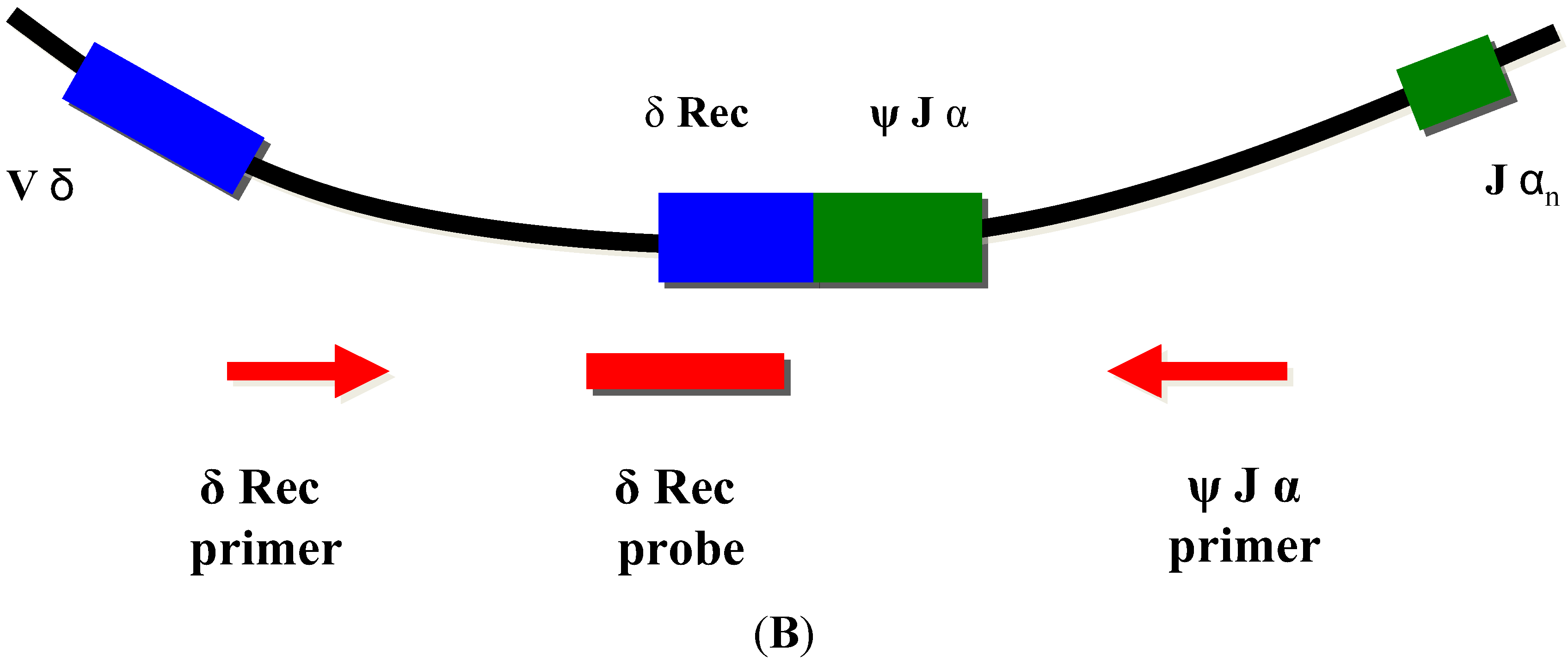
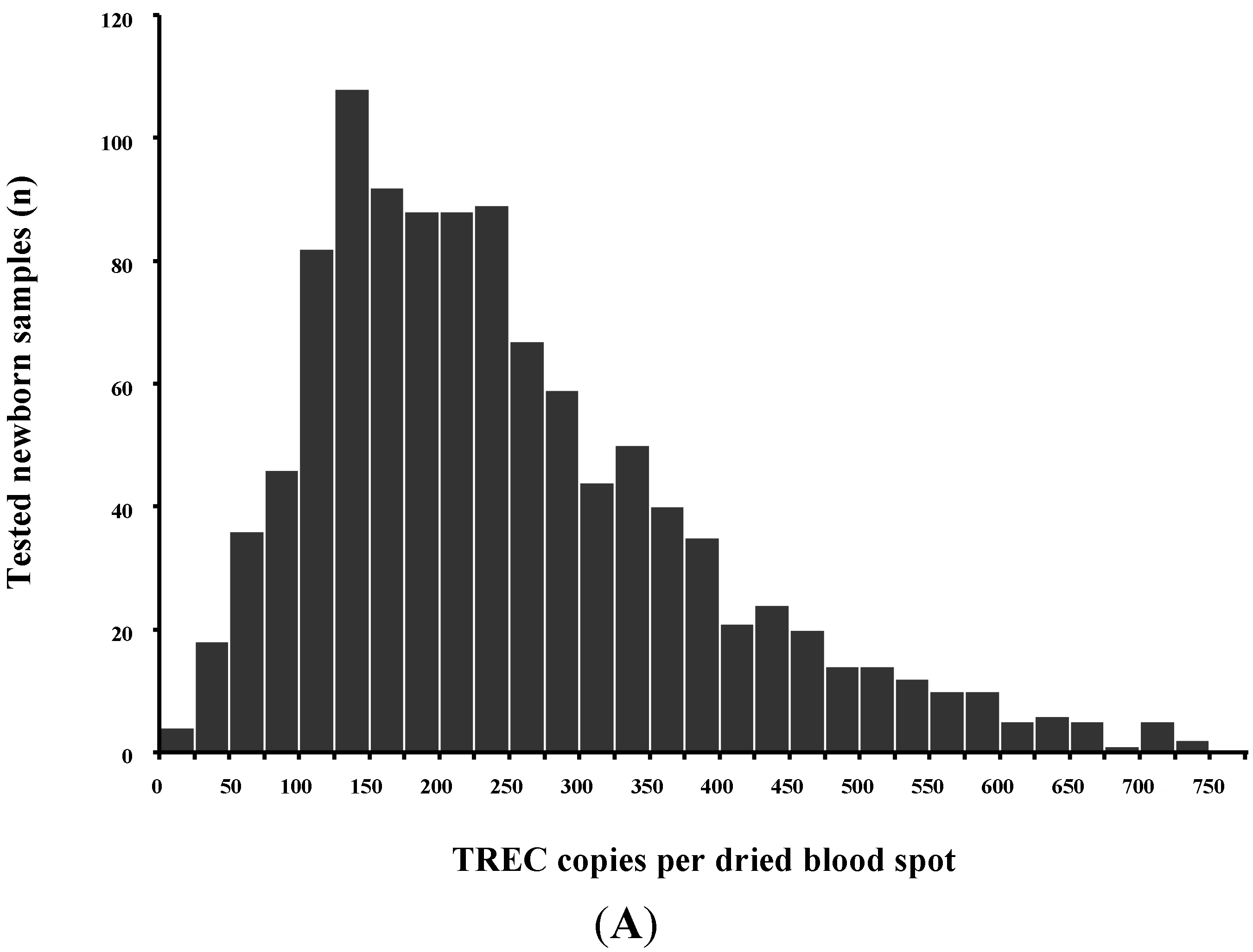
Normal T-cell development requires production of precursor T-cells in the bone marrow and subsequent processing of T-cells in the thymus. Although SCID can arise from a variety of genetic defects, there is an abnormality of T-cell development in the thymus in all cases. During normal thymic processing, T cells undergo receptor gene splicing and rearrangement, leading to intracellular accumulation of DNA by-products known as T-cell receptor excision circles (TRECs) [16,17]. When used in NBS assays, TRECs are a surrogate marker of newborns’ capability to produce T cells, which is severely hampered in SCID patients. Suitable TREC assays are based on DNA extracted from a regular dried blood spot collected at birth and can be determined either by quantitative or end-point PCR (Figure 2).
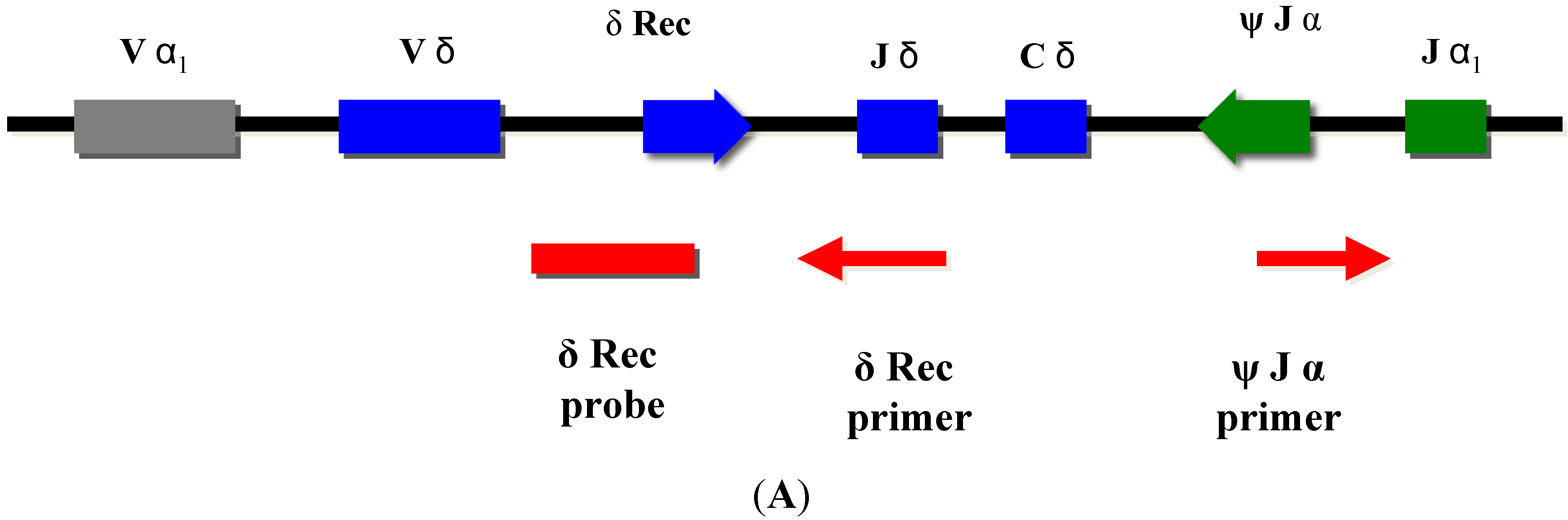
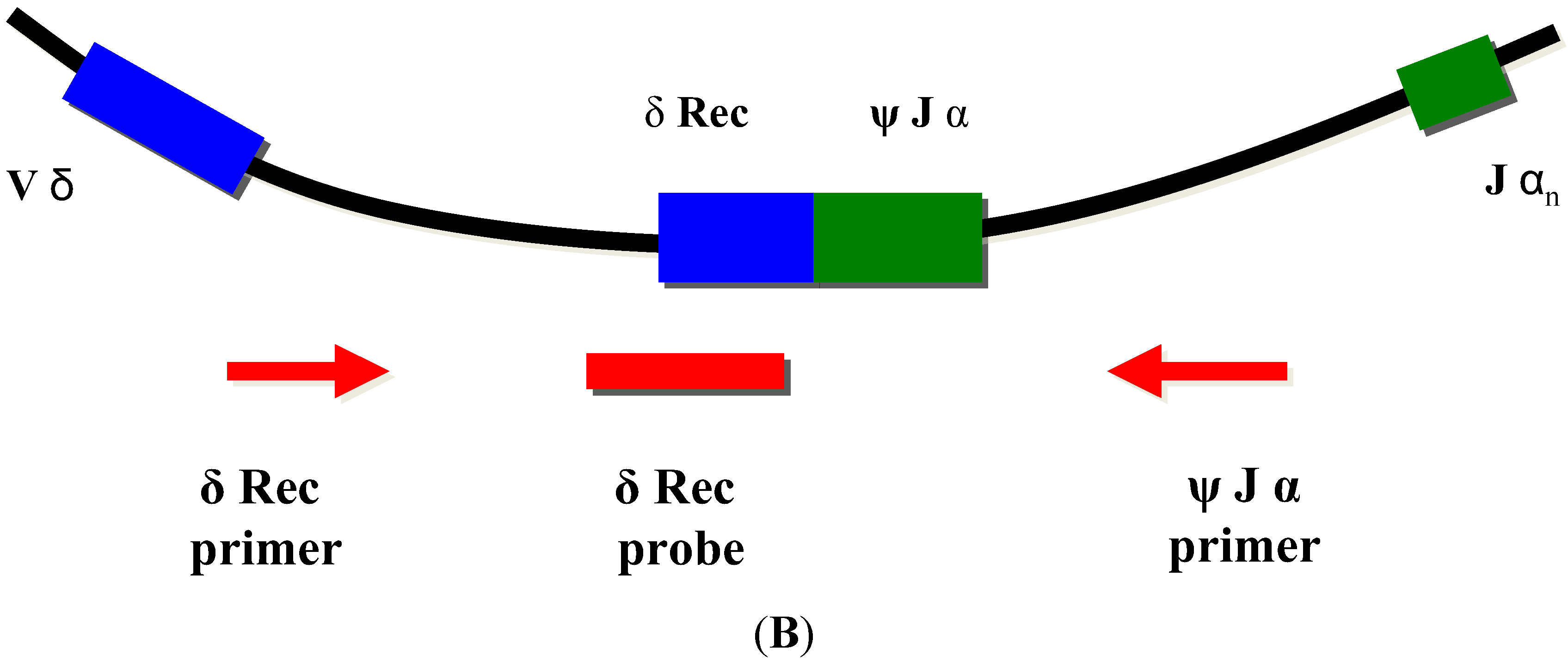
TRECs do not replicate in dividing cells and are diluted out upon cellular division. They are therefore only found in recent thymic emigrant naive T-cells [8,9] (Figure 3A). This aspect is important, as in certain conditions such as engraftment of maternal T-cells or expansion of a few oligoclonal T-cells in Omenn syndrome, a substantial amount of T-cells can be found in an infant with SCID. As these T-cells have undergone multiple rounds of cell division, TRECs are diluted and the TREC value is low despite high numbers of T-cells in peripheral blood [6].
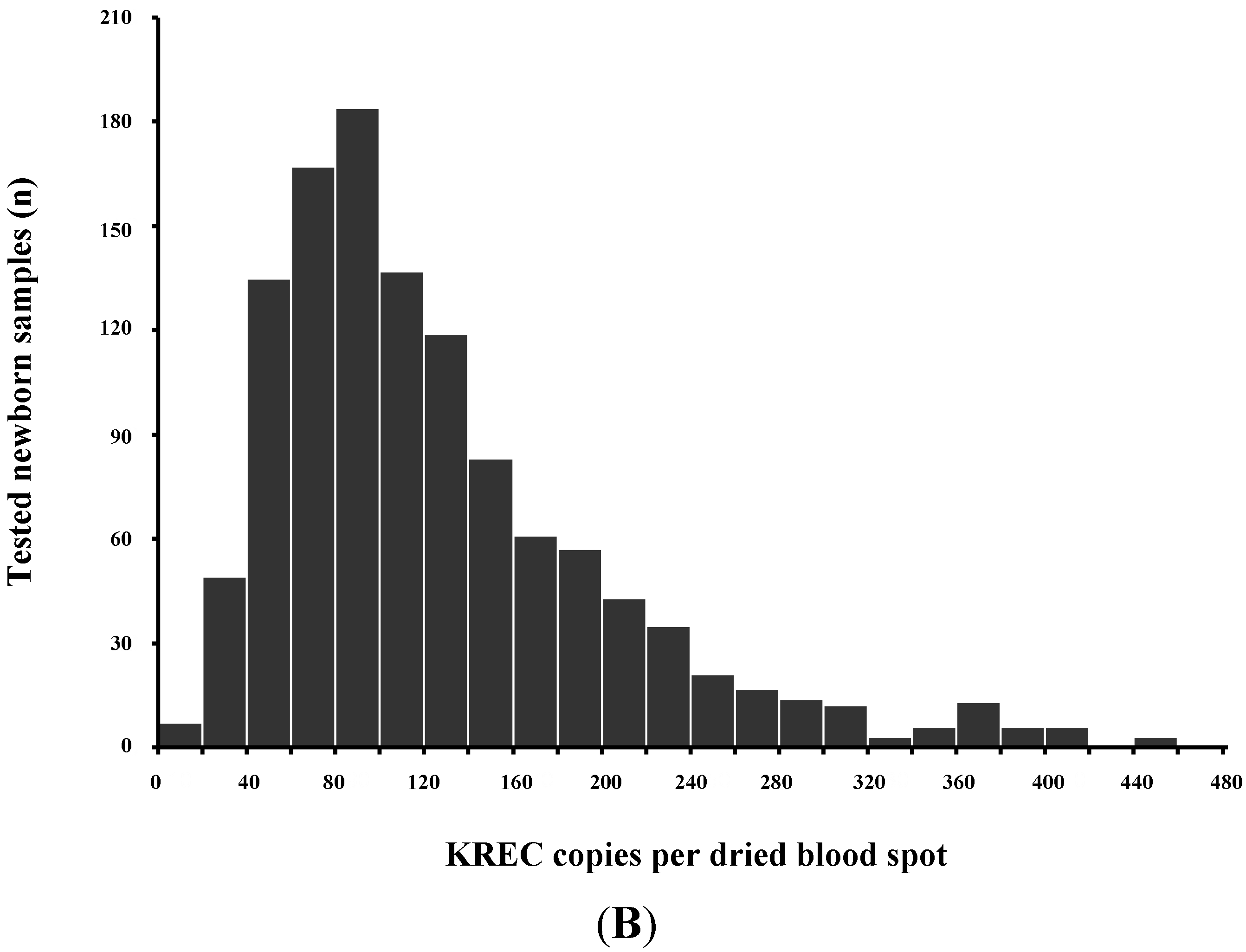
As some leaky, variant, or delayed-onset forms of SCID might not be detected at birth based on the TREC assay, the addition of other screening markers such as KREC [18], which detect defects of B-cell development has been proposed [19,20,21] (Figure 3B).
4. TREC Assay Based Newborn Screening Programs
Population-based screening is the only means to detect SCID before the onset of life-threatening infections in most patients, as more than 80% lack a positive family history [22,23]. In addition, SCID is the first condition being screened for in population-wide newborn screening that can actually be cured. Newborn screening for SCID by the TREC assay in the USA began in 2008 in Wisconsin and has now been extended to 23 states, the District of Columbia and the Navajo Nation [24]. Available data on >3 million newborns from the first 5 years of screening in 11 programs in the USA reported an incidence of 1 in 58,000 live births (95% CI, 1/46,000–1/80,000) with an overall survival of 87%. The majority of cases had typical SCID (42/52 identified SCID cases), 10/52 infants had leaky SCID and 1/52 had Omenn syndrome. No cases of SCID were missed by initial TREC screening but detected later. Survival of identified infants with SCID who received HSCT, GT or enzyme replacement was 92%. Specificity was high at >99.8%, and in addition to 52 cases of typical SCID, leaky SCID and Omenn syndrome, other cases of non-SCID severe T-cell lymphopenia were detected. Identification of non-SCID severe T-cell lymphopenia allows to avoid life vaccines and to install appropriate infection prophylaxis and follow-up [25,26,27].
Non-SCID diseases with severe T-cell deficiency detected by TREC screening can be grouped in (1) congenital syndromes with T-cell impairment; (2) secondary T-cell impairment; (3) preterm birth; (4) variant SCID; (5) unspecified T-cell lymphopenia. In the above report from the USA the incidence of non-SCID T-cell lymphopenia was 1:14,000 using a definition of <1500 T-cells/μL as cut-off and excluding preterm infants [24]. 33% of the infants with non-SCID T-cell lymphopenia had congenital syndromes known to be associated with T-cell lymphopenia: 57% had DiGeorge syndrome/chromosome 22q11.2 deletion, 15% had trisomy 21, 3% each had ataxia telangiectasia or trisomy 18, and 2% had CHARGE syndrome. Non-SCID T-cell lymphopenia was attributed to other medical conditions in 28%, of which 26% had congenital heart disease, followed by multiple other congenital anomalies, loss into third space due to vascular leakage, or gastrointestinal anomalies.
Population-based newborn screening by TRECs has thus provided a new incidence of SCID, higher than the incidence of 1:100,000 estimated from retrospective clinical diagnoses, suggesting that SCID has previously been underdiagnosed in infants with fatal infections. SCID newborn screening has been recommended by the US Department of Health and Human Services Secretary across all states [28], and many other countries worldwide are beginning to include TRECs screening in their screening programs, or are in the phase of pilot-studies or applications of respective proposals to their medical authorities. Other states or provinces currently using the TREC assay for prospective population-wide newborn screening are: Ontario/Canada, Taiwan and Sao Paulo/Brazil.
Newborn screening for PID (TREC-only) is an established test within the state wide screening programs in the most populous states in the USA and in Taiwan. Europe is currently lagging behind but pilot screening programs have recently been initiated in Sweden [19], Germany [20], UK [29], France [30], and Spain [31] and the rationale for newborn screening (originally established by Wilson and Jungner [1]) has recently been outlined for patients with PID [4,9]. Proposals to include SCID diagnostics in population-based newborn screening programs have been submitted to national agencies in Germany, the Netherlands, the UK, and Switzerland.
5. TREC Screening Algorithm
Screening for severe T-cell lymphopenia by TREC in the USA is not standardized and employs different methods, leading to marked differences in cut-offs for the number of newly formed T-cells in the ongoing screening programs in the various states. This, in turn, has resulted in marked differences in the number of patient recalls and diagnostic procedures (fluorescence activated cell sorting, (FACS)), with an almost ten-fold difference between states [24].
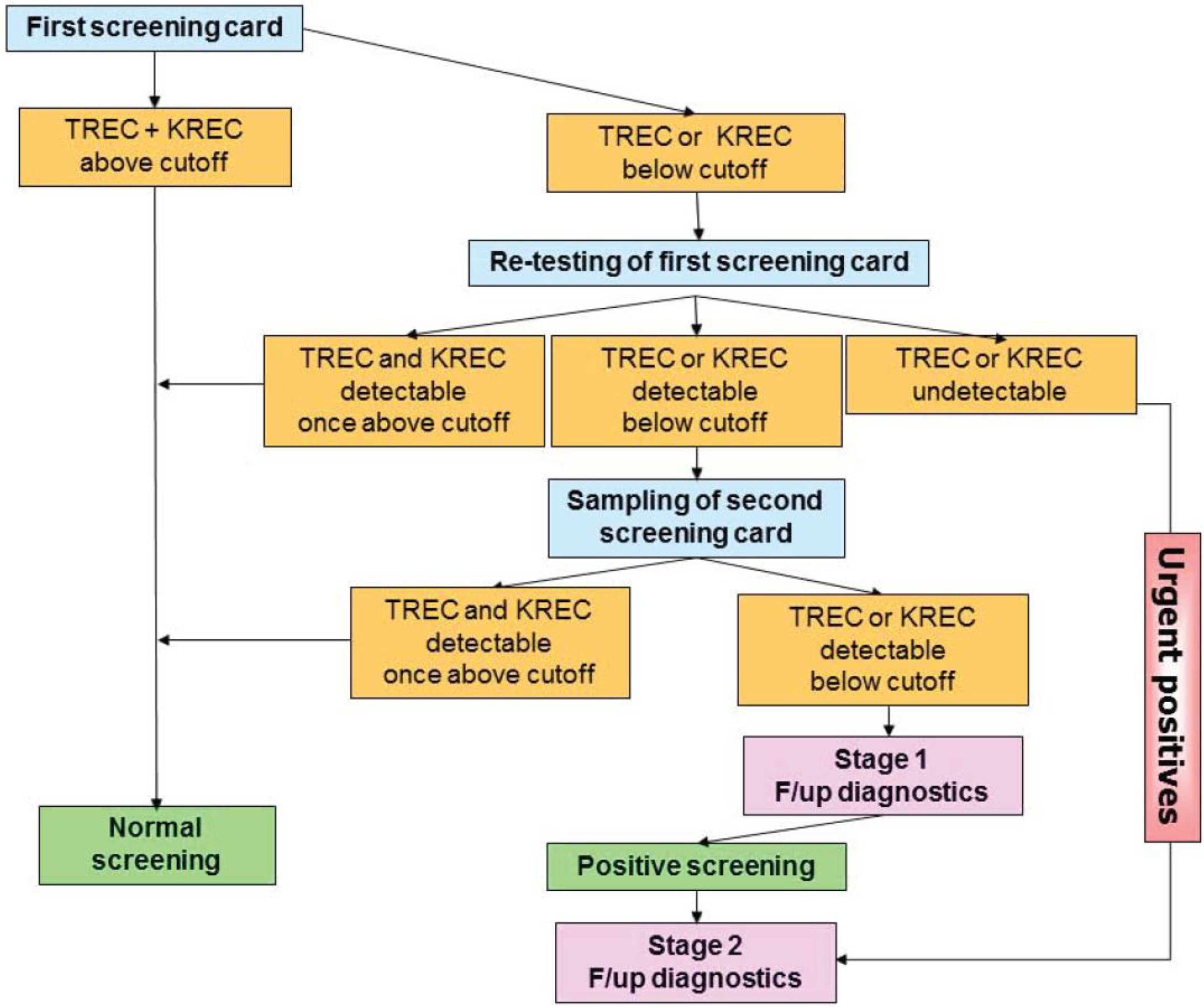
To ensure adequate follow-up of infants with likely SCID identified by TREC screening and to limiting the number of false-positive results at the same time, algorithms have been designed by screening centers together with clinical immunologists. In most cases this will include the following (Figure 4).
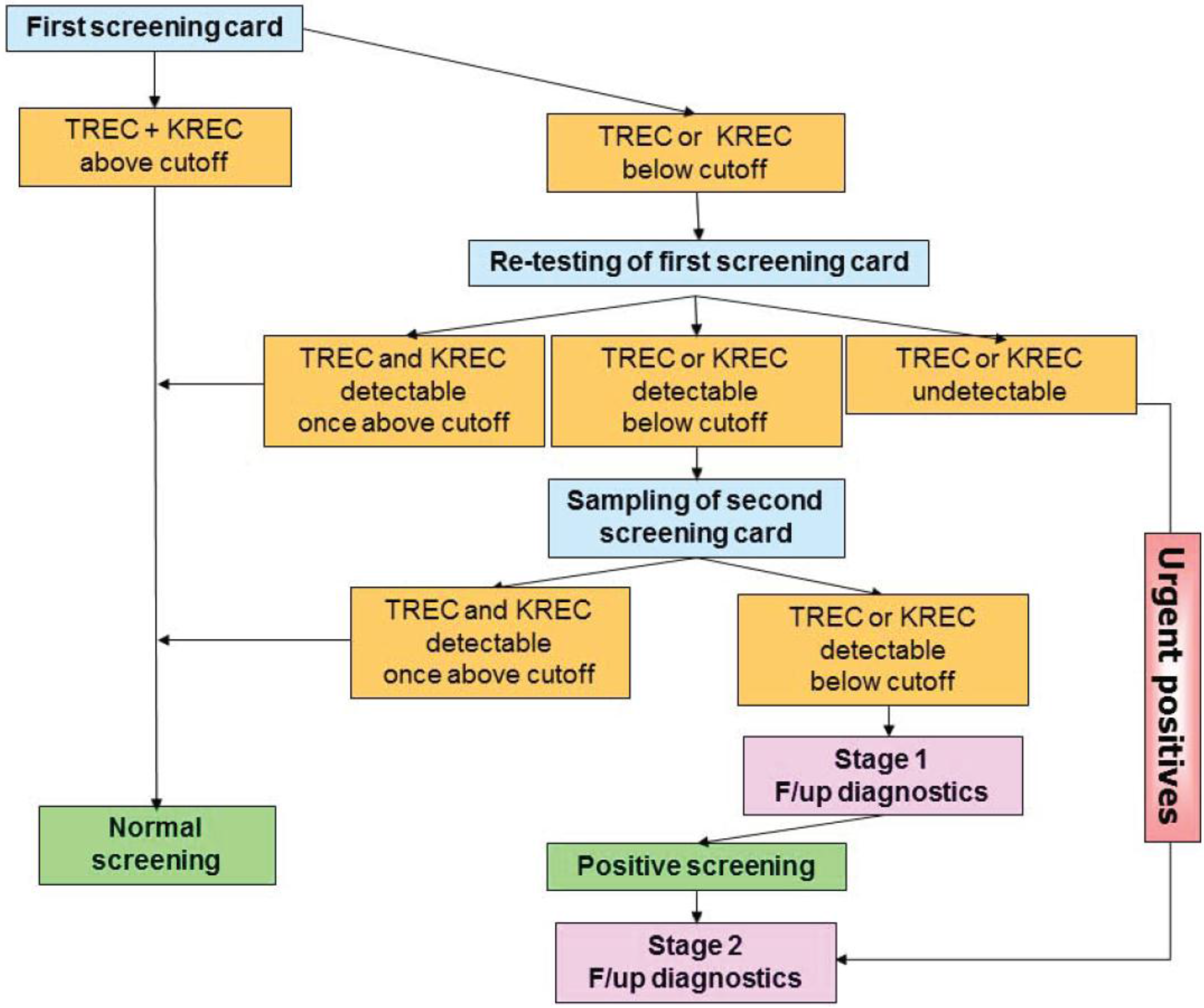
All infants undergo screening by the TREC assay. If normal, no further intervention is recommended. In infants with TREC levels below the cut-off, the first screening card will be retested for TREC as well as DNA amplification by quantifying β-actin or RNase P levels by qRT-PCR. If the β-actin level is normal and TRECs are still below the cut-off the primary care provider is contacted for two scenarios (1) an emergency scenario—if TRECs are undetectable (~1 in 20,000 cases, [24]), the infant needs urgent confirmatory testing by flow cytometry and treatment by a clinical immunologist (2) an intermediate scenario—if TRECs are detectable, yet below the established local cut-off value, re-testing of a second screening card is performed (tracking process): Tracking of newborns is thus initiated in case of repeated abnormal test results for TREC and/or KREC copy numbers after examination of at least three independent dried blood punches of the first dried blood spot submitted.
As first action within the tracking procedure, the obstetric unit (“doctor–doctor” contact) and the parents of the newborn (“doctor–patient” contact) should be contacted to obtain additional information on the status of the child (stage 1). Since repeated abnormal TREC and/or KREC test results can occur especially in preterm infants (<32nd week of pregnancy), children with syndromic diseases (e.g., trisomy 21 or 22q11 microdeletion), metabolic diseases (such as propionyl-CoA carboxylase deficiency or methylmalonic aciduria), and severe congenital infections or malformations with subsequent lymphocyte extravasation (e.g., neonatal sepsis or gastroschisis), it is important to gather and compile additional examination findings to evaluate a suspected diagnosis of a severe congenital immunodeficiency.
While the parents of the newborn should be informed without delay after the examination of the second separate dried blood card, even if the findings are normal, a detailed explanation of the significance of the test results, if they are abnormal again, will be provided only at a specialized immunodeficiency center. The parents will be directed to such a center in the area (stage 2), and the center will be informed about the patient to be expected.
Subsequently, a specialized treatment center should be selected that is close to home: in the case of suspected severe T- and/or B-lymphopenia, intensive hygiene measures and possibly early, strict isolation to prevent opportunistic infections are necessary steps to be taken. In order to perform HSCT or gene therapy, a transfer to a specialized transplant center may be necessary. Only such centers should be selected that have already gained experience in this field in patients with primary immunodeficiencies (optimized conditioning and transplantation protocols).
6. Fiscal Implications of SCID Newborn Screening
A number of USA states have already adopted SCID NBS, based on the TREC assay. The success of these programs so far has led to a nationwide recommendation of NBS for SCID by the USA Department of Health and Human Services [24]. The cost-effectiveness analysis of case finding SCID screening has been shown to be economically favorable in relation to the expenditures on medical healthcare for this group of disorders. As one of the most important determining factors, detection of SCID at birth allows for curative treatment before the occurrence of complications that often require prolonged and expensive intensive care management that markedly increases healthcare costs. The rapid access to curative therapy also reduces the social burden, tremendous impairment in quality-of-life and the inestimable social costs associated with an undiagnosed SCID. Thus far, only five cost analyses of the SCID newborn screening programs have been published based on data from the USA [23,32,33,34] and the UK [35]. They all conclude that in addition to saving lives, newborn screening for SCID results in a reduction in overall health-care costs.
7. Outlook on Expansion of NBS to Detect More Immune Deficiencies
Developing the concept of cost-efficacy in newborn screening further, it has been proposed to multiplex the TREC screening assay with other biomarkers that would allow identification of diseases suitable to be included in population-scale screening programs. Severe inborn immunodeficiency diseases characterized by the absence of B-cells, resembling the clinical diagnosis of primary agammaglobulinemias, can be detected at birth when quantifying KRECs in a multiplex qPCR or endpoint PCR reaction together with TRECs. As both biomarkers can be assessed within the same dried blood spot sample, most infrastructural and technical resources can be used without additional efforts or expenses when the multiplex assay is performed. The additional technical cost fraction of an add-on KREC assessment is less than 6% of the total assay costs. However, analysis of KREC copy numbers in dried blood spot specimen, as a tool for newborn screening, requires further validation to ensure sufficient sensitivity and specificity before broad implementation can be expected.
Acknowledgments
Conclusions presented in the review were made possible by research funds obtained from the German Federal Ministry of Education and Research (BMBF 1315883) to Stephan Borte, Janine Reichenbach was supported by research grants from Gebert Rüf Stiftung, program “Rare Diseases—New Approaches” (grant No. GRS-046/10), Zürich Center for Integrative Human Physiology (ZIHP), EU-FP7 CELL-PID and EU-FP7 NET4CGD.
Author Contributions
Janine Reichenbach performed a structured literature search, created figures, drafted and wrote the review; Stephan Borte shared his experience in prospective screening trials for severe primary immunodeficiencies, created figures and wrote the review.
Conflicts of Interest
Stephan Borte received grants from PerkinElmer (Wallac OY) for comparison studies in the field of newborn screening for SCID.
References
- Wilson, J.M.; Jungner, Y.G. Principles and practice of mass screening for disease. Bol. Oficina Sanit. Panam. 1968, 65, 281–393. [Google Scholar] [PubMed]
- Andermann, A.; Blancquaert, I.; Beauchamp, S.; Dery, V. Revisiting Wilson and Jungner in the genomic age: A review of screening criteria over the past 40 years. Bull. World Health Organ. 2008, 86, 317–319. [Google Scholar] [CrossRef] [PubMed]
- Petros, M. Revisiting the Wilson-Jungner criteria: How can supplemental criteria guide public health in the era of genetic screening? Genet. Med. 2012, 14, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, H.B.; Hammarstrom, L.; Mahlaoui, N.; Borte, M.; Borte, S. The case for mandatory newborn screening for severe combined immunodeficiency (SCID). J. Clin. Immunol. 2014, 34, 393–397. [Google Scholar] [CrossRef] [PubMed]
- Van der Burg, M.; Gennery, A.R. Educational paper. The expanding clinical and immunological spectrum of severe combined immunodeficiency. Eur. J. Pediatr. 2011, 170, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Verbsky, J.; Routes, J. Screening for and treatments of congenital immunodeficiency diseases. Clin. Perinatol. 2014, 41, 1001–1015. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, H.B.; Gilmour, K.C.; Jones, A.M. Severe combined immunodeficiency-molecular pathogenesis and diagnosis. Arch. Dis. Child. 2001, 84, 169–173. [Google Scholar] [CrossRef] [PubMed]
- Rivers, L.; Gaspar, H.B. Severe combined immunodeficiency: Recent developments and guidance on clinical management. Arch. Dis. Child 2015. [Google Scholar] [CrossRef] [PubMed]
- Borte, S.; von Dobeln, U.; Hammarstrom, L. Guidelines for newborn screening of primary immunodeficiency diseases. Curr. Opin. Hematol. 2013, 20, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Brown, L.; Xu-Bayford, J.; Allwood, Z.; Slatter, M.; Cant, A.; Davies, E.G.; Veys, P.; Gennery, A.R.; Gaspar, H.B. Neonatal diagnosis of severe combined immunodeficiency leads to significantly improved survival outcome: The case for newborn screening. Blood 2011, 117, 3243–3246. [Google Scholar] [CrossRef] [PubMed]
- Myers, L.A.; Patel, D.D.; Puck, J.M.; Buckley, R.H. Hematopoietic stem cell transplantation for severe combined immunodeficiency in the neonatal period leads to superior thymic output and improved survival. Blood 2002, 99, 872–878. [Google Scholar] [CrossRef] [PubMed]
- Gennery, A.R.; Slatter, M.A.; Grandin, L.; Taupin, P.; Cant, A.J.; Veys, P.; Amrolia, P.J.; Gaspar, H.B.; Davies, E.G.; Friedrich, W.; et al. Transplantation of hematopoietic stem cells and long-term survival for primary immunodeficiencies in europe: Entering a new century, do we do better? J. Allergy Clin Immunol. 2010, 126, 602–610. [Google Scholar] [CrossRef] [PubMed]
- Buckley, R.H.; Schiff, S.E.; Schiff, R.I.; Markert, L.; Williams, L.W.; Roberts, J.L.; Myers, L.A.; Ward, F.E. Hematopoietic stem-cell transplantation for the treatment of severe combined immunodeficiency. N. Engl. J. Med. 1999, 340, 508–516. [Google Scholar] [CrossRef] [PubMed]
- McGhee, S.A.; Stiehm, E.R.; Cowan, M.; Krogstad, P.; McCabe, E.R. Two-tiered universal newborn screening strategy for severe combined immunodeficiency. Mol. Genet. Metab. 2005, 86, 427–430. [Google Scholar] [CrossRef] [PubMed]
- Janik, D.K.; Lindau-Shepard, B.; Comeau, A.M.; Pass, K.A. A multiplex immunoassay using the guthrie specimen to detect T-cell deficiencies including severe combined immunodeficiency disease. Clin. Chem. 2010, 56, 1460–1465. [Google Scholar] [CrossRef] [PubMed]
- Puck, J.M. Laboratory technology for population-based screening for severe combined immunodeficiency in neonates: The winner is T-cell receptor excision circles. J. Allergy Clin. Immunol. 2012, 129, 607–616. [Google Scholar] [CrossRef] [PubMed]
- Morinishi, Y.; Imai, K.; Nakagawa, N.; Sato, H.; Horiuchi, K.; Ohtsuka, Y.; Kaneda, Y.; Taga, T.; Hisakawa, H.; Miyaji, R.; et al. Identification of severe combined immunodeficiency by T-cell receptor excision circles quantification using neonatal guthrie cards. J. Pediatr. 2009, 155, 829–833. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, N.; Imai, K.; Kanegane, H.; Sato, H.; Yamada, M.; Kondoh, K.; Okada, S.; Kobayashi, M.; Agematsu, K.; Takada, H.; et al. Quantification of kappa-deleting recombination excision circles in guthrie cards for the identification of early B-cell maturation defects. J. Allergy Clin. Immunol. 2011, 128, 223–225. [Google Scholar] [CrossRef] [PubMed]
- Borte, S.; Wang, N.; Oskarsdottir, S.; von Dobeln, U.; Hammarstrom, L. Newborn screening for primary immunodeficiencies: Beyond SCID and XLA. Ann. N. Y. Acad. Sci. 2011, 1246, 118–130. [Google Scholar] [CrossRef] [PubMed]
- Borte, S.; von Dobeln, U.; Fasth, A.; Wang, N.; Janzi, M.; Winiarski, J.; Sack, U.; Pan-Hammarstrom, Q.; Borte, M.; Hammarstrom, L. Neonatal screening for severe primary immunodeficiency diseases using high-throughput triplex real-time PCR. Blood 2012, 119, 2552–2555. [Google Scholar] [CrossRef] [PubMed]
- Speckmann, C.; Neumann, C.; Borte, S.; la Marca, G.; Sass, J.O.; Wiech, E.; Fisch, P.; Schwarz, K.; Buchholz, B.; Schlesier, M.; et al. Delayed-onset adenosine deaminase deficiency: Strategies for an early diagnosis. J. Allergy Clin. Immunol. 2012, 130, 991–994. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.; Puck, J.M. Development of population-based newborn screening for severe combined immunodeficiency. J. Allergy Clin. Immunol. 2005, 115, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.; Scalchunes, C.; Boyle, M.; Puck, J.M. Early vs. delayed diagnosis of severe combined immunodeficiency: A family perspective survey. Clin. Immunol. 2011, 138, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Kwan, A.; Abraham, R.S.; Currier, R.; Brower, A.; Andruszewski, K.; Abbott, J.K.; Baker, M.; Ballow, M.; Bartoshesky, L.E.; Bonilla, F.A.; et al. Newborn screening for severe combined immunodeficiency in 11 screening programs in the United States. JAMA 2014, 312, 729–738. [Google Scholar] [CrossRef] [PubMed]
- Patel, J.P.; Puck, J.M.; Srinivasan, R.; Brown, C.; Sunderam, U.; Kundu, K.; Brenner, S.E.; Gatti, R.A.; Church, J.A. Nijmegen breakage syndrome detected by newborn screening for T cell receptor excision circles (TRECs). J. Clin. Immunol. 2015, 35, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Bayer, D.K.; Martinez, C.A.; Sorte, H.S.; Forbes, L.R.; Demmler-Harrison, G.J.; Hanson, I.C.; Pearson, N.M.; Noroski, L.M.; Zaki, S.R.; Bellini, W.J.; et al. Vaccine-associated varicella and rubella infections in severe combined immunodeficiency with isolated CD4 lymphocytopenia and mutations in IL7R detected by tandem whole exome sequencing and chromosomal microarray. Clin. Exp. Immunol. 2014, 178, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Marciano, B.E.; Huang, C.Y.; Joshi, G.; Rezaei, N.; Carvalho, B.C.; Allwood, Z.; Ikinciogullari, A.; Reda, S.M.; Gennery, A.; Thon, V.; et al. BCG vaccination in patients with severe combined immunodeficiency: Complications, risks, and vaccination policies. J. Allergy Clin. Immunol. 2014, 133, 1134–1141. [Google Scholar] [CrossRef] [PubMed]
- Accetta Pedersen, D.J.; Verbsky, J.; Routes, J.M. Screening newborns for primary T-cell immunodeficiencies: Consensus and controversy. Expert Rev. Clin. Immunol. 2011, 7, 761–768. [Google Scholar] [CrossRef] [PubMed]
- Adams, S.P.; Rashid, S.; Premachandra, T.; Harvey, K.; Ifederu, A.; Wilson, M.C.; Gaspar, H.B. Screening of neonatal UK dried blood spots using a duplex TREC screening assay. J. Clin. Immunol. 2014, 34, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Audrain, M.; Thomas, C.; Mirallie, S.; Bourgeois, N.; Sebille, V.; Rabetrano, H.; Durand-Zaleski, I.; Boisson, R.; Persyn, M.; Pierres, C.; et al. Evaluation of the T-cell receptor excision circle assay performances for severe combined immunodeficiency neonatal screening on guthrie cards in a french single centre study. Clin. Immunol. 2014, 150, 137–139. [Google Scholar] [CrossRef] [PubMed]
- Olbrich, P.; de Felipe, B.; Delgado-Pecellin, C.; Rodero, R.; Rojas, P.; Aguayo, J.; Marquez, J.; Casanovas, J.; Sanchez, B.; Lucena, J.M.; et al. A first pilot study on the neonatal screening of primary immunodeficiencies in spain: TRECS and KRECS identify severe T- and B-cell lymphopenia. An. Pediatr. (Barc.) 2014, 81, 310–317. [Google Scholar] [CrossRef] [PubMed]
- McGhee, S.A.; Stiehm, E.R.; McCabe, E.R. Potential costs and benefits of newborn screening for severe combined immunodeficiency. J. Pediatr. 2005, 147, 603–608. [Google Scholar] [CrossRef] [PubMed]
- Modell, V.; Knaus, M.; Modell, F. An analysis and decision tool to measure cost benefit of newborn screening for severe combined immunodeficiency (SCID) and related T-cell lymphopenia. Immunol. Res. 2014, 60, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Kubiak, C.; Jyonouchi, S.; Kuo, C.; Garcia-Lloret, M.; Dorsey, M.J.; Sleasman, J.; Zbrozek, A.S.; Perez, E.E. Fiscal implications of newborn screening in the diagnosis of severe combined immunodeficiency. J. Allergy Clin. Immunol. Pract. 2014, 2, 697–702. [Google Scholar] [CrossRef] [PubMed]
- Chan, K. Cost-effectiveness analysis of newborn screening for severe combined immunodeficiency (SCID). Available online: http://www.folhamarela.com.br/PDF/scid.pdf.
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borte, S.; Reichenbach, J. Newborn Screening for Primary Immunodeficiencies: Focus on Severe Combined Immunodeficiency (SCID) and Other Severe T-Cell Lymphopenias. Int. J. Neonatal Screen. 2015, 1, 89-100. https://doi.org/10.3390/ijns1030089
Borte S, Reichenbach J. Newborn Screening for Primary Immunodeficiencies: Focus on Severe Combined Immunodeficiency (SCID) and Other Severe T-Cell Lymphopenias. International Journal of Neonatal Screening. 2015; 1(3):89-100. https://doi.org/10.3390/ijns1030089
Chicago/Turabian StyleBorte, Stephan, and Janine Reichenbach. 2015. "Newborn Screening for Primary Immunodeficiencies: Focus on Severe Combined Immunodeficiency (SCID) and Other Severe T-Cell Lymphopenias" International Journal of Neonatal Screening 1, no. 3: 89-100. https://doi.org/10.3390/ijns1030089
APA StyleBorte, S., & Reichenbach, J. (2015). Newborn Screening for Primary Immunodeficiencies: Focus on Severe Combined Immunodeficiency (SCID) and Other Severe T-Cell Lymphopenias. International Journal of Neonatal Screening, 1(3), 89-100. https://doi.org/10.3390/ijns1030089