
A Radiogenomics Ensemble to Predict EGFR and KRAS Mutations in NSCLC

 , ,
, ,  and
and
Abstract
1. Introduction
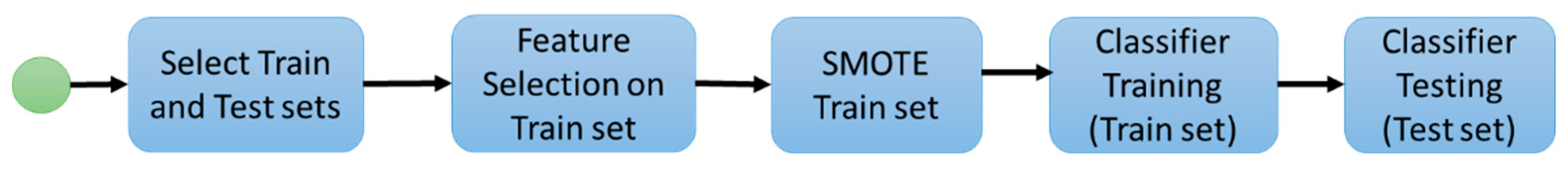
2. Materials and Methods
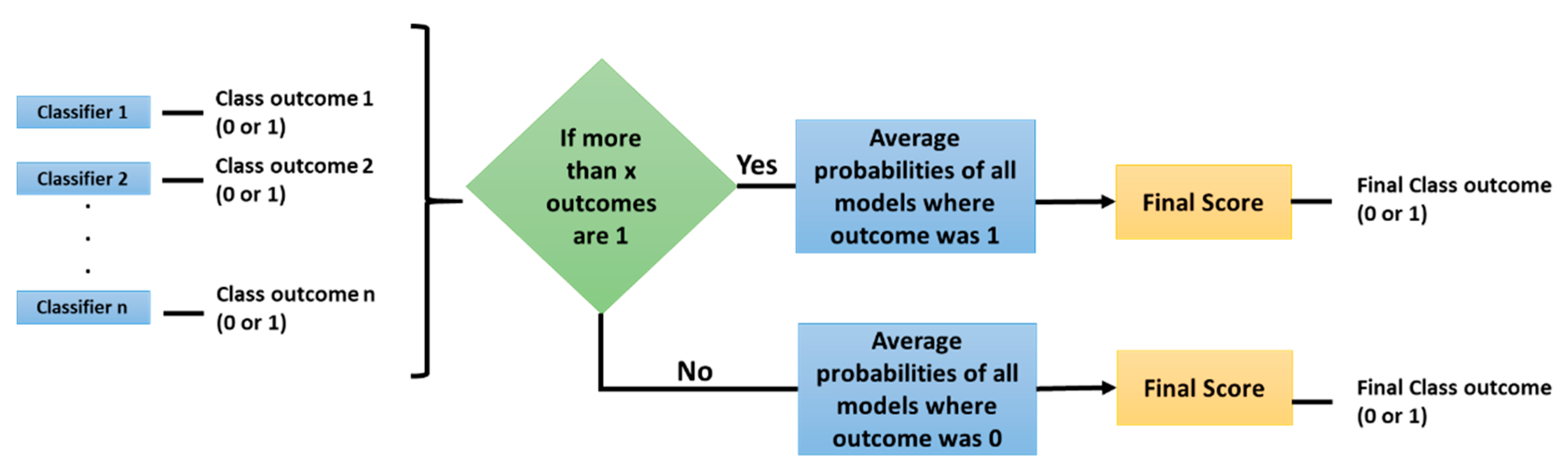
2.1. Experiment 1: Radiomic Features and Machine Learning Classifiers
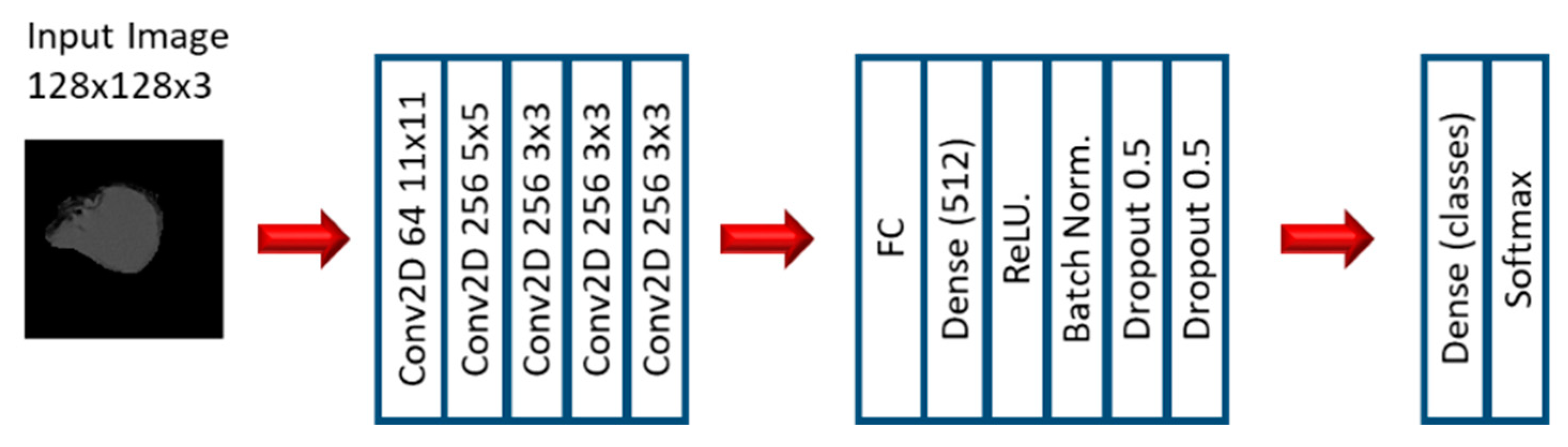
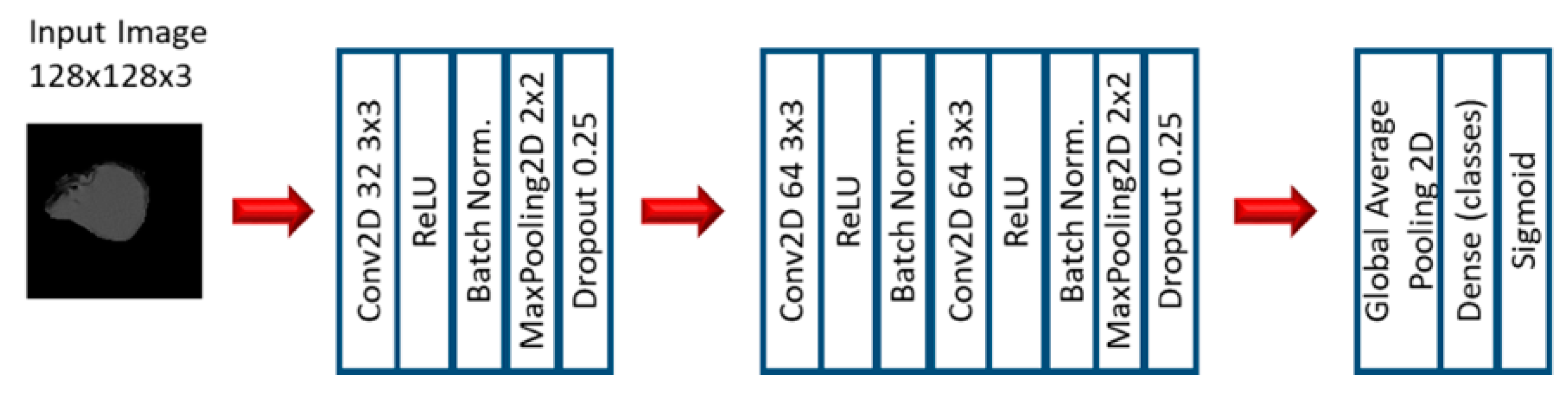
2.2. Experiment 2: Convolutional Neural Networks
3. Results
3.1. Machine Learning Models: EGFR Mutation
3.2. Machine Learning Models: KRAS Mutation
3.3. Convolutional Neural Networks: EGFR Mutation
3.4. Convolutional Neural Networks: KRAS Mutation
4. Discussion
4.1. EGFR Mutation
4.2. KRAS Mutation
4.3. Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- American Cancer Society. Global Cancer Facts & Figures, 4th ed.; American Cancer Society: Atlanta, GA, USA, 2018; pp. 1–76. [Google Scholar]
- Gillies, R.J.; Kinahan, P.E.; Hricak, H. Radiomics: Images are more than pictures, they are data. Radiology 2016, 278, 563–577. [Google Scholar] [CrossRef]
- Fenizia, F.; de Luca, A.; Pasquale, R.; Sacco, A.; Forgione, L.; Lambiase, M.; Iannaccone, A.; Chicchinelli, N.; Franco, R.; Rossi, A.; et al. EGFR mutations in lung cancer: From tissue testing to liquid biopsy. Future Oncol. 2015, 11, 1611–1623. [Google Scholar] [CrossRef]
- Rutman, A.M.; Kuo, M.D. Radiogenomics: Creating a link between molecular diagnostics and diagnostic imaging. Eur. J. Radiol. 2009, 70, 232–241. [Google Scholar] [CrossRef] [PubMed]
- Gevaert, O.; Echegaray, S.; Khuong, A.; Hoang, C.D.; Shrager, J.B.; Jensen, K.C.; Berry, G.J.; Guo, H.H.; Lau, C.; Plevritis, S.K.; et al. Predictive radiogenomics modeling of EGFR mutation status in lung cancer. Sci. Rep. 2017, 7, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Bethune, G.; Bethune, D.; Ridgway, N.; Xu, Z. Epidermal growth factor receptor (EGFR) in lung cancer: An overview and update. J. Thorac. Dis. 2010, 2, 48–51. [Google Scholar] [PubMed]
- Riely, G.J.; Ladanyi, M. KRAS mutations: An old oncogene becomes a new predictive biomarker. J. Mol. Diagn. 2008, 10, 493–495. [Google Scholar] [CrossRef] [PubMed]
- Gaughan, E.M.; Costa, D.B. Genotype-driven therapies for non-small cell lung cancer: Focus on EGFR, KRAS and ALK gene abnormalities. Ther. Adv. Med. Oncol. 2011, 3, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Goldman, J.W.; Mazieres, J.; Barlesi, F.; Dragnev, K.H.; Koczywas, M.; Göskel, T.; Cortot, A.B.; Girard, N.; Wesseler, C.; Bischoff, H.; et al. Randomized Phase III Study of Abemaciclib Versus Erlotinib in Patients with Stage IV Non-small Cell Lung Cancer With a Detectable KRAS Mutation Who Failed Prior Platinum-Based Therapy: JUNIPER. Front. Oncol. 2020, 10, 1–12. [Google Scholar] [CrossRef]
- Pinheiro, G.; Pereira, T.; Dias, C.; Freitas, C.; Hespanhol, V.; Costa, J.L.; Cunha, A.; Oliveira, H.P. Identifying relationships between imaging phenotypes and lung cancer-related mutation status: EGFR and KRAS. Sci. Rep. 2020, 10, 1–9. [Google Scholar]
- Wang, T.; Zhang, T.; Han, X.; Liu, X.; Zhou, N.; Liu, Y. Impact of the international association for the study of lung cancer/american thoracic society/European respiratory society classification of stage IA adenocarcinoma of the lung: Correlation between computed tomography images and EGFR and KRAS gene mutati. Exp. Ther. Med. 2015, 9, 2095–2103. [Google Scholar] [CrossRef]
- Mei, D.; Luo, Y.; Wang, Y.; Gong, J. CT texture analysis of lung adenocarcinoma: Can Radiomic features be surrogate biomarkers for EGFR mutation statuses. Cancer Imaging 2018, 18, 52. [Google Scholar] [CrossRef]
- Shiri, I.; Maleki, H.; Hajianfar, G.; Abdollahi, H.; Ashrafinia, S.; Oghli, M.G.; Hatt, M.; Oveisi, M.; Rahmim, A. PET/CT Radiomic Sequencer for Prediction of EGFR and KRAS Mutation Status in NSCLC Patients. In 2018 IEEE Nuclear Science Symposium and Medical Imaging Conference Proceedings (NSS/MIC); IEEE: Piscatway, NJ, USA, 2018; pp. 1–4. [Google Scholar]
- Koyasu, S.; Nishio, M.; Isoda, H.; Nakamoto, Y.; Togashi, K. Usefulness of gradient tree boosting for predicting histological subtype and EGFR mutation status of non-small cell lung cancer on 18F FDG-PET/CT. Ann. Nucl. Med. 2020, 34, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Kim, J.; Balagurunathan, Y.; Li, Q.; Garcia, A.L.; Stringfield, O.; Ye, Z.; Gillies, R.J. Radiomic Features Are Associated With EGFR Mutation Status in Lung Adenocarcinomas. Clin. Lung Cancer 2016, 17, 441–448.e6. [Google Scholar] [CrossRef]
- Feng, Y.; Yang, F.; Zhou, X.; Guo, Y.; Tang, F.; Ren, F.; Guo, J.; Ji, S. A Deep Learning Approach for Targeted Contrast-Enhanced Ultrasound Based Prostate Cancer Detection. IEEE/ACM Trans. Comput. Biol. Bioinform. 2019, 16, 1794–1801. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Shi, J.; Ye, Z.; Dong, D.; Yu, D.; Zhou, M.; Liu, Y.; Gevaert, O.; Wang, K.; Zhu, Y.; et al. Predicting EGFR mutation status in lung adenocarcinoma on computed tomography image using deep learning. Eur. Respir. J. 2019, 53, 1800986. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Zhang, J.; Cao, Y.; Zhao, Z.; Li, S.; Deng, L.; Zhou, J. Nomogram based on preoperative CT imaging predicts the EGFR mutation status in lung adenocarcinoma. Transl. Oncol. 2021, 14, 100954. [Google Scholar] [CrossRef] [PubMed]
- Paul, R.; Hall, L.; Goldgof, D.; Schabath, M.; Gillies, R. Predicting Nodule Malignancy using a CNN Ensemble Approach. Proc. Int. Jt. Conf. Neural Netw. Int. Jt. Conf. Neural Netw. 2018, 2018. [Google Scholar] [CrossRef]
- Bakr, S.; Gevaert, O.; Echegaray, S.; Ayers, K.; Zhou, M.; Shafiq, M.; Zheng, H.; Zhang, W.; Leung, A.; Kadoch, M.; et al. Data for NSCLC Radiogenomics Collection. The Cancer Imaging Archive. 2017. Available online: https://wiki.cancerimagingarchive.net/display/Public/NSCLC+Radiogenomics (accessed on 20 August 2018).
- Bakr, S.; Gevaert, O.; Echegaray, S.; Ayers, K.; Zhou, M.; Shafiq, M.; Zheng, H.; Benson, J.A.; Zhang, W.; Leung, A.N.C.; et al. A radiogenomic dataset of non-small cell lung cancer. Sci. Data 2018, 5, 180202. [Google Scholar] [CrossRef] [PubMed]
- Balagurunathan, Y.; Gu, Y.; Wang, H.; Kumar, V.; Grove, O.; Hawkins, S.; Kim, J.; Goldgof, D.B.; Hall, L.O.; Gatenby, R.A.; et al. Reproducibility and Prognosis of Quantitative Features Extracted from CT Images. Transl. Oncol. 2014, 7, 72–87. [Google Scholar] [CrossRef]
- Baatz, M.; Zimmermann, J.; Blackmore, C.G. Blackmore, Automated analysis and detailed quantification of biomedical images using Definiens Cognition Network Technology. Comb. Chem. High Throughput Screen. 2009, 12, 908–916. [Google Scholar] [CrossRef]
- Chawla, N.V.; Bowyer, K.W.; Hall, L.O. SMOTE: Synthetic Minority Over-sampling Technique. J. Artif. Intell. Res. 2002, 16, 321–357. [Google Scholar] [CrossRef]
- Mann, H.B.; Whitney, D.R. On a Test of Whether one of Two Random Variables is Stochastically Larger than the Other. Ann. Math. Stat. 1947, 18, 50–60. [Google Scholar] [CrossRef]
- Kira, K.; Rendell, L.A. The Feature Selection Problem: Traditional Methods and a New Algorithm. In Proceedings of the Tenth National Conference on Artificial Intelligence, San Jose, CA, USA, 12–16 July 1992; pp. 129–134. [Google Scholar]
- Ho, T.K. Random decision forests. In Proceedings of the 3rd International Conference on Document Analysis and Recognition, Montreal, QC, Canada, 14–16 August 1995; Volume 1, pp. 278–282. [Google Scholar]
- Cortes, C.; Vapnik, V. Support-vector networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef]
- Friedman, J.H. Stochastic gradient boosting. Comput. Stat. Data Anal. 2002, 38, 367–378. [Google Scholar] [CrossRef]
- Bhadeshia, B. Neural Networks in Materials Science. ISIJ Int. 1999, 39, 966–979. [Google Scholar] [CrossRef]
- Fawcett, T. An introduction to ROC analysis. Pattern Recognit. Lett. 2006, 27, 861–874. [Google Scholar] [CrossRef]
- Romanski, P.; Kotthoff, L. Package ‘FSelector’. 2018. Available online: https://cran.r-project.org/web/packages/FSelector/FSelector.pdf (accessed on 20 September 2018).
- Torgo, L. Package ‘DMwR’. 2015. Available online: https://cran.r-project.org/web/packages/DMwR/DMwR.pdf (accessed on 20 September 2018).
- Kuhn, M.; Wing, J.; Weston, S.; Williams, A.; Keefer, C.; Engelhardt, A.; Cooper, T.; Mayer, Z.; Kenkel, B.; R Core Team; et al. Package ‘Caret’. 2018. Available online: https://cran.r-project.org/web/packages/caret/caret.pdf (accessed on 20 September 2018).
- Krizhevsky, A.; Sutskever, I.; Hinton, G.E. ImageNet classification with deep convolutional neural networks. Adv. Neural Inf. Process. Syst. 2012, 2, 1097–1105. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | Values | Number of Cases (%) |
|---|---|---|
| EGFR Mutation Status | Mutant | 12 (14%) |
| Wildtype | 71 (86%) | |
| Total | 83 (100%) | |
| KRAS Mutation Status | Mutant | 20 (24%) |
| Wildtype | 63 (76%) | |
| Total | 83 (100%) |
| Variable. | Overall Dataset | EGFR Mutant | EGFR Wildtype | KRAS Mutant | KRAS Wildtype |
|---|---|---|---|---|---|
| Median Age (Range) | 69 (46–85) | 72 (55–85) | 69 (46–84) | 68 (50–81) | 69 (46–85) |
| Gender | |||||
| Male | 65 (78%) | 7 (8%) | 58 (70%) | 16 (19%) | 49 (59%) |
| Female | 18 (22%) | 5 (6%) | 13 (16%) | 4 (5%) | 14 (17%) |
| Smoking Status | |||||
| Current | 18 (22%) | 1 (1%) | 17 (21%) | 6 (6%) | 12 (16%) |
| Former | 56 (67%) | 8 (9%) | 48 (58%) | 14 (17%) | 42 (50%) |
| Non-smoker | 9 (11%) | 3 (4%) | 6 (7%) | 0 (0%) | 9 (11%) |
| Pathological T Stage | |||||
| Tis | 3 (4%) | 1 (1%) | 2 (3%) | 0 (0%) | 3 (4%) |
| T1a | 17 (21%) | 1 (1%) | 16 (20%) | 4 (5%) | 13 (16%) |
| T1b | 19 (23%) | 5 (6%) | 14 (17%) | 3 (3%) | 16 (20%) |
| T2a | 26 (31%) | 3 (3%) | 23 (28%) | 7 (8%) | 19 (23%) |
| T2b | 6 (7%) | 1 (1%) | 5 (6%) | 1 (1%) | 5 (6%) |
| T3 | 8 (9%) | 1 (1%) | 7 (8%) | 5 (6%) | 3 (3%) |
| T4 | 4 (5%) | 0 | 4 (5%) | 0 (0%) | 4 (5%) |
| Pathological N Stage | |||||
| N0 | 65 (78%) | 10 (12%) | 55 (66%) | 16 (20%) | 49 (58%) |
| N1 | 8 (10%) | 1 (1%) | 7 (9%) | 1 (1%) | 7 (9%) |
| N2 | 10 (12%) | 1 (1%) | 9 (11%) | 3 (3%) | 7 (9%) |
| Pathological M Stage | |||||
| M0 | 80 (96%) | 12 (14%) | 68 (82%) | 19 (23%) | 61 (73%) |
| M1b | 3 (4%) | 0 0% | 3 (4%) | 1 (1%) | 2 (3%) |
| Histology | |||||
| Adenocarcinoma | 66 (80%) | 12(14%) | 54 (66%) | 19 (23%) | 47 (57%) |
| Squamous cell carcinoma NSCLC NOS | 14 (17%) 3 (3%) | 0 (0%) 0 (0%) | 14 (17%) 3 (3%) | 0 (0%) | 14 (17%) |
| 1 (1%) | 2 (2%) |
| Feature Selection | Classifier | SMOTE | Accuracy | Sensitivity | Specificity | AUC |
|---|---|---|---|---|---|---|
| MW (5 features) | nnet | No | 0.83 | 0.00 | 0.98 | 0.43 |
| ReliefF (15 features) | SVM | Yes | 0.76 | 0.66 | 0.78 | 0.68 |
| ReliefF (10 features) | RF | Yes | 0.76 | 0.41 | 0.82 | 0.67 |
| ReliefF (15 features) | nnet | Yes | 0.76 | 0.58 | 0.79 | 0.67 |
| ReliefF (5 features) | RF | Yes | 0.77 | 0.50 | 0.82 | 0.64 |
| ReliefF (20 features) | RF | Yes | 0.73 | 0.16 | 0.83 | 0.63 |
| ReliefF (20 features) | SVM | Yes | 0.68 | 0.66 | 0.69 | 0.63 |
| ReliefF (5 features) | nnet | Yes | 0.71 | 0.50 | 0.75 | 0.60 |
| ReliefF (5 features) | SVM | Yes | 0.79 | 0.25 | 0.89 | 0.59 |
| ReliefF (15 features) | RF | Yes | 0.72 | 0.25 | 0.80 | 0.57 |
| MW (5 features) | gbm | Yes | 0.68 | 0.16 | 0.78 | 0.53 |
| Ensemble Combination | Classifiers | Accuracy | Sensitivity | Specificity | AUC |
|---|---|---|---|---|---|
| Ensemble SCAV thresh 3 (10 models) | gbm, SVM, nnet | 0.59 | 0.75 | 0.57 | 0.70 |
| Ensemble SCAV thresh 6 (10 models) | gbm, SVM, nnet | 0.80 | 0.33 | 0.89 | 0.68 |
| Ensemble Average (10 models) | gbm, SVM, nnet | 0.78 | 0.16 | 0.89 | 0.68 |
| Ensemble Average (5 models) | All | 0.78 | 0.16 | 0.89 | 0.67 |
| Ensemble Average (5 models) | RF, SVM, nnet | 0.79 | 0.33 | 0.87 | 0.66 |
| Ensemble Maximum (10 models) | gbm, SVM, nnet | 0.75 | 0.41 | 0.82 | 0.59 |
| Feature Selection | Classifier | SMOTE | Accuracy | Sensitivity | Specificity | AUC |
|---|---|---|---|---|---|---|
| MW (10 features) | nnet | No | 0.72 | 0.10 | 0.93 | 0.44 |
| Relief (10 features) | nnet | No | 0.75 | 0.00 | 1.00 | 0.44 |
| ReliefF (5 features) | SVM | Yes | 0.70 | 0.35 | 0.81 | 0.65 |
| MW (15 features) | SVM | Yes | 0.64 | 0.40 | 0.72 | 0.64 |
| ReliefF (5 features) | gbm | Yes | 0.64 | 0.60 | 0.65 | 0.63 |
| ReliefF (20 features) | gbm | Yes | 0.63 | 0.50 | 0.67 | 0.63 |
| MW (10 features) | SVM | Yes | 0.64 | 0.45 | 0.70 | 0.63 |
| MW (20 features) | SVM | Yes | 0.71 | 0.40 | 0.81 | 0.63 |
| ReliefF (15 features) | SVM | Yes | 0.67 | 0.45 | 0.75 | 0.62 |
| MW (5 features) | SVM | Yes | 0.71 | 0.35 | 0.83 | 0.62 |
| MW (15 features) | gbm | Yes | 0.67 | 0.40 | 0.77 | 0.62 |
| ReliefF (15 features) | RF | Yes | 0.62 | 0.40 | 0.70 | 0.60 |
| Ensemble Combination | Classifiers | Accuracy | Sensitivity | Specificity | AUC |
|---|---|---|---|---|---|
| Ensemble SCAV thresh 8 (10 models) | SVM, nnet | 0.72 | 0.20 | 0.89 | 0.71 |
| Ensemble SCAV thresh 6 (10 models) | SVM, nnet | 0.73 | 0.30 | 0.87 | 0.69 |
| Ensemble Average (5 models) | SVM | 0.70 | 0.35 | 0.81 | 0.67 |
| Ensemble Maximum (5 models) | SVM | 0.70 | 0.40 | 0.80 | 0.66 |
| Ensemble Average (10 models) | SVM, nnet | 0.66 | 0.35 | 0.76 | 0.65 |
| Model | Optimizer | Learning Rate | Epochs | Accuracy | Sensitivity | Specificity | AUC |
|---|---|---|---|---|---|---|---|
| Arch. 4 | SGD | 0.0005 | 30 | 0.800 | 0.667 | 0.846 | 0.846 |
| Arch. 6 | SGD | 0.0005 | 30 | 0.771 | 0.222 | 0.961 | 0.752 |
| Arch. 1 | SGD | 0.01 | 8 | 0.400 | 1.000 | 0.192 | 0.688 |
| Arch. 6 | SGD | 0.01 | 10 | 0.657 | 0.666 | 0.654 | 0.675 |
| Arch. 3 | SGD | 0.01 | 7 | 0.543 | 0.778 | 0.461 | 0.671 |
| Arch. 4 | SGD | 0.01 | 10 | 0.543 | 0.778 | 0.461 | 0.628 |
| Arch. 1 | SGD | 0.01 | 30 | 0.514 | 0.778 | 0.423 | 0.623 |
| Arch. 2 | SGD | 0.01 | 30 | 0.542 | 0.667 | 0.538 | 0.571 |
| Arch. 4 | SGD | 0.01 | 20 | 0.600 | 0.444 | 0.654 | 0.559 |
| Model | Accuracy | Sensitivity | Specificity | AUC |
|---|---|---|---|---|
| Ensemble (3 models) SCAV thresh 3 | 0.828 | 0.667 | 0.885 | 0.820 |
| Ensemble (5 models) SCAV thresh 5 | 0.857 | 0.667 | 0.923 | 0.778 |
| Ensemble (3 models) Average | 0.486 | 0.778 | 0.385 | 0.743 |
| Ensemble (5 models) Average | 0.628 | 0.778 | 0.577 | 0.641 |
| Ensemble (3 models) Maximum | 0.371 | 0.778 | 0.231 | 0.624 |
| Model | Optimizer | Learning Rate | Epochs | Accuracy | Sensitivity | Specificity | AUC |
|---|---|---|---|---|---|---|---|
| Arch. 1 | SGD | 0.01 | 60 | 0.667 | 0.000 | 1.000 | 0.739 |
| Arch. 6 | Adam | 0.005 | 10 | 0.333 | 1.000 | 0.000 | 0.607 |
| Arch. 6 | Adam | 0.001 | 10 | 0.667 | 0.000 | 1.000 | 0.593 |
| Arch. 1 | Adam | 0.005 | 15 | 0.722 | 0.250 | 0.958 | 0.566 |
| Arch. 1 | SGD | 0.01 | 90 | 0.667 | 0.000 | 1.000 | 0.555 |
| Arch. 1 | SGD | 0.01 | 10 | 0.555 | 0.667 | 0.500 | 0.531 |
| Model | Accuracy | Sensitivity | Specificity | AUC |
|---|---|---|---|---|
| Ensemble (3 models) Average | 0.722 | 0.250 | 0.958 | 0.778 |
| Ensemble (3 models) SCAV thresh 2 | 0.722 | 0.250 | 0.958 | 0.722 |
| Ensemble (4 models) SCAV thresh 3 | 0.722 | 0.250 | 0.958 | 0.642 |
| Ensemble (7 models) SCAV thresh 4 | 0.694 | 0.416 | 0.833 | 0.618 |
| Ensemble (7 models) SCAV thresh 5 | 0.694 | 0.083 | 1.000 | 0.604 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moreno, S.; Bonfante, M.; Zurek, E.; Cherezov, D.; Goldgof, D.; Hall, L.; Schabath, M. A Radiogenomics Ensemble to Predict EGFR and KRAS Mutations in NSCLC. Tomography 2021, 7, 154-168. https://doi.org/10.3390/tomography7020014
Moreno S, Bonfante M, Zurek E, Cherezov D, Goldgof D, Hall L, Schabath M. A Radiogenomics Ensemble to Predict EGFR and KRAS Mutations in NSCLC. Tomography. 2021; 7(2):154-168. https://doi.org/10.3390/tomography7020014
Chicago/Turabian StyleMoreno, Silvia, Mario Bonfante, Eduardo Zurek, Dmitry Cherezov, Dmitry Goldgof, Lawrence Hall, and Matthew Schabath. 2021. "A Radiogenomics Ensemble to Predict EGFR and KRAS Mutations in NSCLC" Tomography 7, no. 2: 154-168. https://doi.org/10.3390/tomography7020014
APA StyleMoreno, S., Bonfante, M., Zurek, E., Cherezov, D., Goldgof, D., Hall, L., & Schabath, M. (2021). A Radiogenomics Ensemble to Predict EGFR and KRAS Mutations in NSCLC. Tomography, 7(2), 154-168. https://doi.org/10.3390/tomography7020014