
Developing New Peptides and Peptide–Drug Conjugates for Targeting the FGFR2 Receptor-Expressing Tumor Cells and 3D Spheroids


Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
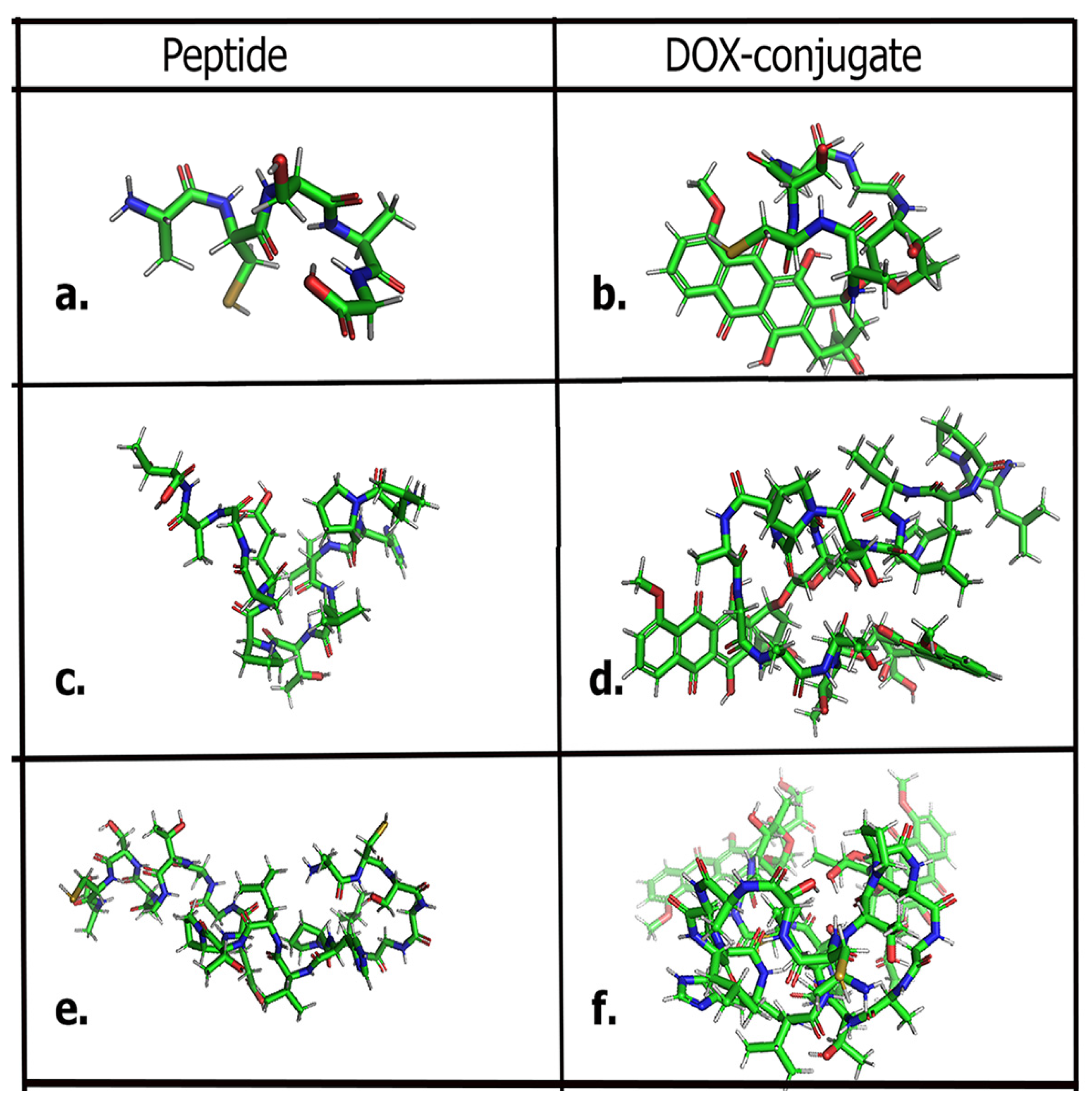
2.2.1. Computational Methods
Peptide Design
Anti-CP Studies
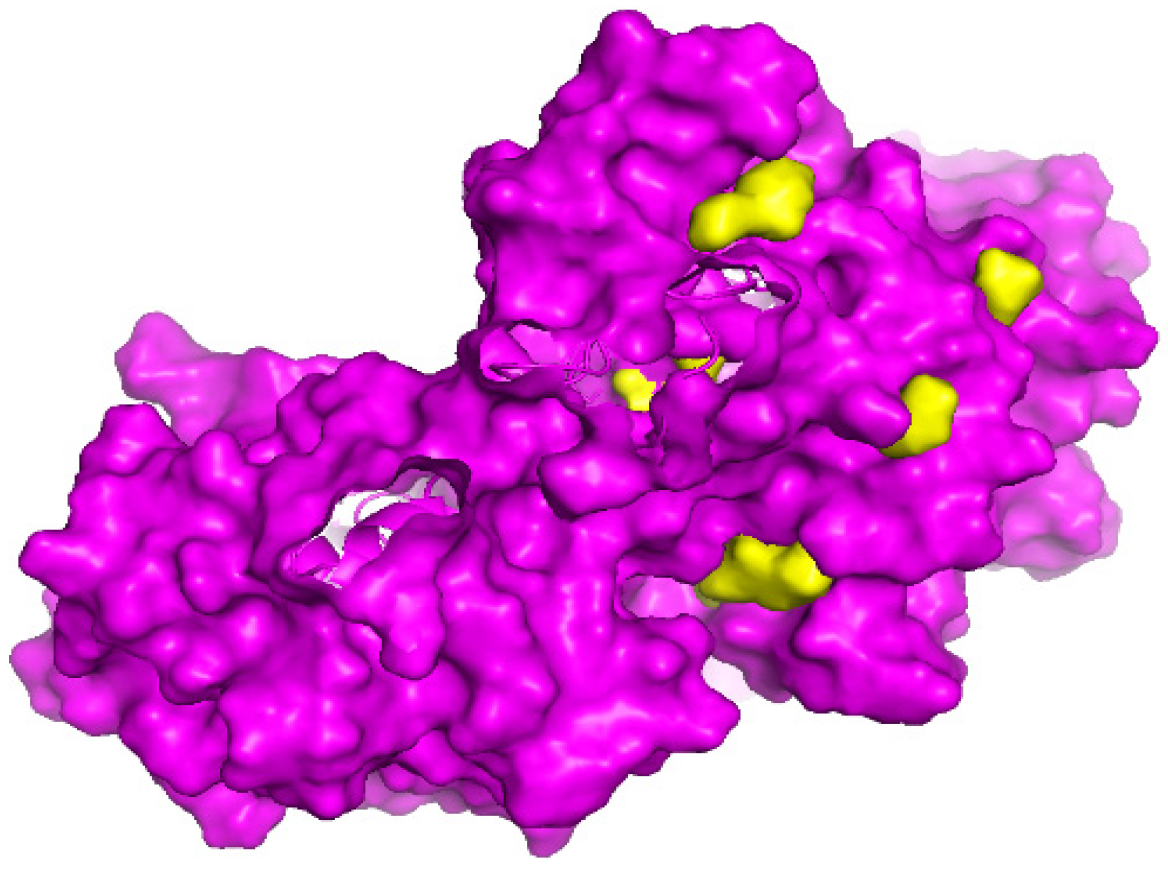
Binding Pocket Analysis
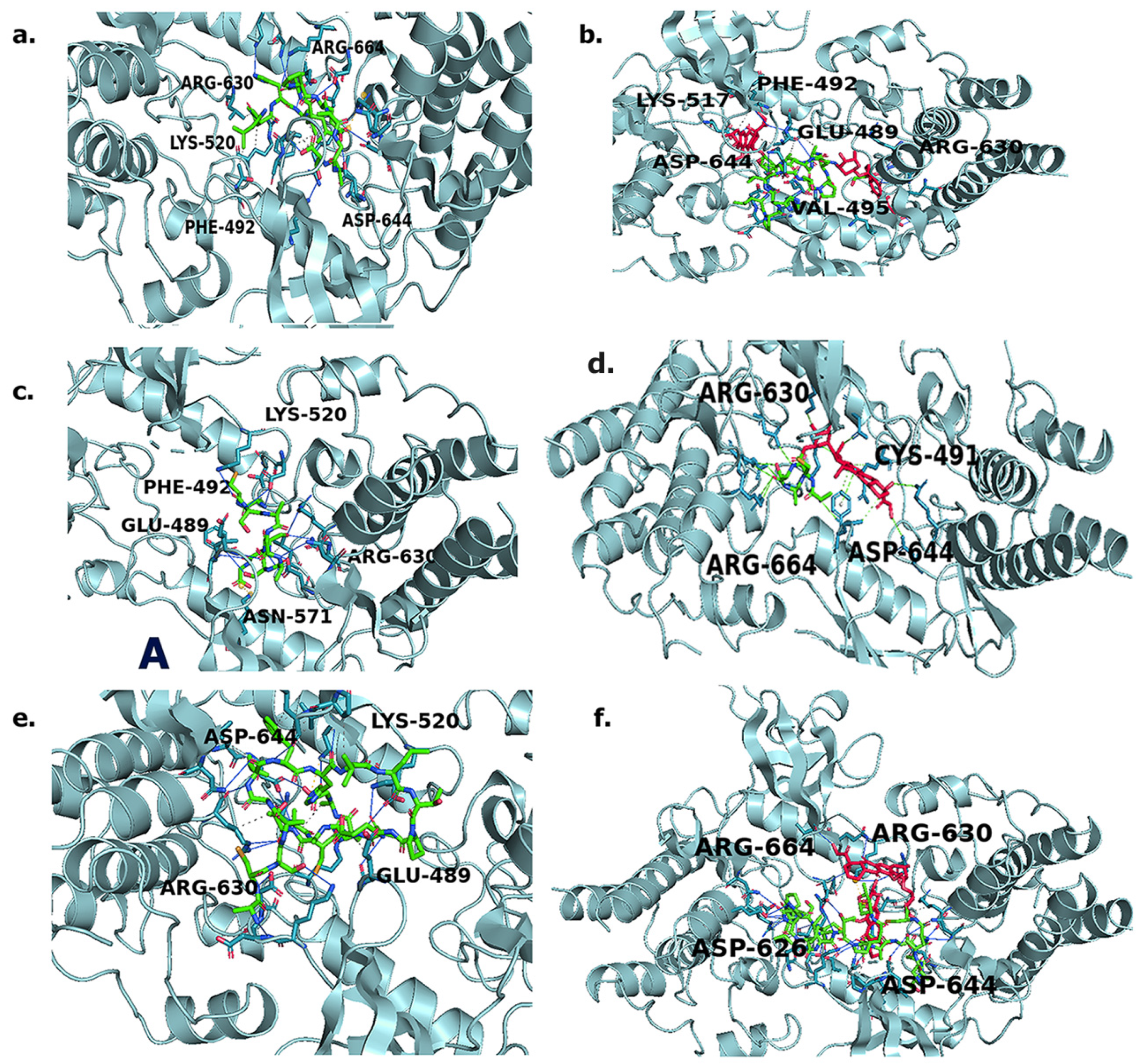
Molecular Docking Studies
Protein–Ligand Interaction Profiler (PLIP) Analysis
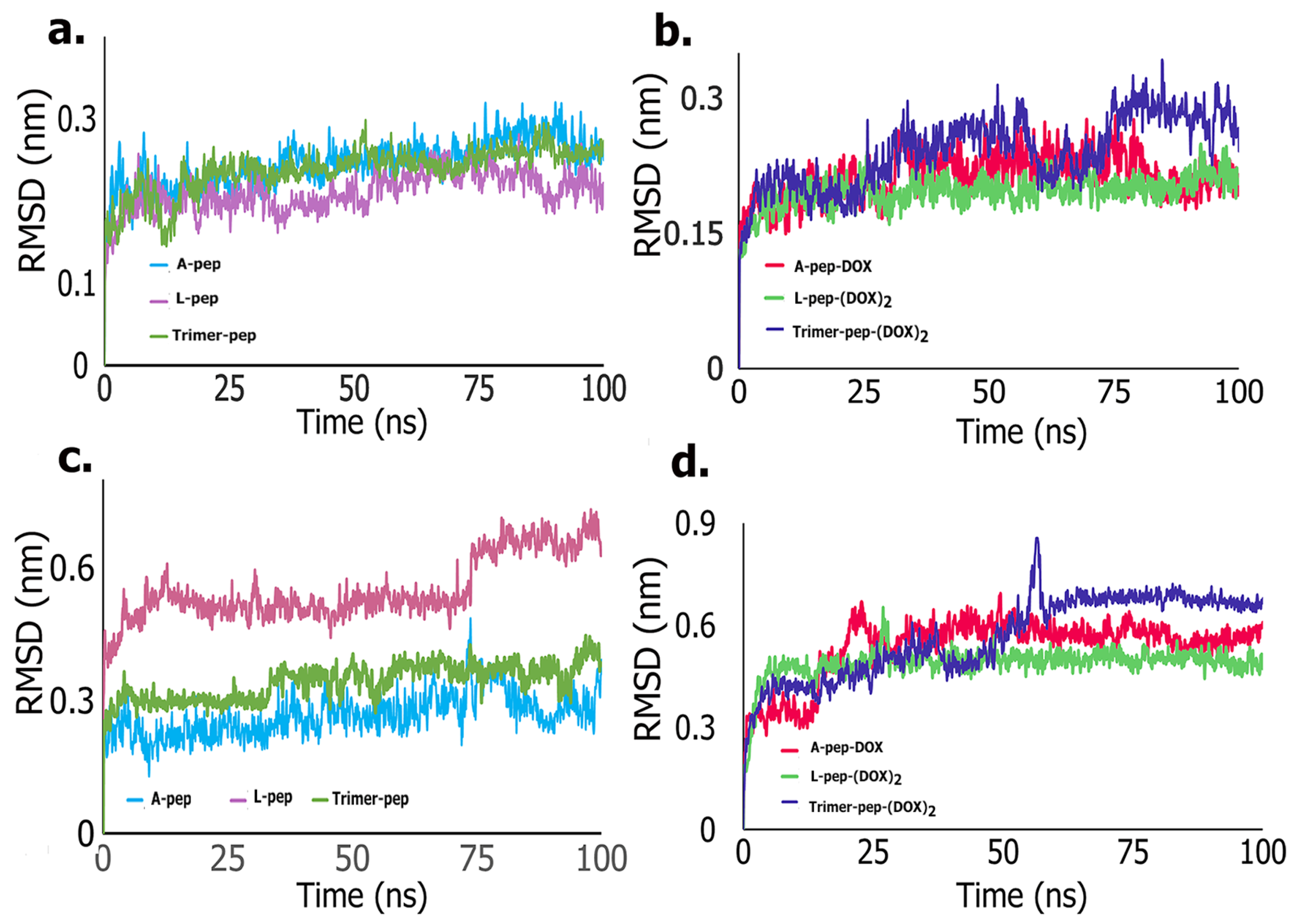
Molecular Dynamics
MM-GBSA Analysis
2.2.2. Laboratory Analysis
Surface Plasmon Resonance
Peptide Conjugation with Doxorubicin (DOX)
2D Cell Cultures
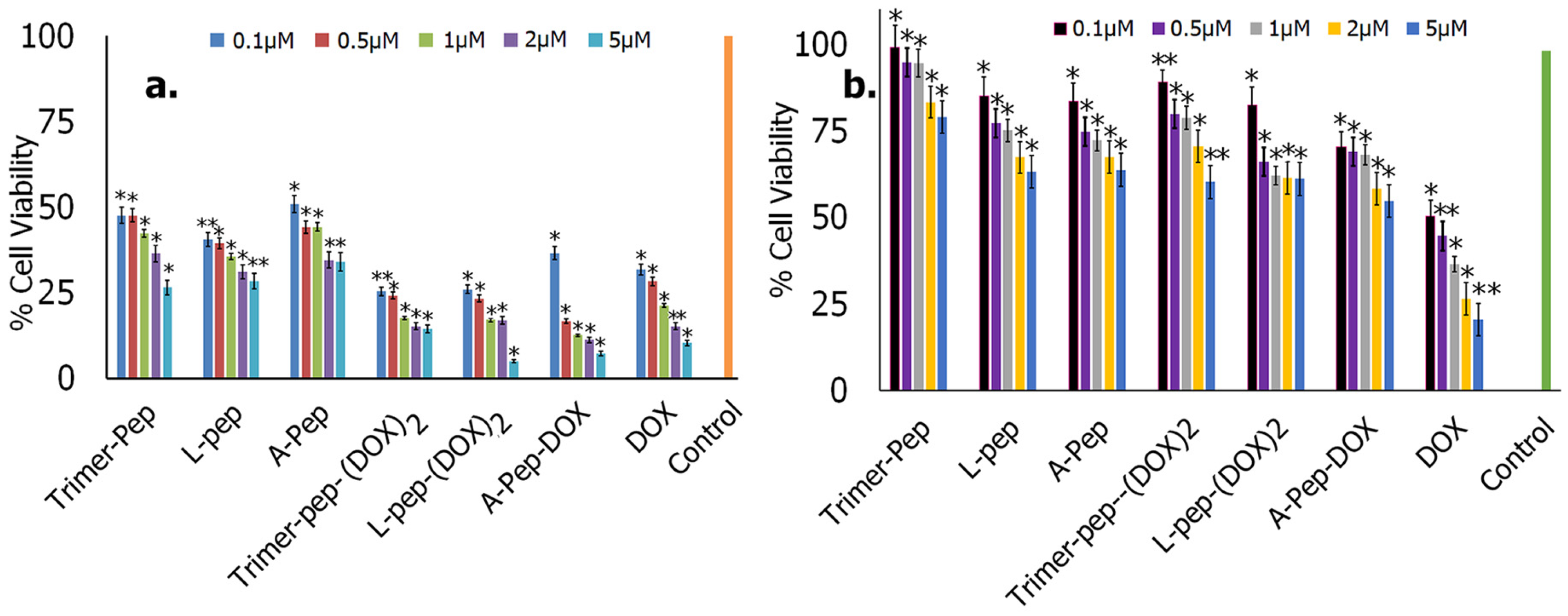
Cytotoxicity Studies
ELISA
Immunofluorescence Studies
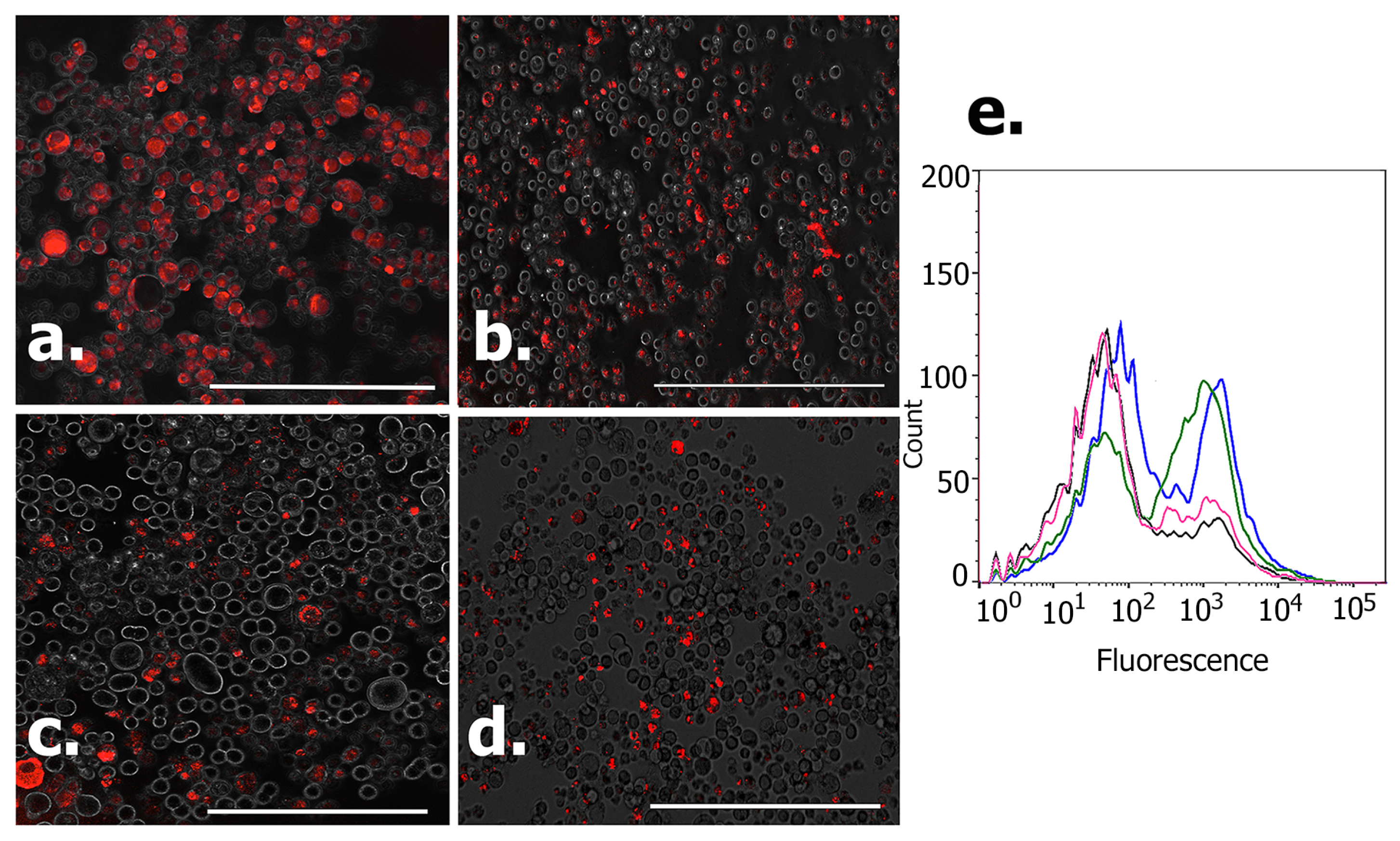
Flow Cytometry
Spheroid Growth
PicoGreen Double Stranded (ds) DNA Assay
Impact of Peptides and Peptide–Drug Conjugates on the Spheroids
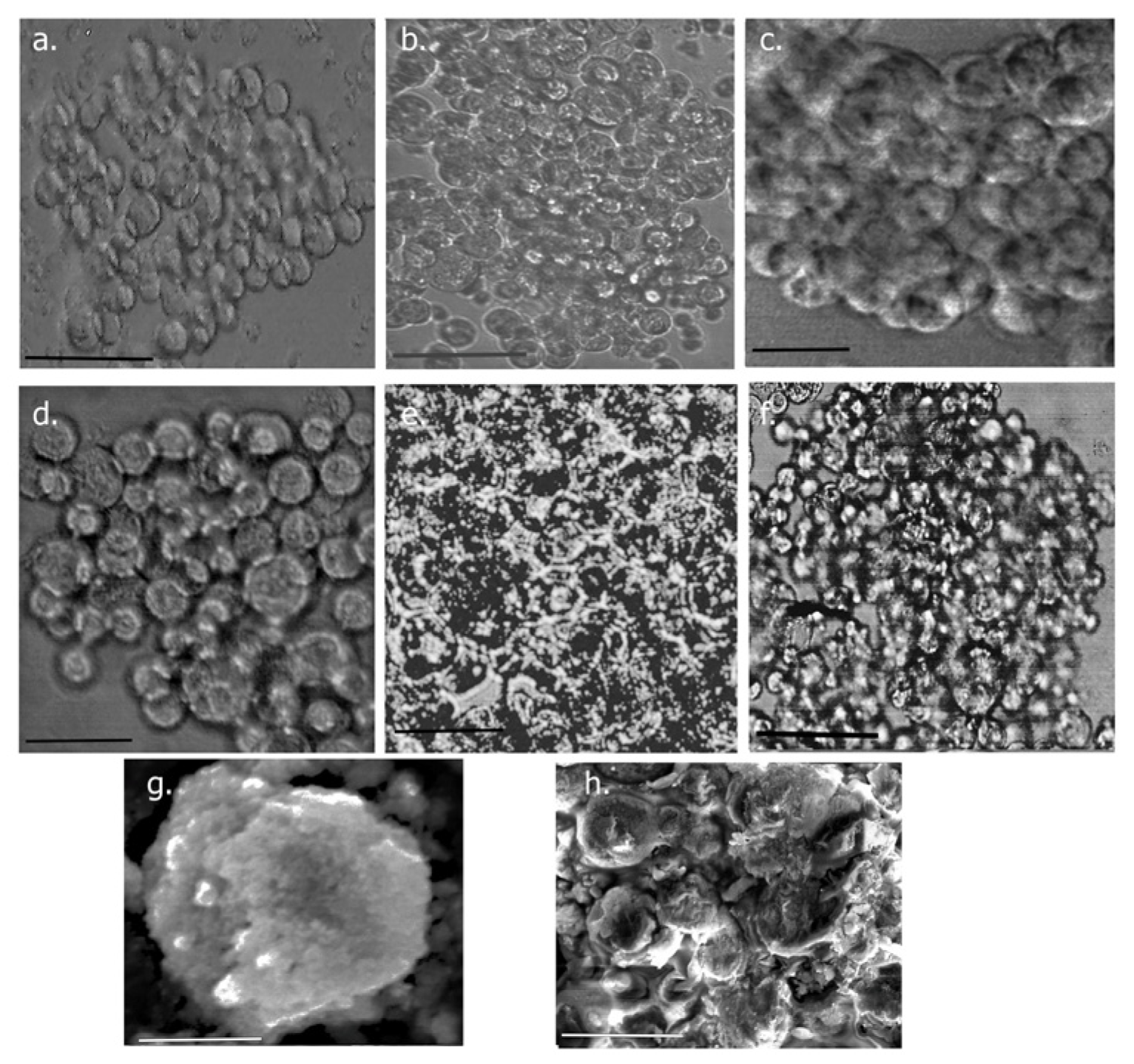
SEM Imaging of Spheroids
2.3. Characterization
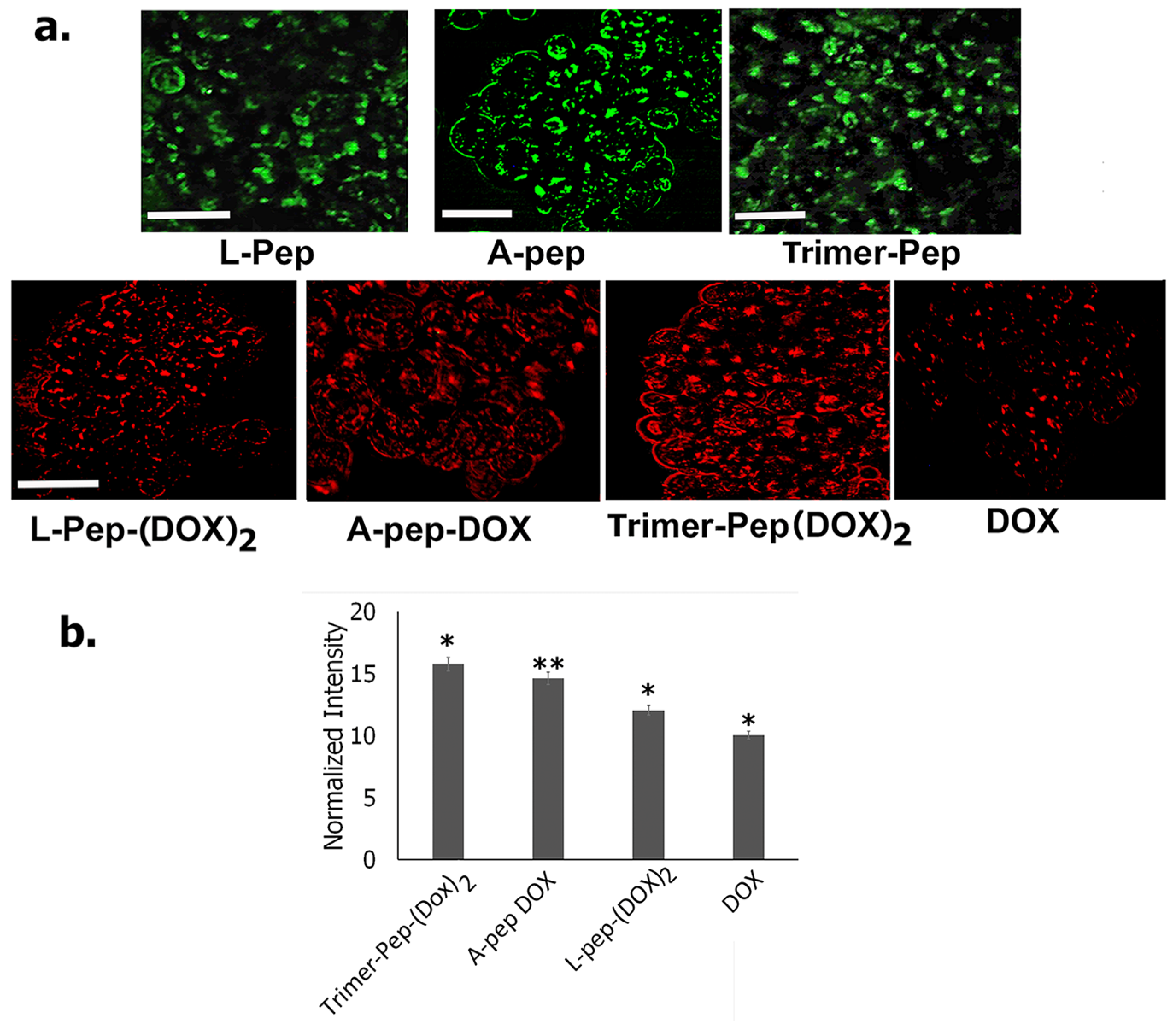
2.3.1. Fluorescence Imaging
2.3.2. SEM Imaging
2.3.3. Fourier Transform Infrared (FTIR) Spectroscopy
2.3.4. Differential Scanning Calorimetry (DSC)
3. Results and Discussion
3.1. Anti-CP Studies
3.2. Binding Pocket Analysis
3.3. Molecular Docking Studies
3.4. Protein–Ligand Interaction Profiler (PLIP) Analysis
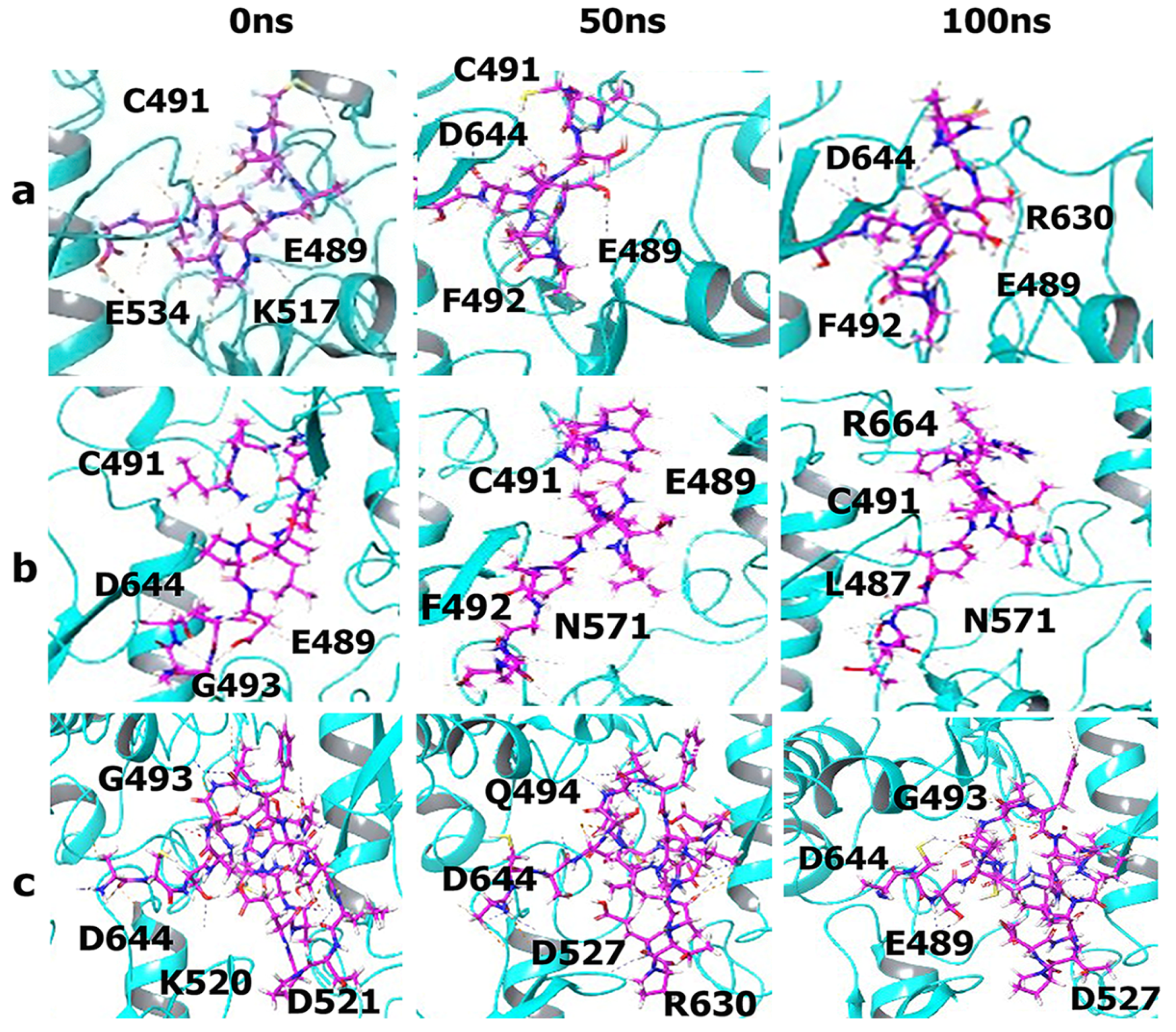
3.5. Molecular Dynamics Simulations
MM-GBSA Analysis
3.6. Laboratory Studies
3.6.1. SPR Analysis
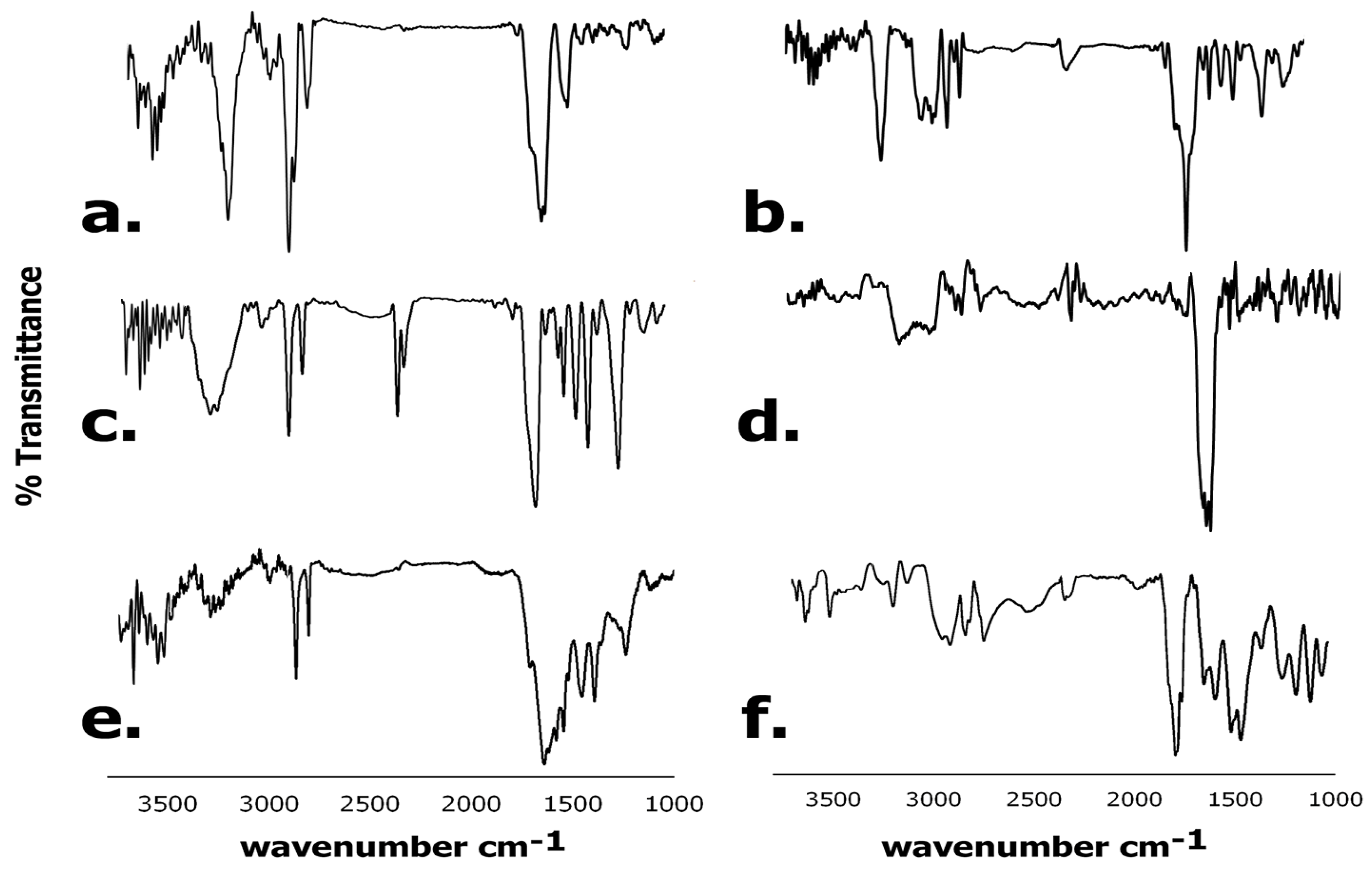
3.6.2. FTIR Spectroscopy
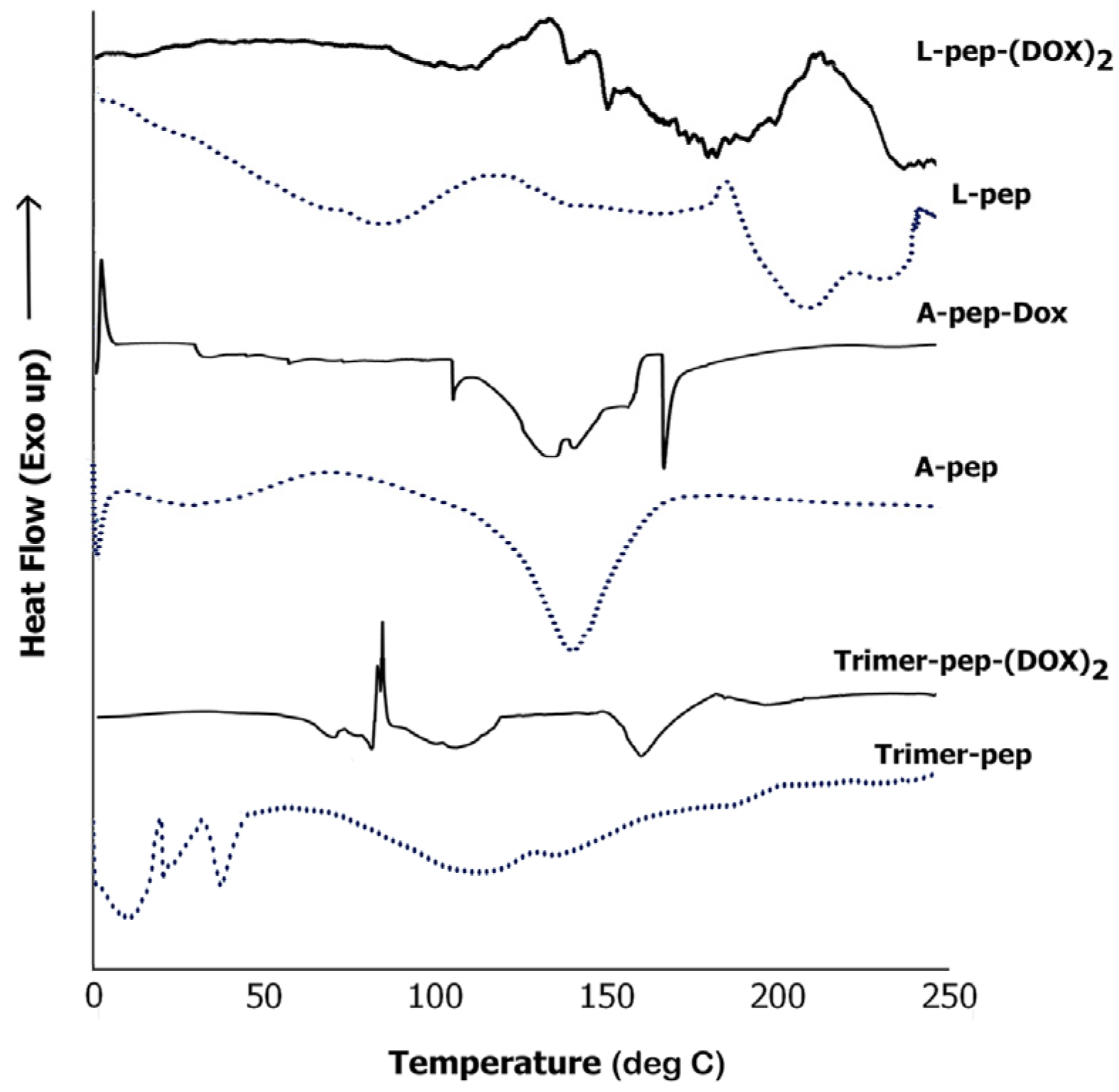
3.6.3. DSC Analysis
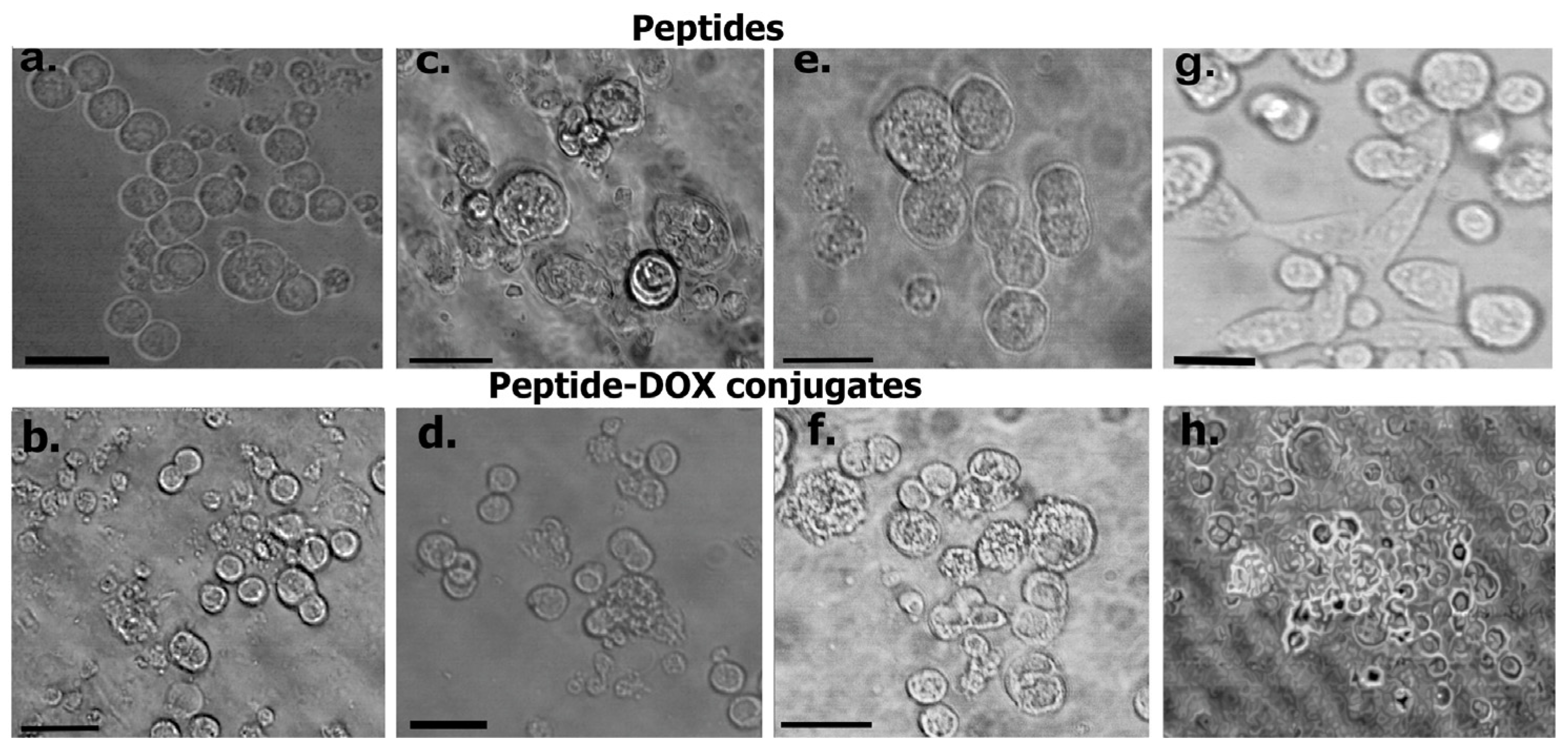
3.6.4. Cell Studies
3.6.5. Growth of Spheroids
Spheroid Imaging
3.7. Permeation of Peptides and DOX Conjugates into 3D Spheroids
3.8. Immunofluorescence Studies
4. Conclusions and Future Directions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Anand, U.; Dey, A.; Singh Chandel, A.K.; Sanyal, R.; Mishra, A.; Kumar Pandey, D.; De Falco, V.; Upadhyay, A.; Kandimalla, R.; Chaudhary, A.; et al. Cancer chemotherapy and beyond: Current status, drug candidates, associated risks and progress in targeted therapeutics. Genes Dis. 2023, 10, 1367–1401. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Gao, Y.; Meng, F.; Luo, L. Recent progress of nanotechnology-based theranostic systems in cancer treatments. Cancer Biol. Med. 2021, 18, 336–351. [Google Scholar] [CrossRef]
- Sharon, J.; Liebman, M.A.; Williams, B.R. Recombinant polyclonal antibodies for cancer therapy. J. Cell. Biochem. 2005, 96, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Melero, I.; Hervas-Stubbs, S.; Glennie, M.; Pardoll, D.M.; Chen, L. Immunostimulatory monoclonal antibodies for cancer therapy. Nat. Rev. Cancer 2007, 7, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Venkatesan, S.; Chanda, K.; Balamurali, M.M. Recent advancements of aptamers in cancer therapy. ACS Omega 2023, 8, 32231–32243. [Google Scholar] [CrossRef] [PubMed]
- Ghaffari, M.; Dehghan, G.; Abedi-Gaballu, F.; Kashanian, S.; Baradaran, B.; Nazhad Dolatabadi, J.; Losic, L. Surface functionalized dendrimers as controlled-release delivery nanosystems for tumor targeting. Eur. J. Pharm. Sci. 2018, 122, 311–330. [Google Scholar] [CrossRef]
- Araste, F.; Abnous, K.; Hashemi, M.; Taghdisi, S.M.; Ramezani, M.; Alibolandi, M. Peptide-based targeted therapeutics: Focus on cancer treatment. J. Control. Release 2018, 292, 141–162. [Google Scholar] [CrossRef]
- Tornesello, A.L.; Borrelli, A.; Buonaguro, L.; Buonaguro, F.M.; Tornesello, M.L. Antimicrobial Peptides as Anticancer Agents: Functional Properties and Biological Activities. Molecules 2020, 25, 2850. [Google Scholar] [CrossRef] [PubMed]
- Krumpe, L.; Mori, T. The use of phage-displayed peptide libraries to develop tumor-targeting drugs. Int. J. Pept. Res. Ther. 2006, 12, 79–91. [Google Scholar] [CrossRef]
- Alberici, L.; Roth, L.; Sugahara, K.N.; Agemy, L.; Kotamraju, V.R.; Teesalu, T.; Bordignon, C.; Traversari, C.; Rizzardi, G.-P.; Ruoslahti, E. De Novo Design of a Tumor-Penetrating Peptide. Cancer Res. 2013, 73, 804–812. [Google Scholar] [CrossRef]
- Pan, X.; Zhao, Y.Q.; Hu, F.Y.; Chi, C.F.; Wang, B. Anticancer activity of a hexapeptide from skate (Raja porosa) cartilage protein hydrolysate in HeLa cells. Mar. Drugs 2016, 14, 153. [Google Scholar] [CrossRef]
- Song, R.; Wei, R.B.; Luo, H.Y.; Yang, Z.S. Isolation and identification of an antiproliferative peptide derived from heated products of peptic hydrolysates of half-fin anchovy (Setipinna taty). J. Funct. Foods 2014, 10, 104–111. [Google Scholar] [CrossRef]
- Yang, Y.; Sarvestani, S.K.; Moeinzadeh, S.; He, X.; Jabbari, E. Effect of CD44 binding peptide conjugated to an engineered inert matrix on maintenance of breast cancer stem cells and tumorsphere formation. PLoS ONE 2013, 8, e59147. [Google Scholar] [CrossRef]
- Mishra, A.; Dey, S. Molecular docking of a cyclic octapeptide-cyclosaplin from sandalwood. Biomolecules 2019, 9, 740. [Google Scholar] [CrossRef]
- Arcangeli, C.; Lico, C.; Baschieri, S.; Mancuso, M. Characterization of blood-brain barrier crossing and tumor homing peptides by molecular dynamics simulations. Int. J. Nanomed. 2019, 14, 10123–10136. [Google Scholar] [CrossRef]
- Michaeli, A.; Mezan, S.; Kühbacher, A.; Finkelmeier, D.; Elias, M.; Zatsepin, M.; Reed, S.G.; Duthie, M.S.; Rupp, S.; Lerner, I.; et al. Computationally Designed Bispecific MD2/CD14 Binding Peptides Show TLR4 Agonist Activity. J. Immunol. 2018, 201, 3383–3391. [Google Scholar] [CrossRef]
- Wang, D.; El-Amouri, S.S.; Dai, M.; Kuan, C.Y.; Hui, D.Y.; Brady, R.O.; Pan, D. Engineering a lysosomal enzyme with a derivative of receptor-binding domain of apoE enables delivery across the blood-brain barrier. Proc. Natl. Acad. Sci. USA 2013, 19, 2999–3004. [Google Scholar] [CrossRef]
- Widyananda, M.H.; Pratama, S.K.; Samoedra, R.S.; Novita, F.; Kharisma, V.D.; Ansori, A.N.M.; Antonius, Y.; Garrido, G. Molecular docking study of sea urchin (Arbacia lixula) peptides as multi-target inhibitor for non-small cell lung cancer (NSCLC) associated proteins. J. Pharm. Pharmacogn. Res. 2021, 9, 1047–1061. [Google Scholar] [CrossRef]
- Oudart, J.-B.; Brassart-Pasco, S.; Vautrin, A.; Sellier, C.; Machado, C.; Dupont-Deshorgue, A.; Brassart, B.; Baud, S.; Dauchez, M.; Monbioisse, J.-C.; et al. Plasmin releases the anti-tumor peptide from NC1 domain of collagen XIX. Oncotarget 2015, 6, 6. [Google Scholar] [CrossRef]
- Wang, L.; Zhao, C.; Lu, L.; Jiang, H.; Wang, F.; Zhang, X. Transcytosable peptide-paclitaxel prodrug nanoparticle for targeted treatment of triple negative breast cancer. Int. J. Mol. Sci. 2023, 24, 4646. [Google Scholar] [CrossRef]
- Das, S.; Al-Toubah, T.; El-Haddad, G.; Strosberg, J. 177Lu-DOTATATE for the treatment of gastroenteropancreatic neuroendocrine tumors. Expert Rev. Gastroenterol. Hepatol. 2019, 13, 1023–1031. [Google Scholar] [CrossRef]
- Kader, S.; Monvarian, M.; Barati, D.; Moeinzadeh, S.; Makris, T.M.; Jabbari, E. Plasmin-cleavable nanoparticles for on-demand release of morphogens in vascularized osteogenesis. Biomacromolecules 2019, 20, 2973–2988. [Google Scholar] [CrossRef]
- Kuo-Chiang, H.; Eunice, C.Y.; Li-Chan, C.L.J. Antiproliferative activity of peptides prepared from enzymatic hydrolysates of tuna dark muscle on human breast cancer cell line MCF-7. Food Chem. 2011, 126, 617–622. [Google Scholar]
- Liu, Y.Q.; Sun, Z.J.; Wang, C.; Li, S.J.; Liu, Y.Z. Purification of a novel antibacterial short peptide in earthworm Eisenia foetida. Acta. Biochim. Biophys. Sin. (Shanghai) 2004, 36, 297–302. [Google Scholar] [CrossRef]
- Albin, J.; Fahrig, L.; Siemanowski, J.; Rehkaemper, J.; Gebauer, F.; Zander, T.; Buettner, R.; Bruns, C.; Shroeder, W.; Alakus, H.; et al. FGFR2-amplified tumor clones are markedly heterogenous distributed carcinomas of the upper gastrointestinal tract. J. Cancer Res. Clin. Oncol. 2023, 149, 5289–5300. [Google Scholar] [CrossRef]
- Lei, H.; Deng, C.-X. Fibroblast growth factor receptor 2 signaling in breast cancer. Int. J. Biol. Sci. 2017, 13, 1163–1171. [Google Scholar] [CrossRef]
- Xie, L.; Su, X.; Zhang, L.; Yin, X.; Tang, L.; Zhang, X.; Xu, Y.; Gao, Z.; Liu, K.; Zhou, M.; et al. FGFR2 gene amplification in gastric cancer predicts sensitivity to the selective FGFR inhibitor AZD4547. Clin. Cancer Res. 2013, 19, 2572–2583. [Google Scholar] [CrossRef]
- Tsimafeyeu, I.; Khasanova, A.; Stepanova, E.; Gordiev, M.; Khochenkov, D.; Naumova, A.; Varlamov, I.; Snegovoy, A.; Demidov, L. FGFR2 overexpression predicts survival outcome in patients with metastatic papillary renal cell carcinoma. Clin. Transl. Oncol. 2017, 19, 264–268. [Google Scholar] [CrossRef]
- Wasunan, P.; Maneewong, C.; Daengprok, W.; Thirabunyanon, M. Bioactive Earthworm Peptides Produced by Novel Protease-Producing Bacillus velezensis PM 35 and Its Bioactivities on Liver Cancer Cell Death via Apoptosis, Antioxidant Activity, Protection Against Oxidative Stress, and Immune Cell Activation. Front. Microbiol. 2022, 13, 892945–892963. [Google Scholar] [CrossRef]
- Tucker, J.A.; Klein, T.; Breed, J.; Breeze, A.L.; Overman, R.; Phillips, C.; Norman, R.A. Structural insights into FGFR kinase isoform selectivity: Diverse binding modes of AZD4547 and ponatinib in complex with FGFR1 and FGFR4. Structure 2014, 22, 1764–1774. [Google Scholar] [CrossRef]
- Çoban, G.; Köse, F.A. Synthesis, biological evaluations and molecular modelling studies of novel indolin-2-ones designing as FGFR inhibitors. Saudi Pharm. J. 2019, 27, 952–967. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, J.; Huang, H.; Ye, M.; Li, X.; Wu, R.; Liu, H.; Song, Y. Metastasis-associated fibroblasts: An emerging target for metastatic cancer. Biomark. Res. 2021, 9, 47. [Google Scholar] [CrossRef]
- The PyMOL Molecular Graphics System, version 2.5; Schrödinger, LLC.: New York, NY, USA, 2021.
- Tyagi, A.; Kapoor, P.; Kumar, R.; Chaudhary, K.; Gautam, A.; Raghava, G.P.S. In Silico models for designing and discovering novel anticancer peptides. Sci. Rep. 2013, 3, 2984. [Google Scholar] [CrossRef]
- Yu, J.; Zhou, Y.; Tanaka, I.; Yao, M. Roll: A new algorithm for the detection of protein pockets and cavities with a rolling probe sphere. Bioinformatics 2010, 26, 46–52. [Google Scholar] [CrossRef]
- Brawn, R.A.; Cook, A.; Omoto, K.; Ke, J.; Karr, C.; Colombo, F.; Virrankoski, M.; Prajapati, S.; Reynolds, D.; Bolduc, D.M.; et al. Discovery of Aminopyrazole Derivatives as Potent Inhibitors of Wild-Type and Gatekeeper Mutant FGFR2 and 3. ACS Med. Chem. Lett. 2021, 12, 93–98. [Google Scholar] [CrossRef]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Adasme, M.F.; Linnemann, K.L.; Bolz, S.N.; Kaiser, F.; Salentin, S.; Haupt, V.J.; Schroeder, M. PLIP 2021: Expanding the scope of the protein–ligand interaction profiler to DNA and RNA. Nucleic Acids Res. 2021, 49, W530–W534. [Google Scholar] [CrossRef]
- Schrodinger Release 2023-2: Desmond Molecular Dynamics Systems, D.E. Shaw Research. Maestro-Desmond Interoperability Tools; Schrodinger: New York, NY, USA, 2023.
- Bowers, K.J.; Chow, D.E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. Scalable algorithms for molecular dynamics simulations on commodity clusters. In Proceedings of the SC’06 Proceedings of the 2006 ACM/IEEE Conference on Supercomputing, Tampa, FL, USA, 11–17 November 2006; Volume 43. [Google Scholar]
- Banks, J.L.; Beard, H.S.; Cao, Y.; Cho, A.E.; Damm, W.; Farid, R.; Felts, A.K.; Halgren, T.A.; Mainz, D.T.; Maple, J.R.; et al. Integrated Modeling Program, Applied Chemical Theory (IMPACT). J. Comput. Chem. 2005, 26, 1752–1780. [Google Scholar] [CrossRef]
- Jacobson, M.P.; Pincus, D.L.; Rapp, C.S.; Day, T.; Honig, B.; Shaw, D.E. A hierarchical approach to all-atom protein loop prediction. Proteins 2004, 55, 351–367. [Google Scholar] [CrossRef]
- Pattar, S.V.; Adhoni, S.A.; Kamanavalli, C.M.; Kumbar, S.S. In silico molecular docking studies and MM/GBSA analysis of coumarin-carbonodithioate hybrid derivatives divulge the anticancer potential against breast cancer. Beni-Suef Univ. J. Basic. Appl. Sci. 2020, 9, 36. [Google Scholar] [CrossRef]
- Wang, S.; Sun, X.; Cui, W.; Yuan, S. MM/PB(GB) SA benchmarks on soluble proteins and membrane proteins. Front. Pharmacol. 2022, 13, 1018351. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.M.; Teoh, J.Y.; Yim, G.K.; Park, Y.; Kim, Y.G.; Kim, J.; Yoo, D. Label-free analysis of multivalent protein binding using bio-responsive nanogels and surface plasmon resonance (SPR). ACS Appl. Mater. Interfaces 2020, 12, 5413–5419. [Google Scholar] [CrossRef]
- Nam, K.; Kimura, T.; Kishida, A. Controlling coupling reaction of EDC and NHS for preparation of collagen gels using ethanol/water co-solvents. Macromol. Biosci. 2008, 8, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Lau, D.K.; Collin, J.P.; Mariadson, J.M. Clinical developments and challenges in treating FGFR2-driven gastric cancer. Biomedicines 2024, 12, 1117. [Google Scholar] [CrossRef]
- Dragan, A.; Casas-Finet, J.; Bishop, E.; Strouse, R.; Schernerman, M.; Geddes, C. Characterization of PicoGreen interaction with dsDNA and the origin of its fluorescence enhancement upon binding. Biophys. J. 2010, 99, 3010–3019. [Google Scholar] [CrossRef]
- Gabernet, G.; Gautschi, D.; Muller, A.T.; Neuhaus, C.S.; Armbrecht, L.; Dittrich, P.S.; Hiss, J.A.; Schneider, G. In Silico design and optimization of selective membranolytic anticancer peptides. Sci. Rep. 2019, 9, 11282. [Google Scholar] [CrossRef]
- Ma, B.; Shatsky, M.; Wolfson, H.J.; Nussinov, R. Multiple diverse ligands binding at a single protein site: A matter of pre-existing populations. Protein Sci. 2002, 11, 184–197. [Google Scholar] [CrossRef]
- Guedes, I.A.; de Magalhaes, C.S.; Dardenne, L.E. Receptor-ligand molecular docking. Biophys. Rev. 2014, 6, 75–87. [Google Scholar] [CrossRef]
- Tan, L.; Wang, J.; Tanizaki, J.; Huang, Z.; Aref, A.R.; Rusan, M.; Zhu, S.J.; Zhang, Y.; Ercan, D.; Liao, R.G.; et al. Development of covalent inhibitors that can overcome resistance to first-generation FGFR kinase inhibitors. Proc. Natl. Acad. Sci. USA 2014, 11, E4869–E4877. [Google Scholar] [CrossRef]
- Dai, S.; Zhou, Z.; Chen, Z.; Xu, G.; Chen, Y. Fibroblast growth factor receptors (FGFRs): Structures and small molecule inhibitors. Cells 2019, 8, 614. [Google Scholar] [CrossRef]
- Zhang, M.; Yasen, M.; Lu, S.; Ma, D.-N. Decoding the conformational selective mechanism of FGFR isoforms: A comparative molecular dynamics simulation. Molecules 2023, 28, 2709. [Google Scholar] [CrossRef] [PubMed]
- Karp, J.M.; Sparks, S.; Cowburn, D. Effects of FGFR2 kinase activation loop dynamics on catalytic activity. PLoS Comput. Biol. 2017, 13, e1005360. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Kwak, Y.; Kim, N.D.; Sim, T. Antitumor effects and molecular mechanisms of ponatinib on endometrial cancer cells harboring activating FGFR2 mutations. Cancer Biol. Ther. 2016, 17, 65–78. [Google Scholar] [CrossRef]
- Cheng, W.; Wang, M.; Tian, X.; Zhang, X. An overview of the binding models of FGFR tyrosine kinases in complex with small molecule inhibitors. Eur. J. Med. Chem. 2017, 126, 476–490. [Google Scholar] [CrossRef]
- Pallerla, S.; Gauthier, T.; Sable, R.; Jois, S.D. Design of a doxorubicin-peptidomimetic conjugate that targets HER-2 positive cancer cells. Eur. J. Med. Chem. 2017, 125, 914–924. [Google Scholar] [CrossRef] [PubMed]
- Sofela, S.O.; Bodun, D.S.; Omoboyowa, D.A.; Ajiboro, P.A.; Nwankwo, D.O.; Ashimiyu-Abdusalam, Z.; Issac, I.B.; Abdulrasheed, B.; Balogun, T.A.; Ajayi, I.H. Virtual screening for novel FGFR2 inhibitors: Exploring Gefitinib-like compounds as promising therapeutic candidates. Inform. Med. Unlocked 2023, 42, 101368. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM-PBSA and MM-GBSA methods to estimate ligand binding affinities. Expert. Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Bansal, R.; Singh, R.; Kaur, K. Quantitative analysis of doxorubicin hydrochloride and arterolate maleate by mid IR spectroscopy using transmission and reflectance modes. BMC Chem. 2021, 15, 27. [Google Scholar] [CrossRef]
- Song, Q.; Chuan, X.; Chen, B.; He, B.; Zhang, H.; Dai, W.; Wang, X.; Zhang, Q. A smart tumor targeting peptide-drug conjugate, pHLIP-SS-DOX: Synthesis and cellular uptake on MCF-7 and MCF-7/Adr cells. Drug Deliv. 2016, 23, 1734–1746. [Google Scholar] [CrossRef]
- Freire, E.; Van Osdol, W.W.; Mayorga, O.L.; Sanchez-Ruiz, J.M. Calorimetrically determined dynamics of complex unfolding transitions in proteins. Annu. Rev. Biophys. Chem. 1990, 19, 159–188. [Google Scholar] [CrossRef] [PubMed]
- Mazurenko, S.; Kunka, A.; Beerens, K.; Johnson, C.M.; Damborsky, J.; Prokop, Z. Exploration of protein unfolding by modelling calorimetric data from reheating. Sci. Rep. 2017, 7, 16321. [Google Scholar] [CrossRef]
- Shankaranarayanan, J.; Kanwar, J.; Al-Juhaishi, A.; Kanwar, R. Doxorubicin conjugated to immunomodulatory anticancer lactoferrin displays improved cytotoxicity overcoming prostate cancer chemo resistance and inhibits tumor development of TRAMP mice. Sci. Rep. 2016, 6, 32062. [Google Scholar] [CrossRef] [PubMed]
- Levya-Porras, C.; Cruz-Alcantar, P.; Espinosa-Solis, V.; Martinez-Guerra, E.; Pinon-Balderrama, C.; Martinez, I.C.; Saavedra-Leos, M. Application of differential scanning calorimetry (DSC) and modulated differential scanning calorimetry (MDSC) in food and drug industries. Polymers 2019, 12, 5. [Google Scholar] [CrossRef] [PubMed]
- Kunii, K.; Davis, L.; Gorenstein, J.; Hatch, H.; Yashiro, M.; Di Bacco, A.; Elbi, C.; Lutterbach, B. FGFR2-amplified gastric cancer lines require FGFR2 and Erbb3 signaling for growth and survival. Cancer Res. 2008, 68, 2340–2348. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.; Pocard, M.; Mirshahi, M. Targeting the differentiation of gastric cancer cells (KATO III) downregulates epithelial-mesenchymal and cancer stem cell markers. Oncol. Rep. 2019, 42, 670–678. [Google Scholar] [CrossRef]
- Villaronga, M.A.; Teijeiro, S.A.; Hermida-Prado, F.; Grazon-Arango, M.; Sanz-Moreno, V.; Garcia-Pedrero, J. Analysis of invasive activity of CAF spheroids into three-dimensional (3D) collagen matrices. Methods Mol. Biol. 2018, 1731, 145–154. [Google Scholar]
- Jabbari, E.; Sarvestani, S.; Daneshian, L.; Moeinzadeh, S. Optimum 3D matrix stiffness for maintenance of cancer stem cells is dependent on tissue origin of cancer cells. PLoS ONE 2015, 10, e0132377. [Google Scholar] [CrossRef]
- Mittler, F.; Obeid, P.; Rulina, A.; Haguet, V.; Gidrol, X.; Balakriev, M. High-content monitoring of drug effects in a 3D spheroid model. Front. Oncol. 2017, 7, 2017.00293. [Google Scholar] [CrossRef]
- Gong, X.; Lin, C.; Chen, J.; Su, J.; Zhao, H.; Liu, T.; Wen, X.; Zhao, P. Generation of multicellular tumor spheroids with microwell-based agarose scaffolds for drug testing. PLoS ONE 2015, 10, e0130348. [Google Scholar] [CrossRef]
- Angelucci, C.; Maulucci, G.; Lama, G.; Proietti, G.; Colabianchi, A.; Papi, M.; Mairona, A.; Spirito, M.; Micera, A.; Balzamino, O.; et al. Epithelial-Stromal Interactions in Human Breast Cancer: Effects on Adhesion, Plasma Membrane Fluidity and Migration Speed and Directness. PLoS ONE 2012, 7, e50804. [Google Scholar] [CrossRef] [PubMed]
- Murphy, R.J.; Gunasingh, G.; Haass, N.K.; Simpson, M.J. Growth and adaptation mechanisms of tumour spheroids with time-dependent oxygen availability. PLoS Comput. Biol. 2023, 19, e1010833. [Google Scholar] [CrossRef] [PubMed]
- Sriraman, S.K.; Aryasomayajula, B.; Torchilin, V.P. Barriers to drug delivery in solid tumors. Tissue Barriers 2014, 2, e29528. [Google Scholar] [CrossRef]
- She, J.-J.; Zhang, P.-G.; Wang, X.; Che, X.-M.; Wang, Z.-M. Side population cells isolated from KATO III human gastric cancer cell line have cancer stem-like characteristics. World J. Gastroenterol. 2012, 18, 4610–4617. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | SVM Score | Anti-CP | Hydrophobicity | Hydropathicity | Hydrophilicity | pI |
|---|---|---|---|---|---|---|
| A-pep | 1.11 | Yes | 0.09 | 0.98 | −0.34 | 5.85 |
| L-pep | 0.68 | Yes | 0.06 | 0.31 | −0.37 | 5.25 |
| Trimer-pep | 0.69 | Yes | 0.07 | 0.61 | −0.35 | 5.25 |
| Rank | Index | Volume | VD Value |
|---|---|---|---|
| 1 | 86 | 293 | 766 |
| 2 | 95 | 92 | 230 |
| 3 | 293 | 41 | 91 |
| 4 | 307 | 25 | 71 |
| 5 | 312 | 23 | 56 |
| Peptide/Conjugate/Drug | Binding Affinity (kcal/mol) |
|---|---|
| ACSAGLPHVLTPEAGATGASCA (Trimer-pep) | −6.7 |
| LPHVLTPEAGAT (L-pep) | −8.2 |
| ACSAG (A-pep) | −6.0 |
| Trimer-pep-(DOX)2 | −8.3 |
| L-pep-(DOX)2 | −9.6 |
| (A-pep)-DOX | −8.6 |
| DOX | −8.4 |
| Peptide/Conjugate | ΔG Bind (kcal/mol) | ΔG Coulomb (kcal/mol) | ΔG Hydrogen Bonds (kcal/mol) | ΔG Lipophilic (kcal/mol) | ΔG Solvation (kcal/mol) | ΔG VdW (kcal/mol) |
|---|---|---|---|---|---|---|
| Trimer-pep | −111.9 | −60.3 | −8.4 | −27.3 | 92.3 | −125.1 |
| L-pep | −78.8 | −43.8 | −4.9 | −17.8 | 59.9 | −77.2 |
| A-pep | −135.5 | 92.8 | −7.8 | −27.5 | −88.2 | −111.5 |
| Trimer-pep-(DOX)2 | −97.6 | −60.3 | −8.8 | −16.4 | 95.2 | −119.0 |
| L-pep-(DOX)2 | −157.3 | 112.6 | −8.6 | −33.6 | −110.7 | −124.1 |
| A-pep-DOX | −153.9 | 104.7 | −8.5 | −32.7 | −103.8 | −119.8 |
| Peptide | KD Values (M) |
|---|---|
| A-pep (ACSAG) | (14.05 ± 2.8) × 10−6 |
| L-pep (LPHVLTPEAGAT) | (22.61 ± 0.8) × 10−6 |
| Trimer-pep (ACSAGLPHVLTPEAGATGASCA) | (7.53 ± 1.2) × 10−6 |
| FGF Receptor Tyrosine Kinase Inhibitor—CAS 192705-79-6 (CONTROL) | (1.9 ± 3.2) × 10−7 |
| Spheroids | DNA Content at 72 h (ng/mL) | DNA Content at 240 h (ng/mL) |
|---|---|---|
| Spheroids grown at 4000 cells/well | 246 | 492.6 |
| Spheroids grown at 2000 cells/well | 164 | 330.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Biggs, M.A.; Das, A.; Goncalves, B.G.; Murray, M.E.; Frantzeskos, S.A.; Hunt, H.L.; Phan, C.A.N.; Banerjee, I.A. Developing New Peptides and Peptide–Drug Conjugates for Targeting the FGFR2 Receptor-Expressing Tumor Cells and 3D Spheroids. Biomimetics 2024, 9, 515. https://doi.org/10.3390/biomimetics9090515
Biggs MA, Das A, Goncalves BG, Murray ME, Frantzeskos SA, Hunt HL, Phan CAN, Banerjee IA. Developing New Peptides and Peptide–Drug Conjugates for Targeting the FGFR2 Receptor-Expressing Tumor Cells and 3D Spheroids. Biomimetics. 2024; 9(9):515. https://doi.org/10.3390/biomimetics9090515
Chicago/Turabian StyleBiggs, Mary A., Amrita Das, Beatriz G. Goncalves, Molly E. Murray, Sophia A. Frantzeskos, Hannah L. Hunt, Chau Ahn N. Phan, and Ipsita A. Banerjee. 2024. "Developing New Peptides and Peptide–Drug Conjugates for Targeting the FGFR2 Receptor-Expressing Tumor Cells and 3D Spheroids" Biomimetics 9, no. 9: 515. https://doi.org/10.3390/biomimetics9090515
APA StyleBiggs, M. A., Das, A., Goncalves, B. G., Murray, M. E., Frantzeskos, S. A., Hunt, H. L., Phan, C. A. N., & Banerjee, I. A. (2024). Developing New Peptides and Peptide–Drug Conjugates for Targeting the FGFR2 Receptor-Expressing Tumor Cells and 3D Spheroids. Biomimetics, 9(9), 515. https://doi.org/10.3390/biomimetics9090515