
Artificial Photosynthesis: Current Advancements and Future Prospects



Abstract
1. Introduction
2. Photoelectrochemical Cells
2.1. Materials in Photochemical Cells
2.1.1. Photosensitizers
2.1.2. Catalysts
2.1.3. Electron Mediators
2.2. Strategies for Enhancing Photochemical Cell Performance
2.2.1. Strategies for Enhancing Photochemical Cell Performance in Artificial Photosynthesis
2.2.2. Challenges of Photochemical Cell Performance in Artificial Photosynthesis
2.2.3. Strategies for Enhancing Photochemical Cell Performance in Artificial Photosynthesis
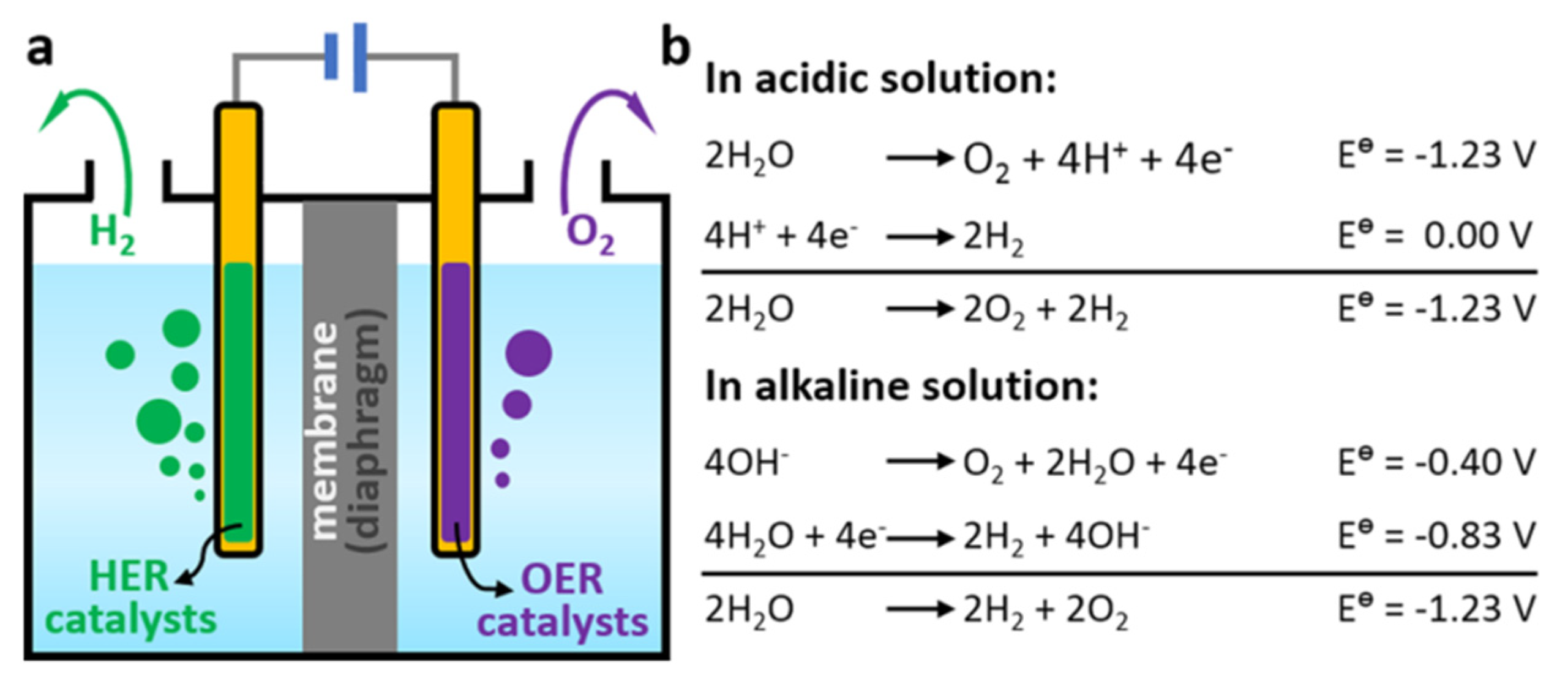
3. Hydrogen and Oxygen Evolution Reactions
3.1. Hydrogen Evolution Reaction (HER)
3.2. Oxygen Evolution Reaction (OER)
4. Catalytic Carbon Dioxide Reduction
4.1. Electrocatalytic Approach
4.2. Photocatalytic Approach
4.3. Photoelectrochemical (PEC) Approach
4.4. Biocatalytic Approach
5. Biomimetic Approaches
5.1. Biohybrids for Enzymatic Catalysis
5.2. Bacteriorhodopsins
5.3. Nanohybrids for Hydrogen Production
5.4. Nanohybrids for CO2 Reduction
5.5. Biomimetic Models Anchored onto Heterogeneous Supports
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kim, J.H.; Hansora, D.; Sharma, P.; Jang, J.-W.; Lee, J.S. Toward Practical Solar Hydrogen Production—An Artificial Photosynthetic Leaf-to-Farm Challenge. Chem. Soc. Rev. 2019, 48, 1908–1971. [Google Scholar] [CrossRef] [PubMed]
- Machín, A.; Fontánez, K.; Arango, J.C.; Ortiz, D.; De León, J.; Pinilla, S.; Nicolosi, V.; Petrescu, F.I.; Morant, C.; Márquez, F. One-Dimensional (1D) Nanostructured Materials for Energy Applications. Materials 2021, 14, 2609. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wu, Q.; Ma, H.; Ma, C.; Yu, Z.; Fu, Y.; Dong, X. Fabrication of PbO2 Tipped Co3O4 Nanowires for Efficient Photoelectrochemical Decolorization of Dye (Reactive Brilliant Blue KN-R) Wastewater. Sol. Energy Mater. Sol. Cells 2019, 191, 381–388. [Google Scholar] [CrossRef]
- Kobayashi, A.; Takizawa, S.; Hirahara, M. Photofunctional Molecular Assembly for Artificial Photosynthesis: Beyond a Simple Dye Sensitization Strategy. Coord. Chem. Rev. 2022, 467, 214624. [Google Scholar] [CrossRef]
- Kalyanasundaram, K.; Graetzel, M. Artificial Photosynthesis: Biomimetic Approaches to Solar Energy Conversion and Storage. Curr. Opin. Biotechnol. 2010, 21, 298–310. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.H.; Lee, Y.-M.; Nam, W.; Fukuzumi, S. Molecular Photocatalytic Water Splitting by Mimicking Photosystems I and II. J. Am. Chem. Soc. 2022, 144, 695–700. [Google Scholar] [CrossRef]
- Shen, L.; Yang, X.; An, J.; Zhang, L.; Yang, K.; Deng, Z. Effect of Different Site Trifluoromethylbenzoic Acid Organic Photosensitizer for Dye-sensitized Solar Cells. ChemistrySelect 2021, 6, 4645–4650. [Google Scholar] [CrossRef]
- Liao, L.; Zhang, Q.; Su, Z.; Zhao, Z.; Wang, Y.; Li, Y.; Lu, X.; Wei, D.; Feng, G.; Yu, Q.; et al. Efficient Solar Water-Splitting Using a Nanocrystalline CoO Photocatalyst. Nat. Nanotech. 2014, 9, 69–73. [Google Scholar] [CrossRef]
- Eren, E.O.; Özkar, S. Recent Advances in Heterogeneous Catalysts for the Effective Electroreduction of Carbon Dioxide to Carbon Monoxide. J. Power Sources 2021, 506, 230215. [Google Scholar] [CrossRef]
- Sukhova, E.; Sukhov, V. Relation of Photochemical Reflectance Indices Based on Different Wavelengths to the Parameters of Light Reactions in Photosystems I and II in Pea Plants. Remote Sens. 2020, 12, 1312. [Google Scholar] [CrossRef]
- Salimi, S.; Zand, Z.; Hołyńska, M.; Allakhverdiev, S.I.; Najafpour, M.M. Nanostructured Manganese Oxide on Carbon for Water Oxidation: New Findings and Challenges. Int. J. Hydrogen Energy 2022, 47, 40943–40951. [Google Scholar] [CrossRef]
- Shi, Y.; Eze, C.; Xiong, B.; He, W.; Zhang, H.; Lim, T.M.; Ukil, A.; Zhao, J. Recent Development of Membrane for Vanadium Redox Flow Battery Applications: A Review. Appl. Energy 2019, 238, 202–224. [Google Scholar] [CrossRef]
- Saygili, Y.; Stojanovic, M.; Flores-Díaz, N.; Zakeeruddin, S.M.; Vlachopoulos, N.; Grätzel, M.; Hagfeldt, A. Metal Coordination Complexes as Redox Mediators in Regenerative Dye-Sensitized Solar Cells. Inorganics 2019, 7, 30. [Google Scholar] [CrossRef]
- Saeed, M.A.; Yoo, K.; Kang, H.C.; Shim, J.W.; Lee, J.-J. Recent Developments in Dye-Sensitized Photovoltaic Cells under Ambient Illumination. Dyes Pigm. 2021, 194, 109626. [Google Scholar] [CrossRef]
- Meng, X.; Li, Z.; Liu, Y.; Wang, Z.; Wang, P.; Zheng, Z.; Dai, Y.; Huang, B.; Cheng, H.; He, J.-H. Enabling Unassisted Solar Water Splitting with Concurrent High Efficiency and Stability by Robust Earth-Abundant Bifunctional Electrocatalysts. Nano Energy 2023, 109, 108296. [Google Scholar] [CrossRef]
- Ye, S.; Ding, C.; Li, C. Artificial Photosynthesis Systems for Catalytic Water Oxidation. In Advances in Inorganic Chemistry; Elsevier: Amsterdam, The Netherlands, 2019; Volume 74, pp. 3–59. ISBN 978-0-12-816082-4. [Google Scholar]
- Mathew, S.; Yella, A.; Gao, P.; Humphry-Baker, R.; Curchod, B.F.E.; Ashari-Astani, N.; Tavernelli, I.; Rothlisberger, U.; Nazeeruddin, M.d.K.; Grätzel, M. Dye-Sensitized Solar Cells with 13% Efficiency Achieved through the Molecular Engineering of Porphyrin Sensitizers. Nat. Chem. 2014, 6, 242–247. [Google Scholar] [CrossRef]
- Nien, Y.-H.; Chen, H.-H.; Hsu, H.-H.; Kuo, P.-Y.; Chou, J.-C.; Lai, C.-H.; Hu, G.-M.; Kuo, C.-H.; Ko, C.-C. Enhanced Photovoltaic Conversion Efficiency in Dye-Sensitized Solar Cells Based on Photoanode Consisting of TiO2/GO/Ag Nanofibers. Vacuum 2019, 167, 47–53. [Google Scholar] [CrossRef]
- Ikpesu, J.E.; Iyuke, S.E.; Daramola, M.; Okewale, A.O. Synthesis of Improved Dye-Sensitized Solar Cell for Renewable Energy Power Generation. Solar Energy 2020, 206, 918–934. [Google Scholar] [CrossRef]
- Zhang, Y.; Wu, G.; Liu, F.; Ding, C.; Zou, Z.; Shen, Q. Photoexcited Carrier Dynamics in Colloidal Quantum Dot Solar Cells: Insights into Individual Quantum Dots, Quantum Dot Solid Films and Devices. Chem. Soc. Rev. 2020, 49, 49–84. [Google Scholar] [CrossRef]
- Zhang, C.; Du, P.; Jiang, Z.; Jin, M.; Chen, G.; Cao, X.; Cui, X.; Zhang, Y.; Li, R.; Abd El-Aty, A.M.; et al. A Simple and Sensitive Competitive Bio-Barcode Immunoassay for Triazophos Based on Multi-Modified Gold Nanoparticles and Fluorescent Signal Amplification. Anal. Chim. Acta 2018, 999, 123–131. [Google Scholar] [CrossRef]
- Yoo, J.J.; Seo, G.; Chua, M.R.; Park, T.G.; Lu, Y.; Rotermund, F.; Kim, Y.-K.; Moon, C.S.; Jeon, N.J.; Correa-Baena, J.-P. Efficient Perovskite Solar Cells via Improved Carrier Management. Nature 2021, 590, 587–593. [Google Scholar] [CrossRef]
- Ilaiyaraja, P.; Rakesh, B.; Das, T.K.; Mocherla, P.S.V.; Sudakar, C. CuInS2 Quantum Dot Sensitized Solar Cells with High VOC ≈ 0.9 V Achieved Using Microsphere-Nanoparticulate TiO2 Composite Photoanode. Sol. Energy Mater. Sol. Cells 2018, 178, 208–222. [Google Scholar] [CrossRef]
- Brunetti, J.; Riolo, G.; Gentile, M.; Bernini, A.; Paccagnini, E.; Falciani, C.; Lozzi, L.; Scali, S.; Depau, L.; Pini, A. Near-Infrared Quantum Dots Labelled with a Tumor Selective Tetrabranched Peptide for in Vivo Imaging. J. Nanobiotechnol. 2018, 16, 21. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Long, G.; Chen, Q.; Zhong, Q. High-Efficiency Layered Sulfur-Doped Reduced Graphene Oxide and Carbon Nanotube Composite Counter Electrode for Quantum Dot Sensitized Solar Cells. J. Power Sources 2019, 430, 95–103. [Google Scholar] [CrossRef]
- Aleixandre-Tudó, J.L.; Castelló-Cogollos, L.; Aleixandre, J.L.; Aleixandre-Benavent, R. Renewable Energies: Worldwide Trends in Research, Funding and International Collaboration. Renew. Energy 2019, 139, 268–278. [Google Scholar] [CrossRef]
- Bates, J.S.; Khamespanah, F.; Cullen, D.A.; Al-Omari, A.A.; Hopkins, M.N.; Martinez, J.J.; Root, T.W.; Stahl, S.S. Molecular Catalyst Synthesis Strategies to Prepare Atomically Dispersed Fe-N-C Heterogeneous Catalysts. J. Am. Chem. Soc. 2022, 144, 18797–18802. [Google Scholar] [CrossRef]
- Oliver, N.; Avramov, A.P.; Nürnberg, D.J.; Dau, H.; Burnap, R.L. From Manganese Oxidation to Water Oxidation: Assembly and Evolution of the Water-Splitting Complex in Photosystem II. Photosynth. Res. 2022, 152, 107–133. [Google Scholar] [CrossRef]
- Ye, S.; Ding, C.; Liu, M.; Wang, A.; Huang, Q.; Li, C. Water Oxidation Catalysts for Artificial Photosynthesis. Adv. Mater. 2019, 31, 1902069. [Google Scholar] [CrossRef]
- Zhang, L.; Mathew, S.; Hessels, J.; Reek, J.N.H.; Yu, F. Homogeneous Catalysts Based on First-Row Transition-Metals for Electrochemical Water Oxidation. ChemSusChem 2021, 14, 234–250. [Google Scholar] [CrossRef]
- Wolff, C.M.; Frischmann, P.D.; Schulze, M.; Bohn, B.J.; Wein, R.; Livadas, P.; Carlson, M.T.; Jäckel, F.; Feldmann, J.; Würthner, F. All-in-One Visible-Light-Driven Water Splitting by Combining Nanoparticulate and Molecular Co-Catalysts on CdS Nanorods. Nat. Energy 2018, 3, 862–869. [Google Scholar] [CrossRef]
- Dogutan, D.K.; Nocera, D.G. Artificial Photosynthesis at Efficiencies Greatly Exceeding That of Natural Photosynthesis. Acc. Chem. Res. 2019, 52, 3143–3148. [Google Scholar] [CrossRef]
- Machín, A.; Cotto, M.; Duconge, J.; Arango, J.C.; Morant, C.; Pinilla, S.; Soto-Vázquez, L.; Resto, E.; Márquez, F. Hydrogen Production via Water Splitting Using Different Au@ZnO Catalysts under UV–Vis Irradiation. J. Photochem. Photobiol. A 2018, 353, 385–394. [Google Scholar] [CrossRef]
- Wang, Y.; Guo, L.; Qi, P.; Liu, X.; Wei, G. Synthesis of Three-Dimensional Graphene-Based Hybrid Materials for Water Purification: A Review. Nanomaterials 2019, 9, 1123. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhu, Y.; Zhao, S.; She, S.; Zhang, F.; Chen, Y.; Williams, T.; Gengenbach, T.; Zu, L.; Mao, H.; et al. Anion Etching for Accessing Rapid and Deep Self-Reconstruction of Precatalysts for Water Oxidation. Matter 2020, 3, 2124–2137. [Google Scholar] [CrossRef]
- Kang, S.; Ham, K.; Lim, H.-K.; Lee, J. Dischargeable Nickel Matrix Charges Iron Species for Oxygen Evolution Electrocatalysis. Electrochim. Acta 2021, 386, 138401. [Google Scholar] [CrossRef]
- Alam, N.; Zahid, M. Pyrite (FES 2)-decorated 1D TIO2 Nanotubes in a Bilayer as a Sustainable Photoanode for Photoelectrochemical Water Splitting Activity. J. Chinese Chemical Soc. 2023, 70, 857–868. [Google Scholar] [CrossRef]
- Agrawal, A.; Siddiqui, S.A.; Soni, A.; Sharma, G.D. Advancements, Frontiers and Analysis of Metal Oxide Semiconductor, Dye, Electrolyte and Counter Electrode of Dye Sensitized Solar Cell. Solar Energy 2022, 233, 378–407. [Google Scholar] [CrossRef]
- Karthika, P.; Ganesan, S.; Thomas, A.; Sheeba Rani, T.M.; Prakash, M. Influence of Synthesized Thiourea Derivatives as a Prolific Additive with Tris(1,10-Phenanthroline)Cobalt(II/III)Bis/Tris(Hexafluorophosphate)/ Hydroxypropyl Cellulose Gel Polymer Electrolytes on Dye-Sensitized Solar Cells. Electrochim. Acta 2019, 298, 237–247. [Google Scholar] [CrossRef]
- Marchini, E.; Caramori, S.; Bignozzi, C.A.; Carli, S. On the Use of PEDOT as a Catalytic Counter Electrode Material in Dye-Sensitized Solar Cells. Appl. Sci. 2021, 11, 3795. [Google Scholar] [CrossRef]
- Michaels, H.; Benesperi, I.; Edvinsson, T.; Muñoz-Garcia, A.; Pavone, M.; Boschloo, G.; Freitag, M. Copper Complexes with Tetradentate Ligands for Enhanced Charge Transport in Dye-Sensitized Solar Cells. Inorganics 2018, 6, 53. [Google Scholar] [CrossRef]
- Lee, C.-P.; Ho, K.-C. Poly(Ionic Liquid)s for Dye-Sensitized Solar Cells: A Mini-Review. Eur. Polym. J. 2018, 108, 420–428. [Google Scholar] [CrossRef]
- Benazzi, E.; Magni, M.; Colombo, A.; Dragonetti, C.; Caramori, S.; Bignozzi, C.A.; Grisorio, R.; Suranna, G.P.; Cipolla, M.P.; Manca, M.; et al. Bis(1,10-Phenanthroline) Copper Complexes with Tailored Molecular Architecture: From Electrochemical Features to Application as Redox Mediators in Dye-Sensitized Solar Cells. Elec. Electrochim. Acta 2018, 271, 180–189. [Google Scholar] [CrossRef]
- Zaine, S.N.A.; Mohamed, N.M.; Khatani, M.; Samsudin, A.E.; Shahid, M.U. Trap State and Charge Recombination in Nanocrystalline Passivized Conductive and Photoelectrode Interface of Dye-Sensitized Solar Cell. Coatings 2020, 10, 284. [Google Scholar] [CrossRef]
- Kwabi, D.G.; Lin, K.; Ji, Y.; Kerr, E.F.; Goulet, M.-A.; De Porcellinis, D.; Tabor, D.P.; Pollack, D.A.; Aspuru-Guzik, A.; Gordon, R.G.; et al. Alkaline Quinone Flow Battery with Long Lifetime at PH 12. Joule 2018, 2, 1894–1906. [Google Scholar] [CrossRef]
- Xia, J.; Wang, Q.; Chen, M.; Li, W.; Liu, J.; Chen, J.; Wu, H.; Fan, S. Bifacial Quasi-Solid-State Dye-Sensitized Solar Cell with Metal Selenide M0.85Se (M = Co, Ni) as Counter Electrode. Electrochim. Acta 2019, 307, 422–429. [Google Scholar] [CrossRef]
- Luciani, G.; Imparato, C.; Vitiello, G. Photosensitive Hybrid Nanostructured Materials: The Big Challenges for Sunlight Capture. Catalysts 2020, 10, 103. [Google Scholar] [CrossRef]
- Xiong, S.; Wang, Y.; Lin, J.; Yu, X.; Tao, J.; Wu, Y.; Yu, G.; Pan, C.; Yamauchi, Y. D-π-A Conjugated Polymer Dyes-Covered TiO2 Compact Layers for Enhancing Photovoltaic Performance of Dye-Sensitized Solar Cells. Synth. Met. 2018, 244, 73–79. [Google Scholar] [CrossRef]
- Chen, C.-H.; Chou, P.-T.; Yin, T.-C.; Chen, K.-F.; Jiang, M.-L.; Chang, Y.J.; Tai, C.-K.; Wang, B.-C. Rational Design of Cost-Effective Dyes for High Performance Dye-Sensitized Cells in Indoor Light Environments. Org. Electron. 2018, 59, 69–76. [Google Scholar] [CrossRef]
- Badawy, S.A.; Su, R.; Fadda, A.A.; Abdel-Latif, E.; El-Shafei, A.; Elmorsy, M.R. Highly Efficient (N-Benzothiazolyl)-Cyanoacetamide Based Co-Sensitizers for High Efficiency Dye-Sensitized Solar Cells. Optik 2022, 249, 168274. [Google Scholar] [CrossRef]
- Tang, Y.; Wang, Y.; Yan, Q.; Zeng, K.; Tang, W.; Zhao, S.; Kong, C.; Xie, Y. Optimization of Porphyrin Dyes with a Bulky Triphenylamine Donor for Developing Efficient Dye-Sensitized Solar Cells. Dyes Pigm. 2021, 187, 109075. [Google Scholar] [CrossRef]
- Cheema, H.; Peddapuram, A.; Adams, R.E.; McNamara, L.; Hunt, L.A.; Le, N.; Watkins, D.L.; Hammer, N.I.; Schmehl, R.H.; Delcamp, J.H. Molecular Engineering of Near Infrared Absorbing Thienopyrazine Double Donor Double Acceptor Organic Dyes for Dye-Sensitized Solar Cells. J. Org. Chem. 2017, 82, 12038–12049. [Google Scholar] [CrossRef]
- Ahmad, Z.; Najeeb, M.A.; Shakoor, R.A.; Al-Muhtaseb, S.A.; Touati, F. Limits and Possible Solutions in Quantum Dot Organic Solar Cells. Renew. Sust. Energ. Rev. 2018, 82, 1551–1564. [Google Scholar] [CrossRef]
- Ho, W.-J.; Feng, S.-K.; Liu, J.-J.; Yang, Y.-C.; Ho, C.-H. Improving Photovoltaic Performance of Silicon Solar Cells Using a Combination of Plasmonic and Luminescent Downshifting Effects. Appl. Surf. Sci. 2018, 439, 868–875. [Google Scholar] [CrossRef]
- Kazanskiy, N.L.; Khonina, S.N.; Butt, M.A. Plasmonic Sensors Based on Metal-Insulator-Metal Waveguides for Refractive Index Sensing Applications: A Brief Review. Physica E Low Dimens. Syst. Nanostruct. 2020, 117, 113798. [Google Scholar] [CrossRef]
- Ghadari, R.; Sabri, A.; Saei, P.-S.; Kong, F.; Mohammadzadeh, Y.; Guzel, E. Plasmon-Enhanced Dye-Sensitized Solar Cells through Porphyrin-Silver Nanoparticle Hybrid Structures: Experimental and Computational Studies. J. Power Sources 2021, 511, 230407. [Google Scholar] [CrossRef]
- Liu, C.C.; Liang, M.S.; Khaw, C.C. Effect of Gold Nanoparticles on the Performances of TiO2 Dye-Sensitised Solar Cell. Ceram. Int. 2018, 44, 5926–5931. [Google Scholar] [CrossRef]
- Nandan Arka, G.; Bhushan Prasad, S.; Singh, S. Comprehensive Study on Dye Sensitized Solar Cell in Subsystem Level to Excel Performance Potential: A Review. Solar Energy 2021, 226, 192–213. [Google Scholar] [CrossRef]
- Liang, J.; Gao, H.; Yi, M.; Shi, W.; Liu, Y.; Zhang, Z.; Mao, Y. β-NaYF4:Yb3+, Tm3+@TiO2 Core-Shell Nanoparticles Incorporated into the Mesoporous Layer for High Efficiency Perovskite Solar Cells. Electrochim. Acta 2018, 261, 14–22. [Google Scholar] [CrossRef]
- Chen, C.; Lou, Y.; Wang, K.; Su, Z.; Dong, C.; Chen, J.; Shi, Y.; Gao, X.; Wang, Z. Ternary Two-Step Sequential Deposition Induced Perovskite Orientational Crystallization for High-Performance Photovoltaic Devices. Adv. Energy Mater. 2021, 11, 2101538. [Google Scholar] [CrossRef]
- Yang, G.; Wang, C.; Lei, H.; Zheng, X.; Qin, P.; Xiong, L.; Zhao, X.; Yan, Y.; Fang, G. Interface Engineering in Planar Perovskite Solar Cells: Energy Level Alignment, Perovskite Morphology Control and High Performance Achievement. J. Mater. Chem. A 2017, 5, 1658–1666. [Google Scholar] [CrossRef]
- Khan, M.I.; Sabir, M.; Mustafa, G.M.; Fatima, M.; Mahmood, A.; Abubshait, S.A.; Abubshait, H.A.; Iqbal, M. 300 KeV Cobalt Ions Irradiations Effect on the Structural, Morphological, Optical and Photovolatic Properties of Zn Doped TiO2 Thin Films Based Dye Sensitized Solar Cells. Ceram. Int. 2020, 46, 16813–16819. [Google Scholar] [CrossRef]
- Ma, Y.; Han, J.; Wang, M.; Chen, X.; Jia, S. Electrophoretic Deposition of Graphene-Based Materials: A Review of Materials and Their Applications. J. Materiomics. 2018, 4, 108–120. [Google Scholar] [CrossRef]
- Geleta, T.A.; Imae, T. Nanocomposite Photoanodes Consisting of P-NiO/n-ZnO Heterojunction and Carbon Quantum Dot Additive for Dye-Sensitized Solar Cells. ACS Appl. Nano Mater. 2021, 4, 236–249. [Google Scholar] [CrossRef]
- Kandasamy, M.; Murugesan, S. Aminosilicate Modified Mesoporous Anatase TiO2@graphene Oxide Nanocomposite for Dye-Sensitized Solar Cells. Solar Energy 2020, 211, 789–798. [Google Scholar] [CrossRef]
- Galindev, O.; Takeguchi, T.; Rahman, M.M. Understanding the Mechanisms and Design Principles for Oxygen Evolution and Oxygen Reduction Activity on Perovskite Catalysts for Alkaline Zinc–Air Batteries. Catal. Sci. Technol. 2021, 11, 5200–5211. [Google Scholar] [CrossRef]
- Xue, Y.; Sun, M. Engineering Hierarchical NiFe-Layered Double Hydroxides Derived Phosphosulfide for High-Efficiency Hydrogen Evolving Electrocatalysis. Int. J. Hydrogen Energy 2019, 44, 16378–16386. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, Y.-C.; Li, X.; Song, C.; Wan, L.; Usman, K.; Fang, J. Recent Advance in Solution-Processed Organic Interlayers for High-Performance Planar Perovskite Solar Cells. Adv. Sci. 2018, 5, 1800159. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Wang, S.; Wang, H.; Zhong, Y.; Hu, Y. An Efficient and Stable Ni–Fe Selenides/Nitrogen-Doped Carbon Nanotubes in Situ-Derived Electrocatalyst for Oxygen Evolution Reaction. J. Mater. Sci. 2020, 55, 13927–13937. [Google Scholar] [CrossRef]
- Lim, D.; Oh, E.; Lim, C.; Shim, S.E.; Baeck, S.-H. Bimetallic NiFe Alloys as Highly Efficient Electrocatalysts for the Oxygen Evolution Reaction. Catal. Today 2020, 352, 27–33. [Google Scholar] [CrossRef]
- Deng, S.; Liu, X.; Huang, T.; Zhao, T.; Lu, Y.; Cheng, J.; Shen, T.; Liang, J.; Wang, D. MoO2 Modulated Electrocatalytic Properties of Ni: Investigate from Hydrogen Oxidation Reaction to Hydrogen Evolution Reaction. Electrochim. Acta 2019, 324, 134892. [Google Scholar] [CrossRef]
- Bao, Z.; Fu, N.; Qin, Y.; Lv, J.; Wang, Y.; He, J.; Hou, Y.; Jiao, C.; Chen, D.; Wu, Y.; et al. Broadband Plasmonic Enhancement of High-Efficiency Dye-Sensitized Solar Cells by Incorporating Au@Ag@SiO 2 Core–Shell Nanocuboids. ACS Appl. Mater. Interfaces 2020, 12, 538–545. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Jian, C.; He, X.; Liu, W. Self-Supported Molybdenum Selenide Nanosheets Grown on Urchin-like Cobalt Selenide Nanowires Array for Efficient Hydrogen Evolution. Int. J. Hydrogen Energy 2020, 45, 13282–13289. [Google Scholar] [CrossRef]
- Bonomo, M.; Di Girolamo, D.; Piccinni, M.; Dowling, D.P.; Dini, D. Electrochemically Deposited NiO Films as a Blocking Layer in P-Type Dye-Sensitized Solar Cells with an Impressive 45% Fill Factor. Nanomaterials 2020, 10, 167. [Google Scholar] [CrossRef] [PubMed]
- Heo, D.Y.; Do, H.H.; Ahn, S.H.; Kim, S.Y. Metal-Organic Framework Materials for Perovskite Solar Cells. Polymers 2020, 12, 2061. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, Q.; Li, L.; Banis, M.N.; Li, J.; Adair, K.; Sun, Y.; Li, R.; Zhao, Z.-J.; Gu, M.; et al. Single Atom Surface Engineering: A New Strategy to Boost Electrochemical Activities of Pt Catalysts. Nano Energy 2022, 93, 106813. [Google Scholar] [CrossRef]
- Ksepko, E.; Lysowski, R. Stable Mixed Fe-Mn Oxides Supported on ZrO2 Oxygen Carriers for Practical Utilization in CLC Processes. Catalysts 2021, 11, 1047. [Google Scholar] [CrossRef]
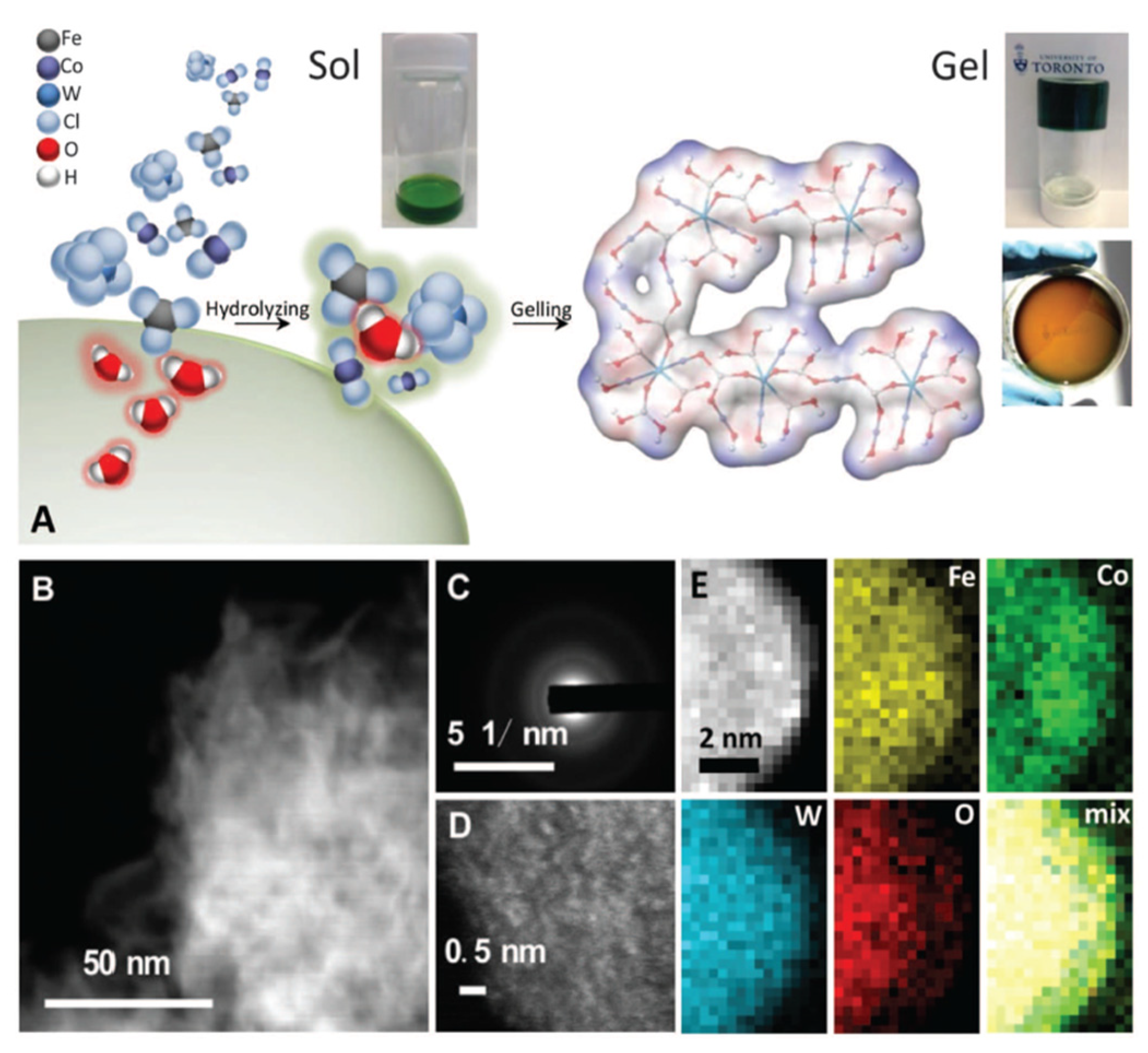
- Zhang, B.; Zheng, X.; Voznyy, O.; Comin, R.; Bajdich, M.; García-Melchor, M.; Han, L.; Xu, J.; Liu, M.; Zheng, L. Homogeneously Dispersed Multimetal Oxygen-Evolving Catalysts. Science 2016, 352, 333–337. [Google Scholar] [CrossRef]
- Machín, A.; Arango, J.C.; Fontánez, K.; Cotto, M.; Duconge, J.; Soto-Vázquez, L.; Resto, E.; Petrescu, F.I.T.; Morant, C.; Márquez, F. Biomimetic Catalysts Based on Au@ZnO–Graphene Composites for the Generation of Hydrogen by Water Splitting. Biomimetics 2020, 5, 39. [Google Scholar] [CrossRef]
- Ahmad, T.; Zhang, D. A Critical Review of Comparative Global Historical Energy Consumption and Future Demand: The Story Told so Far. Energy Rep. 2020, 6, 1973–1991. [Google Scholar] [CrossRef]
- Zhang, P.; Wang, T.; Gong, J. Current Mechanistic Understanding of Surface Reactions over Water-Splitting Photocatalysts. Chem 2018, 4, 223–245. [Google Scholar] [CrossRef]
- Arunachalam, P.; Nagai, K.; Amer, M.S.; Ghanem, M.A.; Ramalingam, R.J.; Al-Mayouf, A.M. Recent Developments in the Use of Heterogeneous Semiconductor Photocatalyst Based Materials for a Visible-Light-Induced Water-Splitting System—A Brief Review. Catalysts 2021, 11, 160. [Google Scholar] [CrossRef]
- Zeng, K.; Tong, Z.; Ma, L.; Zhu, W.-H.; Wu, W.; Xie, Y. Molecular Engineering Strategies for Fabricating Efficient Porphyrin-Based Dye-Sensitized Solar Cells. Energy Environ. Sci. 2020, 13, 1617–1657. [Google Scholar] [CrossRef]
- Wang, Q.; Hisatomi, T.; Jia, Q.; Tokudome, H.; Zhong, M.; Wang, C.; Pan, Z.; Takata, T.; Nakabayashi, M.; Shibata, N. Scalable Water Splitting on Particulate Photocatalyst Sheets with a Solar-to-Hydrogen Energy Conversion Efficiency Exceeding 1%. Nat. Mater. 2016, 15, 611–615. [Google Scholar] [CrossRef]
- Bhullar, V.; Sardana, S.; Mahajan, A. Size Modeling of TiO2 Nanofibers for Efficient TiO2 Sensitized Mesoscopic Solar Cells. Solar Energy 2021, 230, 177–185. [Google Scholar] [CrossRef]
- Machín, A.; Fontánez, K.; García, D.; Sampayo, P.; Colón-Cruz, C.; Claudio-Serrano, G.J.; Soto-Vázquez, L.; Resto, E.; Petrescu, F.I.; Morant, C. Hydrogen Production and Degradation of Ciprofloxacin by Ag@TiO2-MoS2 Photocatalysts. Catalysts 2022, 12, 267. [Google Scholar] [CrossRef]
- Seh, Z.W.; Kibsgaard, J.; Dickens, C.F.; Chorkendorff, I.; Nørskov, J.K.; Jaramillo, T.F. Combining Theory and Experiment in Electrocatalysis: Insights into Materials Design. Science 2017, 355, eaad4998. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, M.H.D.M.; Souza Junior, J.B.; Leite, E.R. The Influence of Magnetic Field and Nanoparticle Concentration on the Thin Film Colloidal Deposition Process of Magnetic Nanoparticles: The Search for High-Efficiency Hematite Photoanodes. Nanomaterials 2022, 12, 1636. [Google Scholar] [CrossRef]
- Victoria, M.; Haegel, N.; Peters, I.M.; Sinton, R.; Jäger-Waldau, A.; Del Cañizo, C.; Breyer, C.; Stocks, M.; Blakers, A.; Kaizuka, I.; et al. Solar Photovoltaics Is Ready to Power a Sustainable Future. Joule 2021, 5, 1041–1056. [Google Scholar] [CrossRef]
- Olaleru, S.A.; Kirui, J.K.; Wamwangi, D.; Roro, K.T.; Mwakikunga, B. Perovskite Solar Cells: The New Epoch in Photovoltaics. Solar Energy 2020, 196, 295–309. [Google Scholar] [CrossRef]
- Feng, D.; Lei, T.; Lukatskaya, M.R.; Park, J.; Huang, Z.; Lee, M.; Shaw, L.; Chen, S.; Yakovenko, A.A.; Kulkarni, A.; et al. Robust and Conductive Two-Dimensional Metal−organic Frameworks with Exceptionally High Volumetric and Areal Capacitance. Nat. Energy 2018, 3, 30–36. [Google Scholar] [CrossRef]
- Di Bartolomeo, A. Emerging 2D Materials and Their Van Der Waals Heterostructures. Nanomaterials 2020, 10, 579. [Google Scholar] [CrossRef]
- Alotaibi, I.; Abido, M.A.; Khalid, M.; Savkin, A.V. A Comprehensive Review of Recent Advances in Smart Grids: A Sustainable Future with Renewable Energy Resources. Energies 2020, 13, 6269. [Google Scholar] [CrossRef]
- Makeev, M.A.; Rajput, N.N. Computational Screening of Electrolyte Materials: Status Quo and Open Problems. Curr. Opin. 2019, 23, 58–69. [Google Scholar] [CrossRef]
- De Pablo, J.J.; Jackson, N.E.; Webb, M.A.; Chen, L.-Q.; Moore, J.E.; Morgan, D.; Jacobs, R.; Pollock, T.; Schlom, D.G.; Toberer, E.S. New Frontiers for the Materials Genome Initiative. Comput. Mater. 2019, 5, 41. [Google Scholar] [CrossRef]
- Yamada, T.; Domen, K. Development of Sunlight Driven Water Splitting Devices towards Future Artificial Photosynthetic Industry. ChemEngineering 2018, 2, 36. [Google Scholar] [CrossRef]
- You, B.; Sun, Y. Innovative Strategies for Electrocatalytic Water Splitting. Acc. Chem. Res. 2018, 51, 1571–1580. [Google Scholar] [CrossRef] [PubMed]
- Machín, A.; Cotto, M.; Ducongé, J.; Arango, J.C.; Morant, C.; Márquez, F. Synthesis and Characterization of Au@TiO2 NWs and Their Catalytic Activity by Water Splitting: A Comparative Study with Degussa P25. AJEAS 2017, 10, 298–311. [Google Scholar] [CrossRef]
- Pinilla, S.; Machín, A.; Park, S.-H.; Arango, J.C.; Nicolosi, V.; Márquez -Linares, F.; Morant, C. TiO2 -Based Nanomaterials for the Production of Hydrogen and the Development of Lithium-Ion Batteries. J. Phys. Chem. B 2018, 122, 972–983. [Google Scholar] [CrossRef]
- Jackson, M.N.; Jung, O.; Lamotte, H.C.; Surendranath, Y. Donor-Dependent Promotion of Interfacial Proton-Coupled Electron Transfer in Aqueous Electrocatalysis. ACS Catal. 2019, 9, 3737–3743. [Google Scholar] [CrossRef]
- Zhang, X.; Li, Y.; Guo, P.; Le, J.-B.; Zhou, Z.-Y.; Cheng, J.; Sun, S.-G. Theory on Optimizing the Activity of Electrocatalytic Proton Coupled Electron Transfer Reactions. J. Catal. 2019, 376, 17–24. [Google Scholar] [CrossRef]
- Anjum, M.A.R.; Jeong, H.Y.; Lee, M.H.; Shin, H.S.; Lee, J.S. Efficient Hydrogen Evolution Reaction Catalysis in Alkaline Media by All-in-One MoS2 with Multifunctional Active Sites. Adv. Mater. 2018, 30, 1707105. [Google Scholar] [CrossRef]
- Murthy, A.P.; Madhavan, J.; Murugan, K. Recent Advances in Hydrogen Evolution Reaction Catalysts on Carbon/Carbon-Based Supports in Acid Media. J. Power Sources 2018, 398, 9–26. [Google Scholar] [CrossRef]
- Pu, Z.; Amiinu, I.S.; Cheng, R.; Wang, P.; Zhang, C.; Mu, S.; Zhao, W.; Su, F.; Zhang, G.; Liao, S.; et al. Single-Atom Catalysts for Electrochemical Hydrogen Evolution Reaction: Recent Advances and Future Perspectives. Nano-Micro Lett. 2020, 12, 21. [Google Scholar] [CrossRef] [PubMed]
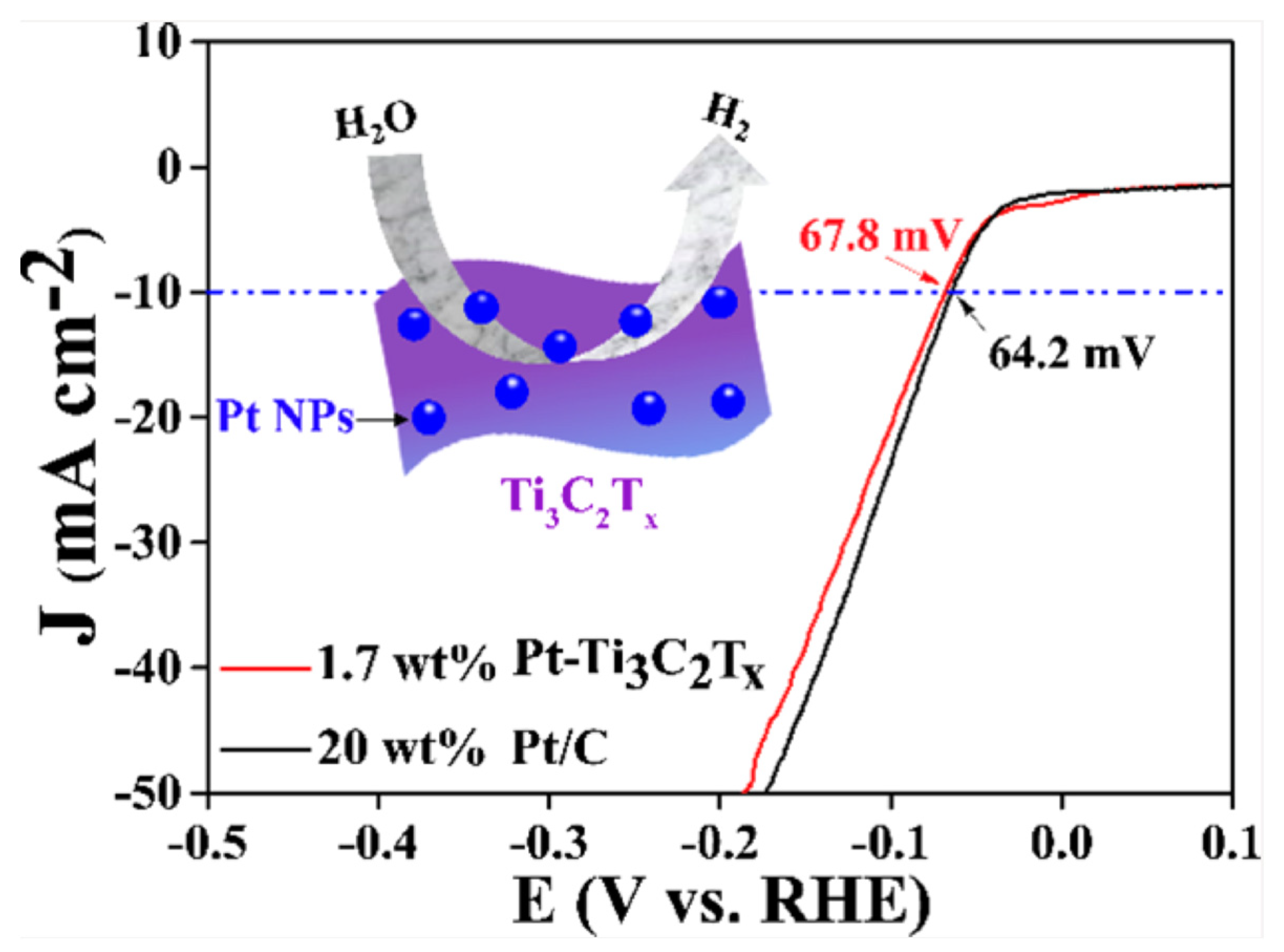
- Zhang, X.; Shao, B.; Sun, Z.; Gao, Z.; Qin, Y.; Zhang, C.; Cui, F.; Yang, X. Platinum Nanoparticle-Deposited Ti3C2Tx MXene for Hydrogen Evolution Reaction. Ind. Eng. Chem. Res. 2020, 59, 1822–1828. [Google Scholar] [CrossRef]
- Jin, H.; Liu, X.; Chen, S.; Vasileff, A.; Li, L.; Jiao, Y.; Song, L.; Zheng, Y.; Qiao, S.-Z. Heteroatom-Doped Transition Metal Electrocatalysts for Hydrogen Evolution Reaction. ACS Energy Lett. 2019, 4, 805–810. [Google Scholar] [CrossRef]
- Wu, H.; Feng, C.; Zhang, L.; Zhang, J.; Wilkinson, D.P. Non-Noble Metal Electrocatalysts for the Hydrogen Evolution Reaction in Water Electrolysis. Electrochem. Energ. Rev. 2021, 4, 473–507. [Google Scholar] [CrossRef]
- Zhao, Z.; Liu, H.; Gao, W.; Xue, W.; Liu, Z.; Huang, J.; Pan, X.; Huang, Y. Surface-Engineered PtNi-O Nanostructure with Record-High Performance for Electrocatalytic Hydrogen Evolution Reaction. J. Am. Chem. Soc. 2018, 140, 9046–9050. [Google Scholar] [CrossRef]
- Li, Z.; Yue, Y.; Peng, J.; Luo, Z. Phase Engineering Two-Dimensional Nanostructures for Electrocatalytic Hydrogen Evolution Reaction. Chin. Chem. Lett. 2023, 34, 107119. [Google Scholar] [CrossRef]
- Kim, J.; Jung, H.; Jung, S.-M.; Hwang, J.; Kim, D.Y.; Lee, N.; Kim, K.-S.; Kwon, H.; Kim, Y.-T.; Han, J.W. Tailoring Binding Abilities by Incorporating Oxophilic Transition Metals on 3D Nanostructured Ni Arrays for Accelerated Alkaline Hydrogen Evolution Reaction. J. Am. Chem. Soc. 2021, 143, 1399–1408. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Liu, X.; Chen, S.; Ma, T.; Li, Y. Interface Engineering and Anion Engineering of Mo-Based Heterogeneous Electrocatalysts for Hydrogen Evolution Reaction. Energy Environ. Mat. 2023, 6, e12310. [Google Scholar] [CrossRef]
- Sun, J.; Tian, F.; Yu, F.; Yang, Z.; Yu, B.; Chen, S.; Ren, Z.; Zhou, H. Robust Hydrogen-Evolving Electrocatalyst from Heterogeneous Molybdenum Disulfide-Based Catalyst. ACS Catal. 2020, 10, 1511–1519. [Google Scholar] [CrossRef]
- Lu, S.; Cao, J.; Zhang, Y.; Lou, F.; Yu, Z. Transition Metal Single-Atom Supported on PC3 Monolayer for Highly Efficient Hydrogen Evolution Reaction by Combined Density Functional Theory and Machine Learning Study. Appl. Surface Sci. 2022, 606, 154945. [Google Scholar] [CrossRef]
- Gao, L.; Cui, X.; Sewell, C.D.; Li, J.; Lin, Z. Recent Advances in Activating Surface Reconstruction for the High-Efficiency Oxygen Evolution Reaction. Chem. Soc. Rev. 2021, 50, 8428–8469. [Google Scholar] [CrossRef] [PubMed]
- Zhu, K.; Shi, F.; Zhu, X.; Yang, W. The Roles of Oxygen Vacancies in Electrocatalytic Oxygen Evolution Reaction. Nano Energy 2020, 73, 104761. [Google Scholar] [CrossRef]
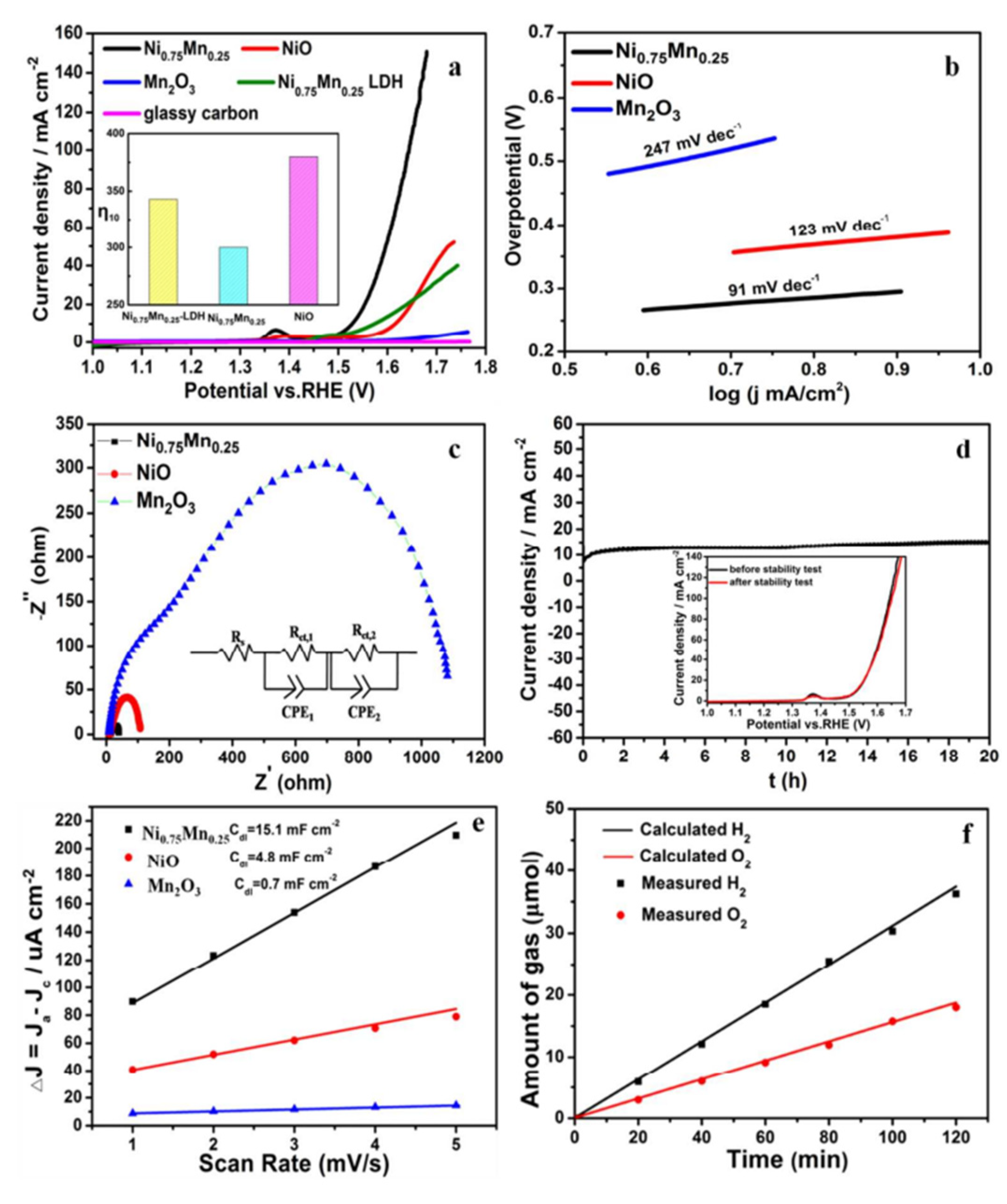
- Tian, T.; Gao, H.; Zhou, X.; Zheng, L.; Wu, J.; Li, K.; Ding, Y. Study of the Active Sites in Porous Nickel Oxide Nanosheets by Manganese Modulation for Enhanced Oxygen Evolution Catalysis. ACS Energy Lett. 2018, 3, 2150–2158. [Google Scholar] [CrossRef]
- Pei, X.; Yi, S.; Zhao, Y.; Mu, Y.; Yu, Y.; Cui, M.; Meng, C.; Huang, C.; Zhang, Y. Nickel Oxide Nanoparticles Dispersed on Biomass–Derived Amorphous Carbon/Cobalt Silicate Support Accelerate the Oxygen Evolution Reaction. J. Colloid Interface Sci. 2022, 616, 476–487. [Google Scholar] [CrossRef]
- Ding, Z.; Bian, J.; Shuang, S.; Liu, X.; Hu, Y.; Sun, C.; Yang, Y. High Entropy Intermetallic–Oxide Core–Shell Nanostructure as Superb Oxygen Evolution Reaction Catalyst. Adv. Sustain. Syst. 2020, 4, 1900105. [Google Scholar] [CrossRef]
- Mahata, A.; Nair, A.S.; Pathak, B. Recent Advancements in Pt-Nanostructure-Based Electrocatalysts for the Oxygen Reduction Reaction. Catal. Sci. Technol. 2019, 9, 4835–4863. [Google Scholar] [CrossRef]
- Song, F.; Bai, L.; Moysiadou, A.; Lee, S.; Hu, C.; Liardet, L.; Hu, X. Transition Metal Oxides as Electrocatalysts for the Oxygen Evolution Reaction in Alkaline Solutions: An Application-Inspired Renaissance. J. Am. Chem. Soc. 2018, 140, 7748–7759. [Google Scholar] [CrossRef]
- Tariq, M.; Zaman, W.Q.; Sun, W.; Zhou, Z.; Wu, Y.; Cao, L.; Yang, J. Unraveling the Beneficial Electrochemistry of IrO2 /MoO3 Hybrid as a Highly Stable and Efficient Oxygen Evolution Reaction Catalyst. ACS Sustain. Chem. Eng. 2018, 6, 4854–4862. [Google Scholar] [CrossRef]
- Hu, L.; Zeng, X.; Wei, X.; Wang, H.; Wu, Y.; Gu, W.; Shi, L.; Zhu, C. Interface Engineering for Enhancing Electrocatalytic Oxygen Evolution of NiFe LDH/NiTe Heterostructures. Appl. Catal. B Environ. 2020, 273, 119014. [Google Scholar] [CrossRef]
- Liu, Y.; Jiang, S.; Li, S.; Zhou, L.; Li, Z.; Li, J.; Shao, M. Interface Engineering of (Ni, Fe)S2@MoS2 Heterostructures for Synergetic Electrochemical Water Splitting. Appl. Catal. B Environ. 2019, 247, 107–114. [Google Scholar] [CrossRef]
- Wu, L.; Guo, T.; Li, T. Machine Learning-Accelerated Prediction of Overpotential of Oxygen Evolution Reaction of Single-Atom Catalysts. iScience 2021, 24, 102398. [Google Scholar] [CrossRef]
- Deng, C.; Su, Y.; Li, F.; Shen, W.; Chen, Z.; Tang, Q. Understanding Activity Origin for the Oxygen Reduction Reaction on Bi-Atom Catalysts by DFT Studies and Machine-Learning. J. Mater. Chem. A 2020, 8, 24563–24571. [Google Scholar] [CrossRef]
- Lu, W.; Zhang, Y.; Zhang, J.; Xu, P. Reduction of Gas CO2 to CO with High Selectivity by Ag Nanocube-Based Membrane Cathodes in a Photoelectrochemical System. Ind. Eng. Chem. Res. 2020, 59, 5536–5545. [Google Scholar] [CrossRef]
- Xu, L.; Xiu, Y.; Liu, F.; Liang, Y.; Wang, S. Research Progress in Conversion of CO2 to Valuable Fuels. Molecules 2020, 25, 3653. [Google Scholar] [CrossRef]
- Song, J.T.; Song, H.; Kim, B.; Oh, J. Towards Higher Rate Electrochemical CO2 Conversion: From Liquid-Phase to Gas-Phase Systems. Catalysts 2019, 9, 224. [Google Scholar] [CrossRef]
- Kjeang, E.; Michel, R.; Harrington, D.A.; Sinton, D.; Djilali, N. An Alkaline Microfluidic Fuel Cell Based on Formate and Hypochlorite Bleach. Electrochim. Acta 2008, 54, 698–705. [Google Scholar] [CrossRef]
- Wu, M.; Xu, B.; Zhang, Y.; Qi, S.; Ni, W.; Hu, J.; Ma, J. Perspectives in Emerging Bismuth Electrochemistry. J. Chem. Eng. 2020, 381, 122558. [Google Scholar] [CrossRef]
- Zheng, X.; De Luna, P.; García De Arquer, F.P.; Zhang, B.; Becknell, N.; Ross, M.B.; Li, Y.; Banis, M.N.; Li, Y.; Liu, M. Sulfur-Modulated Tin Sites Enable Highly Selective Electrochemical Reduction of CO2 to Formate. Joule 2017, 1, 794–805. [Google Scholar] [CrossRef]
- Hoffman, Z.B.; Gray, T.S.; Moraveck, K.B.; Gunnoe, T.B.; Zangari, G. Electrochemical Reduction of Carbon Dioxide to Syngas and Formate at Dendritic Copper–Indium Electrocatalysts. ACS Catal. 2017, 7, 5381–5390. [Google Scholar] [CrossRef]
- Lu, X.; Wu, Y.; Yuan, X.; Wang, H. An Integrated CO2 Electrolyzer and Formate Fuel Cell Enabled by a Reversibly Restructuring Pb–Pd Bimetallic Catalyst. Angew. Chem. Int. Ed. 2019, 58, 4031–4035. [Google Scholar] [CrossRef] [PubMed]
- Dinh, C.-T.; Burdyny, T.; Kibria, M.G.; Seifitokaldani, A.; Gabardo, C.M.; García De Arquer, F.P.; Kiani, A.; Edwards, J.P.; De Luna, P.; Bushuyev, O.S. CO2 Electroreduction to Ethylene via Hydroxide-Mediated Copper Catalysis at an Abrupt Interface. Science 2018, 360, 783–787. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Kuang, Y.; Meng, Y.; Tian, X.; Hung, W.-H.; Zhang, X.; Li, A.; Xu, M.; Zhou, W.; Ku, C.-S.; et al. Electroreduction of CO2 to Formate on a Copper-Based Electrocatalyst at High Pressures with High Energy Conversion Efficiency. J. Am. Chem. Soc. 2020, 142, 7276–7282. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Pang, Y.; Zhang, B.; De Luna, P.; Voznyy, O.; Xu, J.; Zheng, X.; Dinh, C.T.; Fan, F.; Cao, C.; et al. Enhanced Electrocatalytic CO2 Reduction via Field-Induced Reagent Concentration. Nature 2016, 537, 382–386. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Kattel, S.; Jiao, F.; Chen, J.G. Shape—Controlled CO2 Electrochemical Reduction on Nanosized Pd Hydride Cubes and Octahedra. Adv. Energy Mater. 2019, 9, 1802840. [Google Scholar] [CrossRef]
- Chen, C.; Sun, X.; Yan, X.; Wu, Y.; Liu, H.; Zhu, Q.; Bediako, B.B.A.; Han, B. Boosting CO2 Electroreduction on N,P-Co-doped Carbon Aerogels. Angew. Chem. Int. Ed. 2020, 59, 11123–11129. [Google Scholar] [CrossRef]
- Yang, H.B.; Hung, S.-F.; Liu, S.; Yuan, K.; Miao, S.; Zhang, L.; Huang, X.; Wang, H.-Y.; Cai, W.; Chen, R.; et al. Atomically Dispersed Ni(i) as the Active Site for Electrochemical CO2 Reduction. Nat. Energy 2018, 3, 140–147. [Google Scholar] [CrossRef]
- Fan, W.; Duan, Z.; Liu, W.; Mehmood, R.; Qu, J.; Cao, Y.; Guo, X.; Zhong, J.; Zhang, F. Rational Design of Heterogenized Molecular Phthalocyanine Hybrid Single-Atom Electrocatalyst towards Two-Electron Oxygen Reduction. Nat. Commun. 2023, 14, 1426. [Google Scholar] [CrossRef]
- Li, J.; Shen, H.; Ma, C.; Zhang, H.; Luo, P.; Chen, J.; Mu, M.; Yin, X. Assembly and Electrocatalytic CO2 Reduction of Two-Dimensional Bimetallic Porphyrin-Based Conjugated Cobalt Metal-Organic Framework. Electrochim. Acta 2023, 443, 141896. [Google Scholar] [CrossRef]
- Ozawa, H.; Kikunaga, R.; Suzuki, H.; Abe, R.; Sakai, K. Efficient Syngas Production with Controllable CO:H2 Ratios Based on Aqueous Electrocatalytic CO2 Reduction over Mesoporous TiO2 Films Modified with a Cobalt Porphyrin Molecular Catalyst. Sustain. Energy Fuels 2023, 7, 1627–1632. [Google Scholar] [CrossRef]
- Huang, C.; Bao, W.; Huang, S.; Wang, B.; Wang, C.; Han, S.; Lu, C.; Qiu, F. Asymmetric Push–Pull Type Co(II) Porphyrin for Enhanced Electrocatalytic CO2 Reduction Activity. Molecules 2022, 28, 150. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Cai, H.; Cui, L.; Yu, L.; Gao, R.; Shi, C. Molecular Modulating of Cobalt Phthalocyanines on Amino-Functionalized Carbon Nanotubes for Enhanced Electrocatalytic CO2 Conversion. Nano Res. 2023, 16, 3649–3657. [Google Scholar] [CrossRef]
- Boutin, E.; Merakeb, L.; Ma, B.; Boudy, B.; Wang, M.; Bonin, J.; Anxolabéhère-Mallart, E.; Robert, M. Molecular Catalysis of CO2 Reduction: Recent Advances and Perspectives in Electrochemical and Light-Driven Processes with Selected Fe, Ni and Co Aza Macrocyclic and Polypyridine Complexes. Chem. Soc. Rev. 2020, 49, 5772–5809. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Zhan, F.; Wang, Q.; Sun, Y.; Yu, C.; Zhao, X.; Wang, H.; Long, R.; Zhang, G.; Gao, C.; et al. Tracking Mechanistic Pathway of Photocatalytic CO 2 Reaction at Ni Sites Using Operando, Time-Resolved Spectroscopy. J. Am. Chem. Soc. 2020, 142, 5618–5626. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Cheng, B.; Lei, J.; Jiang, L.; Han, Z. Promoting Photocatalytic CO2 Reduction with a Molecular Copper Purpurin Chromophore. Nat. Commun. 2021, 12, 1835. [Google Scholar] [CrossRef]
- Leung, C.-F.; Lau, T.-C. Organic Photosensitizers for Catalytic Solar Fuel Generation. Energy Fuels 2021, 35, 18888–18899. [Google Scholar] [CrossRef]
- Chen, L.; Qin, Y.; Chen, G.; Li, M.; Cai, L.; Qiu, Y.; Fan, H.; Robert, M.; Lau, T.-C. A Molecular Noble Metal-Free System for Efficient Visible Light-Driven Reduction of CO2 to CO. Dalton Trans. 2019, 48, 9596–9602. [Google Scholar] [CrossRef]
- Rao, H.; Lim, C.-H.; Bonin, J.; Miyake, G.M.; Robert, M. Visible-Light-Driven Conversion of CO2 to CH4 with an Organic Sensitizer and an Iron Porphyrin Catalyst. J. Am. Chem. Soc. 2018, 140, 17830–17834. [Google Scholar] [CrossRef]
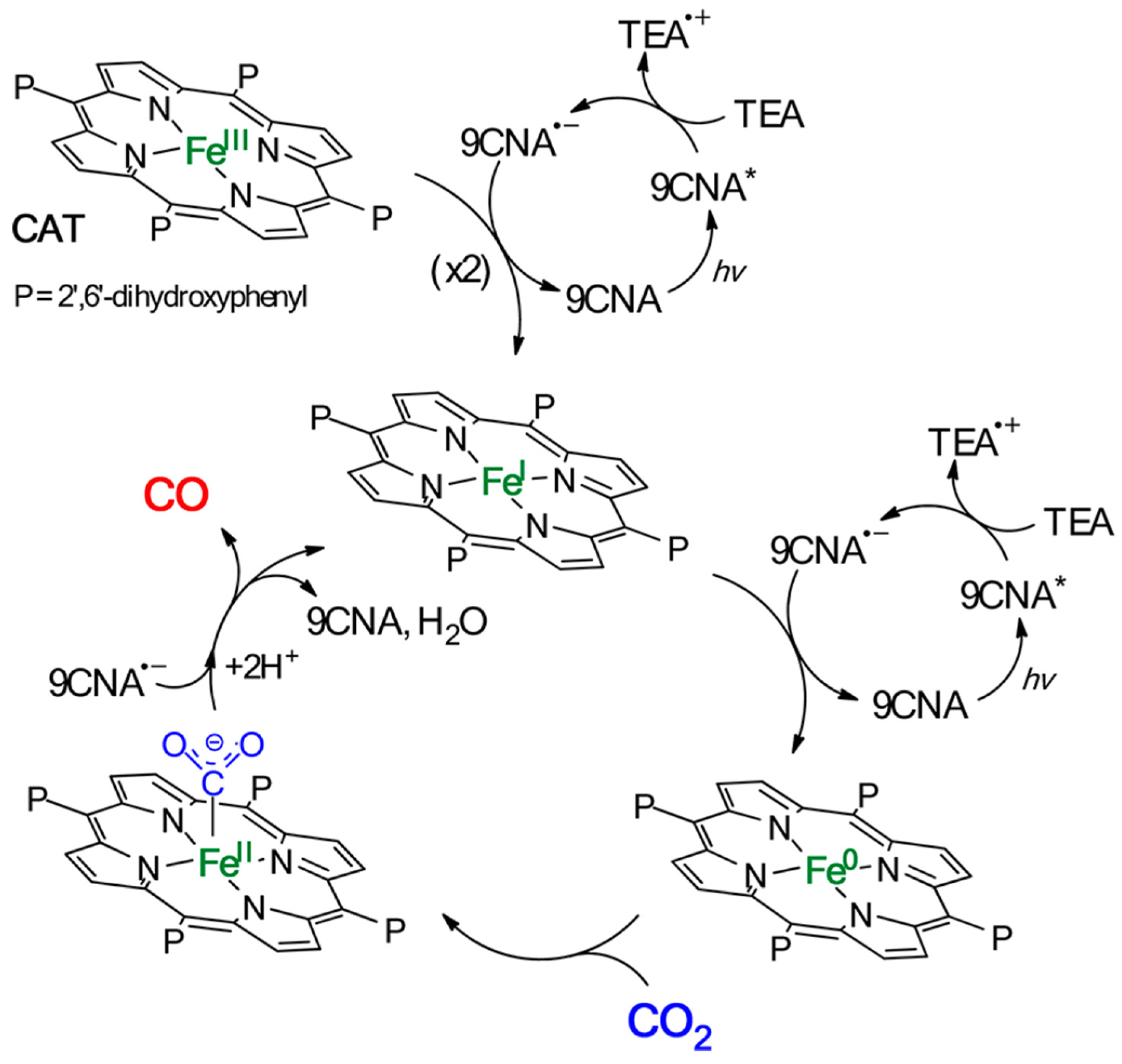
- Bonin, J.; Robert, M.; Routier, M. Selective and Efficient Photocatalytic CO2 Reduction to CO Using Visible Light and an Iron-Based Homogeneous Catalyst. J. Am. Chem. Soc. 2014, 136, 16768–16771. [Google Scholar] [CrossRef]
- Guo, Z.; Cheng, S.; Cometto, C.; Anxolabéhère-Mallart, E.; Ng, S.-M.; Ko, C.-C.; Liu, G.; Chen, L.; Robert, M.; Lau, T.-C. Highly Efficient and Selective Photocatalytic CO2 Reduction by Iron and Cobalt Quaterpyridine Complexes. J. Am. Chem. Soc. 2016, 138, 9413–9416. [Google Scholar] [CrossRef] [PubMed]
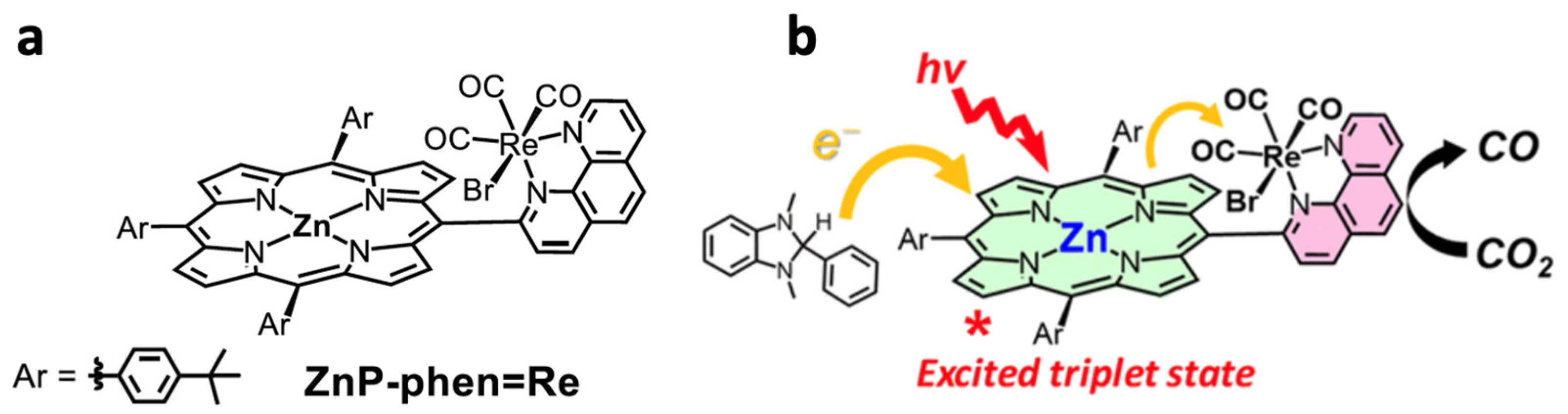
- Kuramochi, Y.; Fujisawa, Y.; Satake, A. Photocatalytic CO2 Reduction Mediated by Electron Transfer via the Excited Triplet State of Zn(II) Porphyrin. J. Am. Chem. Soc. 2020, 142, 705–709. [Google Scholar] [CrossRef] [PubMed]
- Lei, Q.; Yuan, H.; Du, J.; Ming, M.; Yang, S.; Chen, Y.; Lei, J.; Han, Z. Photocatalytic CO2 Reduction with Aminoanthraquinone Organic Dyes. Nat. Commun. 2023, 14, 1087. [Google Scholar] [CrossRef] [PubMed]
- Karthick Raj, A.G.; Murugan, C.; Pandikumar, A. Efficient Photoelectrochemical Reduction of Carbon Dioxide into Alcohols Assisted by Photoanode Driven Water Oxidation with Gold Nanoparticles Decorated Titania Nanotubes. J. CO2 Util. 2021, 52, 101684. [Google Scholar] [CrossRef]
- Merino-Garcia, I.; Albo, J.; Solla-Gullón, J.; Montiel, V.; Irabien, A. Cu Oxide/ZnO-Based Surfaces for a Selective Ethylene Production from Gas-Phase CO2 Electroconversion. J. CO2 Util. 2019, 31, 135–142. [Google Scholar] [CrossRef]
- Zhang, W.; Qin, Q.; Dai, L.; Qin, R.; Zhao, X.; Chen, X.; Ou, D.; Chen, J.; Chuong, T.T.; Wu, B.; et al. Electrochemical Reduction of Carbon Dioxide to Methanol on Hierarchical Pd/SnO2 Nanosheets with Abundant Pd-O-Sn Interfaces. Angew. Chem. Int. Ed. 2018, 57, 9475–9479. [Google Scholar] [CrossRef]
- Chu, S.; Ou, P.; Rashid, R.T.; Ghamari, P.; Wang, R.; Tran, H.N.; Zhao, S.; Zhang, H.; Song, J.; Mi, Z. Decoupling Strategy for Enhanced Syngas Generation from Photoelectrochemical CO2 Reduction. iScience 2020, 23, 101390. [Google Scholar] [CrossRef]
- Liu, B.; Wang, T.; Wang, S.; Zhang, G.; Zhong, D.; Yuan, T.; Dong, H.; Wu, B.; Gong, J. Back-Illuminated Photoelectrochemical Flow Cell for Efficient CO2 Reduction. Nat. Commun. 2022, 13, 7111. [Google Scholar] [CrossRef]
- Pawar, A.U.; Kim, C.W.; Nguyen-Le, M.-T.; Kang, Y.S. General Review on the Components and Parameters of Photoelectrochemical System for CO2 Reduction with in Situ Analysis. ACS Sustain. Chem. Eng. 2019, 7, 7431–7455. [Google Scholar] [CrossRef]
- Thangamuthu, M.; Ruan, Q.; Ohemeng, P.O.; Luo, B.; Jing, D.; Godin, R.; Tang, J. Polymer Photoelectrodes for Solar Fuel Production: Progress and Challenges. Chem. Rev. 2022, 122, 11778–11829. [Google Scholar] [CrossRef]
- Chen, P.; Zhang, Y.; Zhou, Y.; Dong, F. Photoelectrocatalytic Carbon Dioxide Reduction: Fundamental, Advances and Challenges. Nano Mat. Sci. 2021, 3, 344–367. [Google Scholar] [CrossRef]
- Kumaravel, V.; Bartlett, J.; Pillai, S.C. Photoelectrochemical Conversion of Carbon Dioxide (CO2) into Fuels and Value-Added Products. ACS Energy Lett. 2020, 5, 486–519. [Google Scholar] [CrossRef]
- Tang, P.; Han, L.; Hegner, F.S.; Paciok, P.; Biset-Peiró, M.; Du, H.; Wei, X.; Jin, L.; Xie, H.; Shi, Q.; et al. Boosting Photoelectrochemical Water Oxidation of Hematite in Acidic Electrolytes by Surface State Modification. Adv. Energy Mater. 2019, 9, 1901836. [Google Scholar] [CrossRef]
- Ros, C.; Andreu, T.; Morante, J.R. Photoelectrochemical Water Splitting: A Road from Stable Metal Oxides to Protected Thin Film Solar Cells. J. Mater. Chem. A 2020, 8, 10625–10669. [Google Scholar] [CrossRef]
- Irtem, E.; Hernández-Alonso, M.D.; Parra, A.; Fàbrega, C.; Penelas-Pérez, G.; Morante, J.R.; Andreu, T. A Photoelectrochemical Flow Cell Design for the Efficient CO2 Conversion to Fuels. Electrochim. Acta 2017, 240, 225–230. [Google Scholar] [CrossRef]
- Kim, C.; Choi, S.; Choi, M.-J.; Lee, S.A.; Ahn, S.H.; Kim, S.Y.; Jang, H.W. Photoelectrochemical Reduction of CO2 to Syngas by Reduced Ag Catalysts on Si Photocathodes. Appl. Sci. 2020, 10, 3487. [Google Scholar] [CrossRef]
- Chu, S.; Ou, P.; Rashid, R.T.; Pan, Y.; Liang, D.; Zhang, H.; Song, J. Efficient Photoelectrochemical Conversion of CO2 to Syngas by Photocathode Engineering. Green Energy Environ. 2022, 7, 545–553. [Google Scholar] [CrossRef]
- Oliveira, J.A.; Silva, R.R.M.; Da Silva, G.T.S.T.; Torres, J.A.; Vali, A.; Ribeiro, C.; Rajeshwar, K.; Ruotolo, L.A.M. Copper Vanadates: Targeted Synthesis of Two Pure Phases and Use in a Photoanode/Cathode Setup for Selective Photoelectrochemical Conversion of Carbon Dioxide to Liquid Fuel. Mater. Res. Bull. 2022, 149, 111716. [Google Scholar] [CrossRef]
- Bhattacharjee, S.; Rahaman, M.; Andrei, V.; Miller, M.; Rodríguez-Jiménez, S.; Lam, E.; Pornrungroj, C.; Reisner, E. Photoelectrochemical CO2-to-Fuel Conversion with Simultaneous Plastic Reforming. Nat. Synth. 2023, 2, 182–192. [Google Scholar] [CrossRef]
- Bierbaumer, S.; Nattermann, M.; Schulz, L.; Zschoche, R.; Erb, T.J.; Winkler, C.K.; Tinzl, M.; Glueck, S.M. Enzymatic Conversion of CO2: From Natural to Artificial Utilization. Chem. Rev. 2023, 123, 5702–5754. [Google Scholar] [CrossRef]
- Nabavi Zadeh, P.S.; Zezzi Do Valle Gomes, M.; Åkerman, B.; Palmqvist, A.E.C. Förster Resonance Energy Transfer Study of the Improved Biocatalytic Conversion of CO2 to Formaldehyde by Coimmobilization of Enzymes in Siliceous Mesostructured Cellular Foams. ACS Catal. 2018, 8, 7251–7260. [Google Scholar] [CrossRef]
- Oliveira, A.R.; Mota, C.; Mourato, C.; Domingos, R.M.; Santos, M.F.A.; Gesto, D.; Guigliarelli, B.; Santos-Silva, T.; Romão, M.J.; Cardoso Pereira, I.A. Toward the Mechanistic Understanding of Enzymatic CO2 Reduction. ACS Catal. 2020, 10, 3844–3856. [Google Scholar] [CrossRef]
- Zezzi Do Valle Gomes, M.; Masdeu, G.; Eiring, P.; Kuhlemann, A.; Sauer, M.; Åkerman, B.; Palmqvist, A.E.C. Improved Biocatalytic Cascade Conversion of CO2 to Methanol by Enzymes Co-Immobilized in Tailored Siliceous Mesostructured Cellular Foams. Catal. Sci. Technol. 2021, 11, 6952–6959. [Google Scholar] [CrossRef]
- Cobb, S.J.; Badiani, V.M.; Dharani, A.M.; Wagner, A.; Zacarias, S.; Oliveira, A.R.; Pereira, I.A.C.; Reisner, E. Fast CO2 Hydration Kinetics Impair Heterogeneous but Improve Enzymatic CO2 Reduction Catalysis. Nat. Chem. 2022, 14, 417–424. [Google Scholar] [CrossRef] [PubMed]
- Chafik, A.; El Hassani, K.; Essamadi, A.; Çelik, S.Y.; Mavi, A. Efficient Sequestration of Carbon Dioxide into Calcium Carbonate Using a Novel Carbonic Anhydrase Purified from Liver of Camel (Camelus Dromedarius). J. CO2 Util. 2020, 42, 101310. [Google Scholar] [CrossRef]
- Sharma, T.; Sharma, A.; Xia, C.L.; Lam, S.S.; Khan, A.A.; Tripathi, S.; Kumar, R.; Gupta, V.K.; Nadda, A.K. Enzyme Mediated Transformation of CO2 into Calcium Carbonate Using Purified Microbial Carbonic Anhydrase. Environ. Res. 2022, 212, 113538. [Google Scholar] [CrossRef]
- Sharkey, T.D. The Discovery of Rubisco. J. Exp. B. 2023, 74, 510–519. [Google Scholar] [CrossRef]
- Pang, J.-J.; Shin, J.-S.; Li, S.-Y. The Catalytic Role of RuBisCO for in Situ CO2 Recycling in Escherichia Coli. Front. Bioeng. Biotechnol. 2020, 8, 543807. [Google Scholar] [CrossRef]
- Chen, T.; Riaz, S.; Davey, P.; Zhao, Z.; Sun, Y.; Dykes, G.F.; Zhou, F.; Hartwell, J.; Lawson, T.; Nixon, P.J. Producing Fast and Active Rubisco in Tobacco to Enhance Photosynthesis. Plant J. 2023, 35, 795–807. [Google Scholar] [CrossRef]
- Behera, D.; Swain, A.; Karmakar, S.; Dash, M.; Swain, P.; Baig, M.J.; Molla, K.A. Overexpression of Setaria Italica Phosphoenolpyruvate Carboxylase Gene in Rice Positively Impacts Photosynthesis and Agronomic Traits. Plant Physiol. Biochem. 2023, 194, 169–181. [Google Scholar] [CrossRef]
- Hu, J.; Wang, D.; Chen, H.; Wang, Q. Advances in Genetic Engineering in Improving Photosynthesis and Microalgal Productivity. Int. J. Mol. Sci. 2023, 24, 1898. [Google Scholar] [CrossRef]
- Cardona, T.; Shao, S.; Nixon, P.J. Enhancing Photosynthesis in Plants: The Light Reactions. Essays Biochem. 2018, 62, 85–94. [Google Scholar] [CrossRef]
- Perrine, Z.; Negi, S.; Sayre, R.T. Optimization of Photosynthetic Light Energy Utilization by Microalgae. Algal Res. 2012, 1, 134–142. [Google Scholar] [CrossRef]
- Trovão, M.; Schüler, L.M.; Machado, A.; Bombo, G.; Navalho, S.; Barros, A.; Pereira, H.; Silva, J.; Freitas, F.; Varela, J. Random Mutagenesis as a Promising Tool for Microalgal Strain Improvement towards Industrial Production. Mar. Drugs 2022, 20, 440. [Google Scholar] [CrossRef] [PubMed]
- Yau, M.C.M.; Hayes, M.; Kalathil, S. Biocatalytic Conversion of Sunlight and Carbon Dioxide to Solar Fuels and Chemicals. RSC Adv. 2022, 12, 16396–16411. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, F.A.; Hirst, J. Reversibility and Efficiency in Electrocatalytic Energy Conversion and Lessons from Enzymes. Proc. Natl. Acad. Sci. USA 2011, 108, 14049–14054. [Google Scholar] [CrossRef]
- Vogt, L.; Vinyard, D.J.; Khan, S.; Brudvig, G.W. Oxygen-Evolving Complex of Photosystem II: An Analysis of Second-Shell Residues and Hydrogen-Bonding Networks. Curr. Opin. Chem. Biol. 2015, 25, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Vasiliu, T.; Li, F.; Laaksonen, A.; Mocci, F.; Ji, X. Electrochemically Driven Efficient Enzymatic Conversion of CO2 to Formic Acid with Artificial Cofactors. J. CO2 Util. 2021, 52, 101679. [Google Scholar] [CrossRef]
- Di Spiridione, C.; Aresta, M.; Dibenedetto, A. Improving the Enzymatic Cascade of Reactions for the Reduction of CO2 to CH3OH in Water: From Enzymes Immobilization Strategies to Cofactor Regeneration and Cofactor Suppression. Molecules 2022, 27, 4913. [Google Scholar] [CrossRef]
- Song, J.; Lin, H.; Zhao, G.; Huang, X. Photocatalytic Material-Microorganism Hybrid System and Its Application—A Review. Micromachines 2022, 13, 861. [Google Scholar] [CrossRef]
- Ananyev, G.; Dismukes, G.C. How Fast Can Photosystem II Split Water? Kinetic Performance at High and Low Frequencies. Photosynth. Res. 2005, 84, 355–365. [Google Scholar] [CrossRef]
- Zhang, P.; Dai, S.Y.; Yuan, J.S. Producing the “Molecules of Life” from CO2 through Hybrid Catalytic Relay. Chem 2021, 7, 3200–3202. [Google Scholar] [CrossRef]
- Lee, S.H.; Choi, D.S.; Kuk, S.K.; Park, C.B. Photobiocatalysis: Activating Redox Enzymes by Direct or Indirect Transfer of Photoinduced Electrons. Angew. Chem. Int. Ed. 2018, 57, 7958–7985. [Google Scholar] [CrossRef]
- Cai, R.; Minteer, S.D. Nitrogenase Bioelectrocatalysis: From Understanding Electron-Transfer Mechanisms to Energy Applications. ACS Energy Lett. 2018, 3, 2736–2742. [Google Scholar] [CrossRef]
- Proppe, A.H.; Li, Y.C.; Aspuru-Guzik, A.; Berlinguette, C.P.; Chang, C.J.; Cogdell, R.; Doyle, A.G.; Flick, J.; Gabor, N.M.; van Grondelle, R. Bioinspiration in Light Harvesting and Catalysis. Nat. Rev. Mater. 2020, 5, 828–846. [Google Scholar] [CrossRef]
- Li, J.; Zhu, Y.; Chen, W.; Lu, Z.; Xu, J.; Pei, A.; Peng, Y.; Zheng, X.; Zhang, Z.; Chu, S. Breathing-Mimicking Electrocatalysis for Oxygen Evolution and Reduction. Joule 2019, 3, 557–569. [Google Scholar] [CrossRef]
- Maciá-Agulló, J.A.; Corma, A.; Garcia, H. Photobiocatalysis: The Power of Combining Photocatalysis and Enzymes. Chem. Eur. J. 2015, 21, 10940–10959. [Google Scholar] [CrossRef] [PubMed]
- Tucker, J.W.; Stephenson, C.R.J. Shining Light on Photoredox Catalysis: Theory and Synthetic Applications. J. Org. Chem. 2012, 77, 1617–1622. [Google Scholar] [CrossRef]
- Alfredo, N.V.; Jalapa, N.E.; Morales, S.L.; Ryabov, A.D.; Le Lagadec, R.; Alexandrova, L. Light-Driven Living/Controlled Radical Polymerization of Hydrophobic Monomers Catalyzed by Ruthenium(II) Metalacycles. Macromolecules 2012, 45, 8135–8146. [Google Scholar] [CrossRef]
- Guo, J.-T.; Yang, D.-C.; Guan, Z.; He, Y.-H. Chlorophyll-Catalyzed Visible-Light-Mediated Synthesis of Tetrahydroquinolines from N, N -Dimethylanilines and Maleimides. J. Org. Chem. 2017, 82, 1888–1894. [Google Scholar] [CrossRef]
- Gajewska, B.; Raccio, S.; Rodriguez, K.J.; Bruns, N. Chlorophyll Derivatives as Catalysts and Comonomers for Atom Transfer Radical Polymerizations. Polym. Chem. 2019, 10, 125–135. [Google Scholar] [CrossRef]
- Gust, D.; Moore, T.A.; Moore, A.L. Solar Fuels via Artificial Photosynthesis. Acc. Chem. Res. 2009, 42, 1890–1898. [Google Scholar] [CrossRef] [PubMed]
- Steinberg-Yfrach, G.; Rigaud, J.-L.; Durantini, E.N.; Moore, A.L.; Gust, D.; Moore, T.A. Light-Driven Production of ATP Catalysed by F0F1-ATP Synthase in an Artificial Photosynthetic Membrane. Nature 1998, 392, 479–482. [Google Scholar] [CrossRef] [PubMed]
- Gandomkar, S.; Dennig, A.; Dordic, A.; Hammerer, L.; Pickl, M.; Haas, T.; Hall, M.; Faber, K. Biocatalytic Oxidative Cascade for the Conversion of Fatty Acids into α-Ketoacids via Internal H2O2 Recycling. Angew. Chem. Int. Ed. 2018, 57, 427–430. [Google Scholar] [CrossRef]
- Choi, D.S.; Lee, H.; Tieves, F.; Lee, Y.W.; Son, E.J.; Zhang, W.; Shin, B.; Hollmann, F.; Park, C.B. Bias-Free In Situ H2O2 Generation in a Photovoltaic-Photoelectrochemical Tandem Cell for Biocatalytic Oxyfunctionalization. ACS Catal. 2019, 9, 10562–10566. [Google Scholar] [CrossRef]
- Ernst, O.P.; Lodowski, D.T.; Elstner, M.; Hegemann, P.; Brown, L.S.; Kandori, H. Microbial and Animal Rhodopsins: Structures, Functions, and Molecular Mechanisms. Chem. Rev. 2014, 114, 126–163. [Google Scholar] [CrossRef]
- Rakovich, A.; Donegan, J.F.; Oleinikov, V.; Molinari, M.; Sukhanova, A.; Nabiev, I.; Rakovich, Y.P. Linear and Nonlinear Optical Effects Induced by Energy Transfer from Semiconductor Nanoparticles to Photosynthetic Biological Systems. J. Photochem. Photobiol. C Photochem. Rev. 2014, 20, 17–32. [Google Scholar] [CrossRef]
- Ge, X.; Gunner, M.R. Unraveling the Mechanism of Proton Translocation in the Extracellular Half-Channel of Bacteriorhodopsin: Proton Translocation in Bacteriorhodopsin. Proteins 2016, 84, 639–654. [Google Scholar] [CrossRef]
- Das, S.; Wu, C.; Song, Z.; Hou, Y.; Koch, R.; Somasundaran, P.; Priya, S.; Barbiellini, B.; Venkatesan, R. Bacteriorhodopsin Enhances Efficiency of Perovskite Solar Cells. ACS Appl. Mater. Interfaces 2019, 11, 30728–30734. [Google Scholar] [CrossRef]
- Mohammadpour, R.; Janfaza, S. Efficient Nanostructured Biophotovoltaic Cell Based on Bacteriorhodopsin as Biophotosensitizer. ACS Sustain. Chem. Eng. 2015, 3, 809–813. [Google Scholar] [CrossRef]
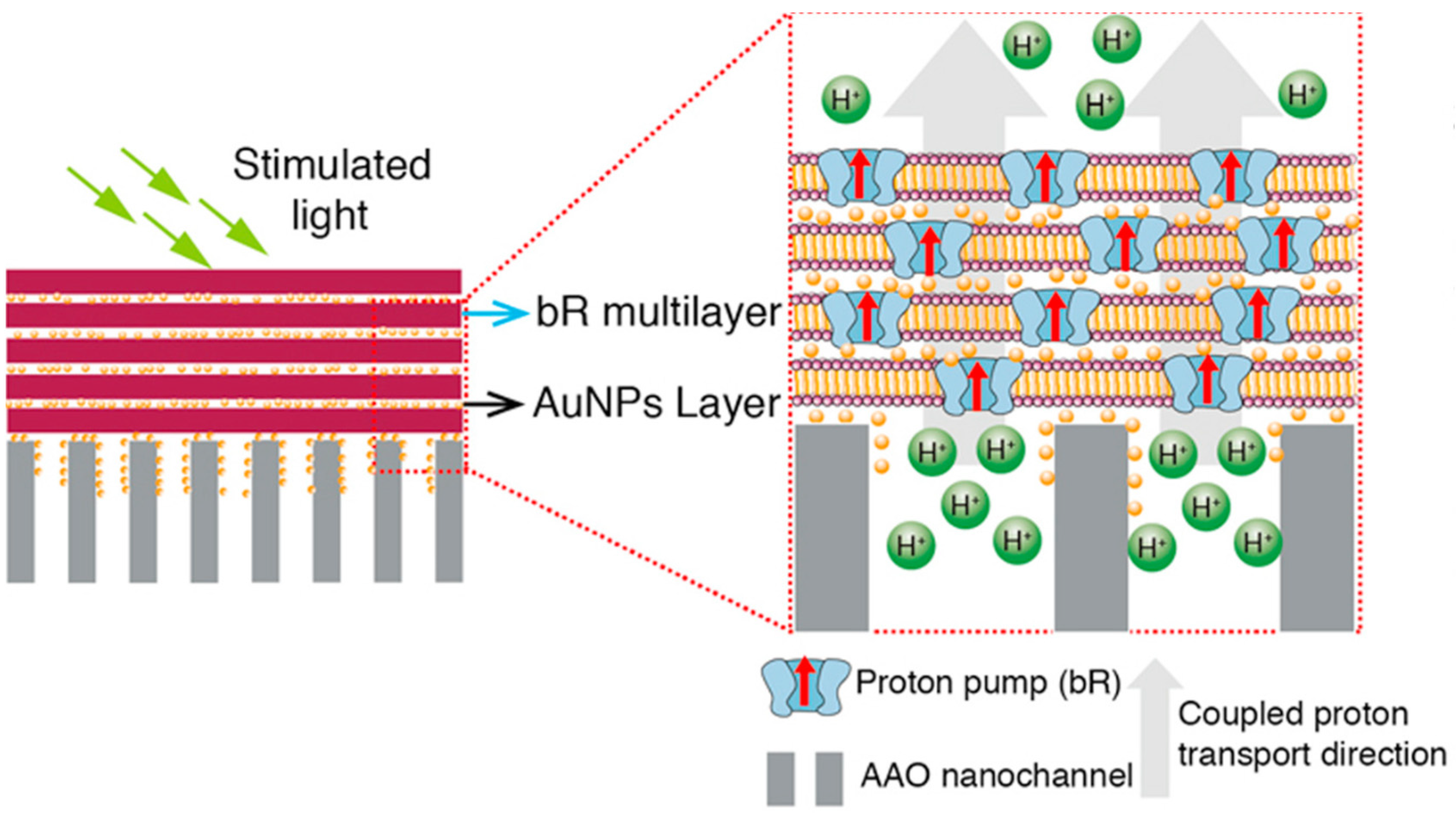
- Molaeirad, A.; Rezaeian, N. Oriented Assembly of Bacteriorhodopsin on ZnO Nanostructured Electrode for Enhanced Photocurrent Generation: Enhanced Photocurrent Generation by BR LB Film. Biotechnol. Appl. Biochem. 2015, 62, 489–493. [Google Scholar] [CrossRef]
- Johnson, K.E.; Gakhar, S.; Risbud, S.H.; Longo, M.L. Development and Characterization of Titanium Dioxide Gel with Encapsulated Bacteriorhodopsin for Hydrogen Production. Langmuir 2018, 34, 7488–7496. [Google Scholar] [CrossRef] [PubMed]
- Berhanu, S.; Ueda, T.; Kuruma, Y. Artificial Photosynthetic Cell Producing Energy for Protein Synthesis. Nat. Commun. 2019, 10, 1325. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Liang, D.; Rao, S.; Xiang, Y. Heterogeneous Bacteriorhodopsin/Gold Nanoparticle Stacks as a Photovoltaic System. Nano Energy 2015, 11, 654–661. [Google Scholar] [CrossRef]
- Krassen, H.; Schwarze, A.; Friedrich, B.; Ataka, K.; Lenz, O.; Heberle, J. Photosynthetic Hydrogen Production by a Hybrid Complex of Photosystem I and [NiFe]-Hydrogenase. ACS Nano 2009, 3, 4055–4061. [Google Scholar] [CrossRef]
- Appel, J.; Hueren, V.; Boehm, M.; Gutekunst, K. Cyanobacterial in Vivo Solar Hydrogen Production Using a Photosystem I–Hydrogenase (PsaD-HoxYH) Fusion Complex. Nat. Energy 2020, 5, 458–467. [Google Scholar] [CrossRef]
- Kanygin, A.; Smith, A.; Nagy, V.; Tóth, S.Z.; Redding, K.E. Interplay between Hydrogen Production and Photosynthesis in a Green Alga Expressing an Active Photosystem I-Hydrogenase Chimera. Int. J. Hydrogen Energy 2022, 47, 21969–21983. [Google Scholar] [CrossRef]
- Zhao, F.; Wang, P.; Ruff, A.; Hartmann, V.; Zacarias, S.; Pereira, I.A.C.; Nowaczyk, M.M.; Rögner, M.; Conzuelo, F.; Schuhmann, W. A Photosystem I Monolayer with Anisotropic Electron Flow Enables Z-Scheme like Photosynthetic Water Splitting. Energy Environ. Sci. 2019, 12, 3133–3143. [Google Scholar] [CrossRef]
- Xu, Z.; Qi, J.; Wang, S.; Liu, X.; Li, M.; Mann, S.; Huang, X. Algal Cell Bionics as a Step towards Photosynthesis-Independent Hydrogen Production. Nat. Commun. 2023, 14, 1872. [Google Scholar] [CrossRef]
- Nagakawa, H.; Takeuchi, A.; Takekuma, Y.; Noji, T.; Kawakami, K.; Kamiya, N.; Nango, M.; Furukawa, R.; Nagata, M. Efficient Hydrogen Production Using Photosystem I Enhanced by Artificial Light Harvesting Dye. Photochem. Photobiol. Sci. 2019, 18, 309–313. [Google Scholar] [CrossRef]
- Amaro-Gahete, J.; Pavliuk, M.V.; Tian, H.; Esquivel, D.; Romero-Salguero, F.J.; Ott, S. Catalytic systems mimicking the [FeFe]-hydrogenase active site for visible-light-driven hydrogen production. Coord. Chem. Rev. 2021, 448, 214172. [Google Scholar] [CrossRef]
- Liu, C.; Colón, B.C.; Ziesack, M.; Silver, P.A.; Nocera, D.G. Water Splitting–Biosynthetic System with CO2 Reduction Efficiencies Exceeding Photosynthesis. Science 2016, 352, 1210–1213. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Jiang, Z.; Yu, J.C.; Wang, J.; Wong, P.K. Enhanced CO2 Reduction and Valuable C2+ Chemical Production by a CdS-Photosynthetic Hybrid System. Nanoscale 2019, 11, 9296–9301. [Google Scholar] [CrossRef] [PubMed]
- Alcala-Torano, R.; Halloran, N.; Gwerder, N.; Sommer, D.J.; Ghirlanda, G. Light-Driven CO2 Reduction by Co-Cytochrome B562. Front. Mol. Biosci. 2021, 8, 609654. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Kolb, M.J.; Göttle, A.J.; Koper, M.T.M. DFT Study on the Mechanism of the Electrochemical Reduction of CO2 Catalyzed by Cobalt Porphyrins. J. Phys. Chem. C 2016, 120, 15714–15721. [Google Scholar] [CrossRef]
- Call, A.; Cibian, M.; Yamamoto, K.; Nakazono, T.; Yamauchi, K.; Sakai, K. Highly Efficient and Selective Photocatalytic CO2 Reduction to CO in Water by a Cobalt Porphyrin Molecular Catalyst. ACS Catal. 2019, 9, 4867–4874. [Google Scholar] [CrossRef]
- Zhang, P.; Chen, K.; Xu, B.; Li, J.; Hu, C.; Yuan, J.S.; Dai, S.Y. Chem-Bio Interface Design for Rapid Conversion of CO2 to Bioplastics in an Integrated System. Chem 2022, 8, 3363–3381. [Google Scholar] [CrossRef]
- Cai, T.; Sun, H.; Qiao, J.; Zhu, L.; Zhang, F.; Zhang, J.; Tang, Z.; Wei, X.; Yang, J.; Yuan, Q.; et al. Cell-Free Chemoenzymatic Starch Synthesis from Carbon Dioxide. Science 2021, 373, 1523–1527. [Google Scholar] [CrossRef] [PubMed]
- Pullen, S.; Fei, H.; Orthaber, A.; Cohen, S.M.; Ott, S. Enhanced Photochemical Hydrogen Production by a Molecular Diiron Catalyst Incorporated into a Metal–Organic Framework. J. Am. Chem. Soc. 2013, 135, 16997–17003. [Google Scholar] [CrossRef]
- Roy, S.; Pascanu, V.; Pullen, S.; Miera, G.G.; Martín-Matute, B.; Ott, S. Catalyst accessibility to chemical reductants in metal–organic frameworks. Chem. Commun. 2017, 53, 3257. [Google Scholar] [CrossRef]
- Wang, W.; Yu, T.; Zeng, Y.; Chen, J.; Yang, G.; Li, Y. Enhanced photocatalytic hydrogen production from an MCM-41-immobilized photosensitizer–[Fe–Fe] hydrogenase mimic dyad. Photochem. Photobiol. Sci. 2014, 13, 1590–1597. [Google Scholar] [CrossRef]
- Cao, M.; Wang, Z.; Zhang, J.; Xu, S.; Zhang, S.; Dai, X.; Jiang, X. Preparation, characterization and photocatalytic properties of diiron mimic modified Nano Silica. Inorg. Chim. Acta 2018, 469, 402–407. [Google Scholar] [CrossRef]
- Himiyama, T.; Waki, M.; Esquivel, D.; Onoda, A.; Hayashi, T.; Van Der Voort, P.; Inagaki, S. A Heterogeneous Hydrogen-Evolution Catalyst Based on a Mesoporous Organosilica with a Diiron Catalytic Center Modelling [FeFe]-Hydrogenase. ChemCatChem 2018, 10, 4894–4899. [Google Scholar] [CrossRef]
- Navarro, M.Á.; Cosano, D.; Bhunia, A.; Simonelli, L.; Martin-Diaconescu, V.; Romero-Salguero, F.J.; Esquivel, D. Cobaloxime tethered pyridine-functionalized ethylene-bridged periodic mesoporous organosilica as an efficient HER catalyst. Sustain. Energy Fuels 2022, 6, 398–407. [Google Scholar] [CrossRef]
- Rojas-Luna, R.; Castillo-Rodríguez, M.; Ruiz, J.R.; Jiménez-Sanchidrián, C.; Esquivel, D.; Romero-Salguero, F.J. Ru- and Ir-complex decorated periodic mesoporous organosilicas as sensitizers for artificial photosynthesis. Dalton Trans. 2022, 51, 18708–18721. [Google Scholar] [CrossRef] [PubMed]
- Amaro-Gahete, J.; Esquivel, D.; Pavliuk, M.V.; Jiménez-Sanchidrián, C.; Tian, H.; Ott, S.; Romero-Salguero, F.J. Hydroxyl-Decorated Diiron Complex as a [FeFe]-Hydrogenase Active Site Model Complex: Light-Driven Photocatalytic Activity and Heterogenization on Ethylene-Bridged Periodic Mesoporous Organosilica. Catalysts 2022, 12, 254. [Google Scholar] [CrossRef]
- Rojas-Luna, R.; Amaro-Gahete, J.; Gil-Gavilán, D.G.; Castillo-Rodríguez, M.; Jiménez-Sanchidrián, C.; Ruiz, J.R.; Esquivel, D.; Romero-Salguero, F.J. Visible-light-harvesting basolite-A520 metal organic framework for photocatalytic hydrogen evolution. Microporous Mesoporous Mater. 2023, 355, 112565. [Google Scholar] [CrossRef]
- Liang, S.; Zhong, X.; Zhong, Z.; Han, B.; Chen, W.; Song, K.; Deng, H.; Lin, Z. Biomimetic inspired porphyrin-based nanoframes for highly efficient photocatalytic CO2 reduction. Chem. Eng. J. 2021, 411, 128414. [Google Scholar] [CrossRef]
























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Natural Photosynthesis | Artificial Photosynthesis | Reference | |
|---|---|---|---|
| Energy Source | Sunlight | Sunlight | [1,2] |
| Reaction Center | Chlorophyll in photosystem | Photo-electrochemical cells | [1,2,3] |
| Energy Storage | Glucose (a carbohydrate) | Hydrogen or other solar fuels | [2,3,4] |
| Oxygen Evolution | Yes, from water | Yes, from water | [1,2,3,4,5,6] |
| Carbon Fixation | Yes, carbon dioxide into glucose | Potentially, carbon dioxide into carbon-based fuels | [3,4,5,6,7] |
| Efficiency | 3–6% | Variable, still under development | [2,4] |
| Product Utility | Mainly food and biomass | Mainly fuels for energy and industry | [1,2,7,8] |
| Environmental Impact | No negative impact, reduces CO2 | No negative impact, could reduce CO2 | [3,5] |
| Catalysts | Enzymes | Man-made catalysts | [1,2,3,8] |
| Rate of Reaction | Relatively slow because of enzymatic constraints | Potentially faster with optimized catalysts | [1,3,9] |
| Operating Conditions | Ambient temperature and pressure | Variable, can be optimized for reaction | [1,2,5,7] |
| Evolution and Optimization | Billions of years of natural selection | Still under development, ongoing optimization | [1,2,3,4,5,6] |
| Dependence on Water | High, water is electron donor | High, water often used for proton/electron source | [6,7,8,10] |
| Lifetime/Durability | Limited by organism’s lifespan | Potentially long, dependent on material degradation | [8,9,10] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Machín, A.; Cotto, M.; Ducongé, J.; Márquez, F. Artificial Photosynthesis: Current Advancements and Future Prospects. Biomimetics 2023, 8, 298. https://doi.org/10.3390/biomimetics8030298
Machín A, Cotto M, Ducongé J, Márquez F. Artificial Photosynthesis: Current Advancements and Future Prospects. Biomimetics. 2023; 8(3):298. https://doi.org/10.3390/biomimetics8030298
Chicago/Turabian StyleMachín, Abniel, María Cotto, José Ducongé, and Francisco Márquez. 2023. "Artificial Photosynthesis: Current Advancements and Future Prospects" Biomimetics 8, no. 3: 298. https://doi.org/10.3390/biomimetics8030298
APA StyleMachín, A., Cotto, M., Ducongé, J., & Márquez, F. (2023). Artificial Photosynthesis: Current Advancements and Future Prospects. Biomimetics, 8(3), 298. https://doi.org/10.3390/biomimetics8030298