Bioisosteres of Carbohydrate Functional Groups in Glycomimetic Design


Abstract
1. Introduction
1.1. General Strategies for Glycomimetic Synthesis
1.2. Affinity Enhancement Versus Enzyme Activity
2. Modifications to the O-Glycoside Linkage
3. Replacement of the Endocyclic O Atom
4. Replacement of OH Functional Groups
4.1. Deoxygenation
4.2. Deoxyfluorination
4.3. Methyl Etherification
4.4. SH and SeH Substitution
4.5. Other Modifications
5. Replacement of H Atoms
5.1. Fluorination
5.2. Other Modifications
6. Replacement of NHAc Substituents
6.1. C-Derivatives
6.2. Other Substitutions
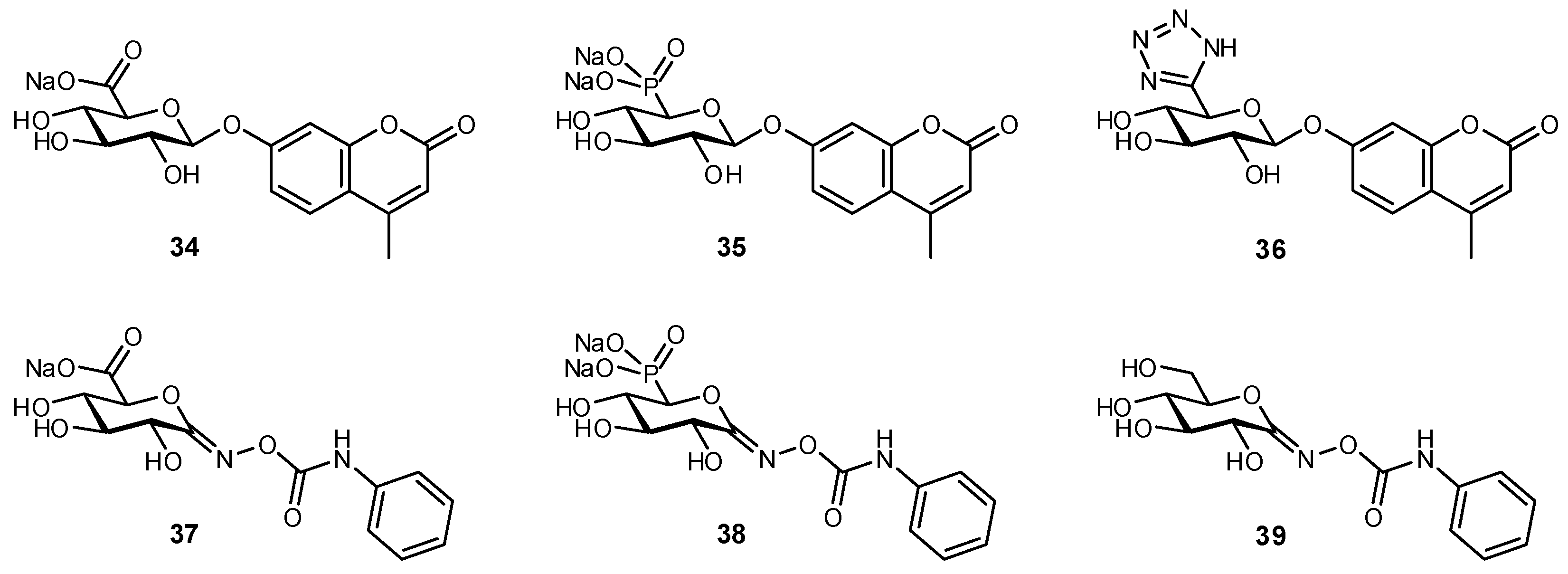
7. Replacement of Charged Substituents
8. Conclusions
Funding
Conflicts of Interest
References
- Nieuwdorp, M.; Meuwese, M.C.; Mooij, H.L.; Ince, C.; Broekhuizen, L.N.; Kastelein, J.J.P.; Stroes, E.S.G.; Vink, H. Measuring endothelial glycocalyx dimensions in humans: A potential novel tool to monitor vascular vulnerability. J. Appl. Physiol. 2008, 104, 845–852. [Google Scholar] [CrossRef] [PubMed]
- Van Teeffelen, J.W.; Brands, J.; Stroes, E.S.; Vink, H. Endothelial glycocalyx: Sweet shield of blood vessels. Trends Cardiovasc. Med. 2007, 17, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Reily, C.; Stewart, T.J.; Renfrow, M.B.; Novak, J. Glycosylation in health and disease. Nat. Rev. Nephrol. 2019, 15, 346–366. [Google Scholar] [CrossRef] [PubMed]
- Ohtsubo, K.; Marth, J.D. Glycosylation in cellular mechanisms of health and disease. Cell 2006, 126, 855–867. [Google Scholar] [CrossRef] [PubMed]
- Ashkani, J.; Naidoo, K.J. Glycosyltransferase gene expression profiles classify cancer types and propose prognostic subtypes. Sci. Rep. 2016, 6, 26451. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Abrahams, J.P.; Skinner, R.; Petitou, M.; Pike, R.N.; Carrell, R.W. The anticoagulant activation of antithrombin by heparin. Proc. Natl. Acad. Sci. USA 1997, 94, 14683–14688. [Google Scholar] [CrossRef] [PubMed]
- Quiocho, F.A. Probing the atomic interactions between proteins and carbohydrates. Biochem. Soc. Trans. 1993, 21, 442–448. [Google Scholar] [CrossRef]
- Sears, P.; Wong, C.-H. Carbohydrate mimetics: A new strategy for tackling the problem of carbohydrate-mediated biological recognition. Angew. Chem. Int. Ed. 1999, 38, 2300–2324. [Google Scholar] [CrossRef]
- Schön, A.; Freire, E. Thermodynamics of intersubunit interactions in cholera toxin upon binding to the oligosaccharide portion of its cell surface receptor, ganglioside GM1. Biochemistry 1989, 28, 5019–5024. [Google Scholar] [CrossRef]
- Mammen, M.; Choi, S.-K.; Whitesides, G.M. Polyvalent interactions in biological systems: Implications for design and use of multivalent ligands and inhibitors. Angew. Chem. Int. Ed. 1998, 37, 2754–2794. [Google Scholar] [CrossRef]
- Müller, C.; Despras, G.; Lindhorst, T.K. Organizing multivalency in carbohydrate recognition. Chem. Soc. Rev. 2016, 45, 3275–3302. [Google Scholar] [CrossRef]
- Multivalency: Concepts, Research & Applications; Huskens, J., Prins, L.J., Haag, R., Ravoo, B.J., Eds.; Wiley: Hoboken, NJ, USA, 2018. [Google Scholar]
- Kiessling, L.L.; Young, T.; Gruber, T.D.; Mortell, K.H. Multivalency in protein-carbohydrate recognition. In Glycoscience: Chemistry and Chemical Biology, 2nd ed.; Fraser-Reid, B.O., Tatsuta, K., Thiem, J., Eds.; Springer: Berlin, Germany, 2008; Volume 1, pp. 2483–2524. [Google Scholar]
- Smith, D.C.; Lord, J.M.; Roberts, L.M.; Johannes, L. Glycosphingolipids as toxin receptors. Semin. Cell Dev. Biol. 2004, 15, 397–408. [Google Scholar] [CrossRef]
- Gamblin, S.J.; Skehel, J.J. Influenza hemagglutinin and neuraminidase membrane glycoproteins. J. Biol. Chem. 2010, 285, 28403–28409. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliver. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Ernst, B.; Magnani, J.L. From carbohydrate leads to glycomimetic drugs. Nat. Rev. Drug Discov. 2009, 8, 661–677. [Google Scholar] [CrossRef]
- Carbohydrate-Based Drug Discovery; Wong, C.-H., Ed.; Wiley VCH: Weinheim, Germany, 2003. [Google Scholar] [CrossRef]
- Kim, C.U.; Lew, W.; Williams, M.A.; Liu, H.; Zhang, L.; Swaminathan, S.; Bischofberger, N.; Chen, M.S.; Mendel, D.B.; Tai, C.Y.; et al. Influenza neuraminidase inhibitors possessing a novel hydrophobic interaction in the enzyme active site: Design, synthesis, and structural analysis of carbocyclic sialic acid analogues with potent anti-influenza activity. J. Am. Chem. Soc. 1997, 119, 681–690. [Google Scholar] [CrossRef]
- McClellan, K.; Perry, C.M. Oseltamivir - a review of its use in influenza. Drugs 2001, 61, 263–283. [Google Scholar] [CrossRef]
- von Itzstein, M.; Wu, W.-Y.; Kok, G.B.; Pegg, M.S.; Dyason, J.C.; Jin, B.; Phan, T.V.; Smythe, M.L.; White, H.F.; Oliver, S.W.; et al. Rational design of potent sialidase-based inhibitors of influenza virus replication. Nature 1993, 363, 418–423. [Google Scholar] [CrossRef]
- Hevey, R. Strategies for the development of glycomimetic drug candidates. Pharmaceuticals 2019, 12, 55. [Google Scholar] [CrossRef]
- Chang, J.; Patton, J.T.; Sarkar, A.; Ernst, B.; Magnani, J.L.; Frenette, P.S. GMI-1070, a novel pan-selectin antagonist, reverses acute vascular occlusions in sickle cell mice. Blood 2010, 116, 1779–1786. [Google Scholar] [CrossRef]
- Dugger, S.A.; Platt, A.; Goldstein, D.B. Drug development in the era of precision medicine. Nat. Rev. Drug Discov. 2018, 17, 183–196. [Google Scholar] [CrossRef]
- Kolb, H.C.; Ernst, B. Development of tools for the design of selectin antagonists. Chem. Eur. J. 1997, 3. [Google Scholar] [CrossRef]
- Thoma, G.; Magnani, J.L.; Patton, J.T.; Ernst, B.; Jahnke, W. Preorganization of the bioactive conformation of sialyl LewisX analogues correlates with their affinity to E-selectin. Angew. Chem. 2001, 113, 1995–1999. [Google Scholar] [CrossRef]
- García-Moreno, M.I.; Ortega-Caballero, F.; Rísquez-Cuadro, R.; Ortiz Mellet, C.; García Fernandez, J.M. The impact of heteromultivalency in lectin recognition and glycosidase inhibition: An integrated mechanistic study. Chem. Eur. J. 2017, 23, 6295–6304. [Google Scholar] [CrossRef]
- Ordanini, S.; Varga, N.; Porkolab, V.; Thépaut, M.; Belvisi, L.; Bertaglia, A.; Palmioli, A.; Berzi, A.; Trabattoni, D.; Clerici, M.; et al. Designing nanomolar antagonists of DC-SIGN-mediated HIV infection: Ligand presentation using molecular rods. Chem. Commun. 2015, 51, 3816–3819. [Google Scholar] [CrossRef]
- Berzi, A.; Ordanini, S.; Joosten, B.; Trabattoni, D.; Cambi, A.; Bernardi, A.; Clerici, M. Pseudo-mannosylated DC-SIGN ligands as immunomodulants. Sci. Rep. 2016, 6, 35373. [Google Scholar] [CrossRef]
- Compain, P. Multivalent effect in glycosidase inhibition: The end of the beginning. Chem. Rec. 2019, 19, 1–14. [Google Scholar] [CrossRef]
- Hevey, R.; Ling, C.-C. Recent advances in developing synthetic carbohydrate-based vaccines for cancer immunotherapies. Future Med. Chem. 2012, 4, 545–584. [Google Scholar] [CrossRef]
- Biffinger, J.C.; Kim, H.W.; DiMagno, S.G. The polar hydrophobicity of fluorinated compounds. ChemBioChem 2004, 5, 622–627. [Google Scholar] [CrossRef]
- Withers, S.G.; MacLennan, D.J.; Street, I.P. The synthesis and hydrolysis of a series of deoxyfluoro-D-glucopyranosyl phosphates. Carbohydr. Res. 1986, 154, 127–144. [Google Scholar] [CrossRef]
- Han, Z.; Pinkner, J.S.; Ford, B.; Obermann, R.; Nolan, W.; Wildman, S.A.; Hobbs, D.; Ellenberger, T.; Cusumano, C.K.; Hultgren, S.J.; et al. Structure-based drug design and optimization of mannose bacterial FimH antagonists. J. Med. Chem. 2010, 53, 4779–4792. [Google Scholar] [CrossRef]
- Bouckaert, J.; Berglund, J.; Schembri, M.; De Genst, E.; Cools, L.; Wuhrer, M.; Hung, C.-S.; Pinkner, J.; Slättegård, R.; Zavialov, A.; et al. Receptor binding studies disclose a novel class of high-affinity inhibitors of the Escherichia coli FimH adhesin. Mol. Microbiol. 2005, 55, 441–455. [Google Scholar] [CrossRef]
- Sperling, O.; Fuchs, A.; Lindhorst, T.K. Evaluation of the carbohydrate recognition domain of the bacterial adhesin FimH: Design, synthesis and binding properties of mannoside ligands. Org. Biomol. Chem. 2006, 4, 3913–3922. [Google Scholar] [CrossRef]
- Klein, T.; Abgottspon, D.; Wittwer, M.; Rabbani, S.; Herold, J.; Jiang, X.; Kleeb, S.; Lüthi, C.; Scharenberg, M.; Bezençon, J.; et al. FimH antagonists for the oral treatment of urinary tract infections: From design and synthesis to in vitro and in vivo evaluation. J. Med. Chem. 2010, 53, 8627–8641. [Google Scholar] [CrossRef]
- Hevey, R.; Ling, C.-C. Conjugation strategies used for the preparation of carbohydrate-conjugate vaccines. In Chemistry of Bioconjugates: Synthesis, Characterization, and Biomedical Applications; Narain, R., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013. [Google Scholar] [CrossRef]
- Krug, L.M.; Ragupathi, G.; Ng, K.K.; Hood, C.; Jennings, H.J.; Guo, Z.; Kris, M.G.; Miller, V.; Pizzo, B.; Tyson, L.; et al. Vaccination of small cell lung cancer patients with polysialic acid or N-propionylated polysialic acid conjugated to keyhole limpet hemocyanin. Clin. Cancer Res. 2004, 10, 916–923. [Google Scholar] [CrossRef]
- Sahabuddin, S.; Chang, T.-C.; Lin, C.-C.; Jan, F.-D.; Hsiao, H.-Y.; Huang, K.-T.; Chen, J.-H.; Horng, J.-C.; Ho, J.A.; Lin, C.-C. Synthesis of N-modified sTn analogs and evaluation of their immunogenicities by microarray-based immunoassay. Tetrahedron 2010, 66, 7510–7519. [Google Scholar] [CrossRef]
- Yang, Y.; Yu, B. Recent advances in the chemical synthesis of C-glycosides. Chem. Rev. 2017, 117, 12281–12356. [Google Scholar] [CrossRef]
- Ati, J.; Lafite, P.; Daniellou, R. Enzymatic synthesis of glycosides: From natural O- and N-glycosides to rare C- and S-glycosides. Beilstein J. Org. Chem. 2017, 13, 1857–1865. [Google Scholar] [CrossRef]
- Horne, G.; Wilson, F.X. Therapeutic applications of iminosugars: Current perspectives and future opportunities. In Progress in Medicinal Chemistry; Lawton, G., Witty, D.R., Eds.; Elsevier: Oxford, UK, 2011; Volume 50, pp. 135–176. [Google Scholar]
- Sánchez-Fernández, E.M.; Rísquez-Cuadro, R.; Ortiz Mellet, C.; García Fernández, J.M.; Nieto, P.M.; Angulo, J. sp2-Iminosugar O-, S-, and N-glycosides as conformational mimics of α-linked disaccharides; Implications for glycosidase inhibition. Chem. Eur. J. 2012, 18, 8527–8539. [Google Scholar] [CrossRef]
- Dondoni, A.; Catozzi, N.; Marra, A. Concise and practical synthesis of C-glycosyl ketones from sugar benzothiazoles and their transformation into chiral tertiary alcohols. J. Org. Chem. 2005, 70, 9257–9268. [Google Scholar] [CrossRef]
- Mydock-McGrane, L.; Cusumano, Z.; Han, Z.; Binkley, J.; Kostakioti, M.; Hannan, T.; Pinkner, J.S.; Klein, R.; Kalas, V.; Crowley, J.; et al. Antivirulence C-mannosides as antibiotic-sparing, oral therapeutics for urinary tract infections. J. Med. Chem. 2016, 59, 9390–9408. [Google Scholar] [CrossRef]
- Chalopin, T.; Alvarez Dorta, D.; Sivignon, A.; Caudan, M.; Dumych, T.I.; Bilyy, R.O.; Deniaud, D.; Barnich, N.; Bouckaert, J.; Gouin, S.G. Second generation of thiazolylmannosides, FimH antagonists for E. coli-induced Crohn’s disease. Org. Biomol. Chem. 2016, 14, 3913–3925. [Google Scholar] [CrossRef]
- Illyés, T.-Z.; Balla, S.; Bényei, A.; Kumar, A.A.; Timári, I.; Kövér, K.E.; Szilágyi, L. Exploring the syntheses of novel glycomimetics. Carbohydrate derivatives with Se-S- or Se-Se- glycosidic linkages. ChemistrySelect 2016, 1, 2383–2388. [Google Scholar] [CrossRef]
- Zhu, F.; O’Neill, S.; Rodriguez, J.; Walczak, M.A. Stereoretentive reactions at the anomeric position: Synthesis of selenoglycosides. Angew. Chem. Int. Ed. 2018, 57, 7091–7095. [Google Scholar] [CrossRef]
- Hirai, G.; Watanabe, T.; Yamaguchi, K.; Miyagi, T.; Sodeoka, M. Stereocontrolled and convergent entry to CF2-sialosides: Synthesis of CF2-linked ganglioside GM4. J. Am. Chem. Soc. 2007, 129, 15420–15421. [Google Scholar] [CrossRef]
- Asensio, J.L.; Espinosa, J.F.; Dietrich, H.; Cañada, F.J.; Schmidt, R.R.; Martín-Lomas, M.; André, S.; Gabius, H.-J.; Jiménez-Barbero, J. Bovine heart galectin-1 selects a unique (syn) conformation of C-lactose, a flexible lactose analogue. J. Am. Chem. Soc. 1999, 121, 8995–9000. [Google Scholar] [CrossRef]
- Asensio, J.L.; Cañada, F.J.; Cheng, X.; Khan, N.; Mootoo, D.R.; Jiménez-Barbero, J. Conformational differences between O- and C-glycosides: The α-O-Man-(1➝1)-β-Gal/α-C-Man-(1➝1)-β-Gal case - a decisive demonstration of the importance of the exo-anomeric effect on the conformation of glycosides. Chem. Eur. J. 2000, 6, 1035–1041. [Google Scholar] [CrossRef]
- Espinosa, J.-F.; Bruix, M.; Jarreton, O.; Skrydstrup, T.; Beau, J.-M.; Jiménez-Barbero, J. Conformational differences between C- and O-glycosides: The α-C-mannobiose/α-O-mannobiose case. Chem. Eur. J. 1999, 5, 442–448. [Google Scholar] [CrossRef]
- O’’Hagan, D.; Rzepa, H.S. Some influences of fluorine in bioorganic chemistry. Chem. Commun. 1997, 1997, 645–652. [Google Scholar] [CrossRef]
- Berber, H.; Brigaud, T.; Lefebvre, O.; Plantier-Royon, R.; Portella, C. Reactions of difluoroenoxysilanes with glycosyl donors: Synthesis of difluoro-C-glycosides and difluoro-C-disaccharides. Chem. Eur. J. 2001, 7, 903–909. [Google Scholar] [CrossRef]
- Moreno, B.; Quehen, C.; Rose-Hélène, M.; Leclerc, E.; Quirion, J.-C. Addition of difluoromethyl radicals to glycals: A new route to alpha-CF2-D-glycosides. Org. Lett. 2007, 9, 2477–2480. [Google Scholar] [CrossRef]
- Poulain, F.; Serre, A.-L.; Lalot, J.; Leclerc, E.; Quirion, J.-C. Synthesis of α-CF2-mannosides and their conversion to fluorinated pseudoglycopeptides. J. Org. Chem. 2008, 73, 2435–2438. [Google Scholar] [CrossRef]
- Mukherjee, C.; Ghosh, S.; Nandi, P.; Sen, P.C.; Misra, A.K. Efficient synthesis of (6-deoxy-glycopyranosid-6-yl) sulfone derivatives and their effect on Ca2+-ATPase. Eur. J. Med. Chem. 2010, 45, 6012–6019. [Google Scholar] [CrossRef]
- Cumpstey, I.; Ramstadius, C.; Akhtar, T.; Goldstein, I.J.; Winter, H.C. Non-glycosidically linked pseudodisaccharides: Thioethers, sulfoxides, sulfones, ethers, selenoethers, and their binding to lectins. Eur. J. Org. Chem. 2010, 2010, 1951–1970. [Google Scholar] [CrossRef]
- Barbaud, C.; Bols, M.; Lundt, I. Synthesis of the first pseudosugar-C-disaccharide. A potential antigen for eliciting glycoside-bond forming antibodies with catalytic groups. Tetrahedron 1995, 51, 9063–9078. [Google Scholar] [CrossRef]
- Céspedes Dávila, M.F.; Schneider, J.P.; Godard, A.; Hazelard, D.; Compain, P. One-pot, highly stereoselective synthesis of dithioacetal-α,α-diglycosides. Molecules 2018, 23, 914. [Google Scholar] [CrossRef]
- Borges de Melo, E.; da Silveira Gomes, A.; Carvalho, I. α- and β-glucosidase inhibitors: Chemical structure and biological activity. Tetrahedron 2006, 62, 10277–10302. [Google Scholar] [CrossRef]
- Drueckhammer, D.G.; Wong, C.-H. Chemoenzymatic syntheses of fluoro sugar phosphates and analogues. J. Org. Chem. 1985, 50, 5912–5913. [Google Scholar] [CrossRef]
- Mehta, S.; Andrews, J.S.; Johnston, B.D.; Pinto, B.M. Novel heteroanalogues of methyl maltoside containing sulfur and selenium as potential glycosidase inhibitors. J. Am. Chem. Soc. 1994, 116, 1569–1570. [Google Scholar] [CrossRef]
- Mehta, S.; Andrews, J.S.; Johnston, B.D.; Svensson, B.; Pinto, B.M. Synthesis and enzyme inhibitory activity of novel glycosidase inhibitors containing sulfur and selenium. J. Am. Chem. Soc. 1995, 117, 9783–9790. [Google Scholar] [CrossRef]
- Johnston, B.D.; Pinto, B.M. Synthesis of heteroanalogues of disaccharides as potential inhibitors of the processing mannosidase Class I enzymes. Carbohydr. Res. 1998, 310, 17–25. [Google Scholar] [CrossRef]
- Iminosugars: From Synthesis to Therapeutic Applications; Compain, P., Martin, O.R., Eds.; Wiley-VCH: Weinheim, Germany, 2007. [Google Scholar]
- Arjona, O.; Gómez, A.M.; López, J.C.; Plumet, J. Synthesis and conformational and biological aspects of carbasugars. Chem. Rev. 2007, 107, 1919–2036. [Google Scholar] [CrossRef]
- Witczak, Z.J.; Culhane, J.M. Thiosugars: New perspectives regarding availability and potential biochemical and medicinal applications. Appl. Microbiol. Biotechnol. 2005, 69, 237–244. [Google Scholar] [CrossRef]
- Paulsen, H. Carbohydrates containing nitrogen or sulfur in the “hemiacetal” ring. Angew. Chem. Int. Ed. 1966, 5, 495–510. [Google Scholar] [CrossRef]
- Gu, X.; Gupta, V.; Yang, Y.; Zhu, J.-Y.; Carlson, E.J.; Kingsley, C.; Tash, J.S.; Schönbrunn, E.; Hawkinson, J.; Georg, G.I. Structure-activity studies of N-butyl-1-deoxynojirimycin (NB-DNJ) analogues: Discovery of potent and selective aminocyclopentitol inhibitors of GBA1 and GBA2. ChemMedChem 2017, 12, 1977–1984. [Google Scholar] [CrossRef]
- Inouye, S.; Tsuruoka, T.; Ito, T.; Niida, T. Structure and synthesis of nojirimycin. Tetrahedron 1968, 23, 2125–2144. [Google Scholar] [CrossRef]
- He, P.; Zhao, C.; Lu, J.; Zhang, Y.; Fang, M.; Du, Y. Synthesis of 5-thio-α-GalCer analogues with fluorinated acyl chain on lipid residue and their biological evaluation. ACS Med. Chem. Lett. 2019, 10, 221–225. [Google Scholar] [CrossRef]
- Liao, X.; Větvička, V.; Crich, D. Synthesis and evaluation of 1,5-dithia-D-laminaribiose, triose, and tetraose as truncated β-(1→3)-glucan mimetics. J. Org. Chem. 2018, 83, 14894–14904. [Google Scholar] [CrossRef]
- Ferry, A.; Malik, G.; Guinchard, X.; Vĕtvička, V.; Crich, D. Synthesis and evaluation of di- and trimeric hydroxylamine-based β-(1→3)-glucan mimetics. J. Am. Chem. Soc. 2014, 136, 14852–14857. [Google Scholar] [CrossRef]
- Santana, A.G.; Jiménez-Moreno, E.; Gómez, A.M.; Corzana, F.; González, C.; Jiménez-Oses, G.; Jiménez-Barbero, J.; Asensio, J.L. A dynamic combinatorial approach for the analysis of weak carbohydrate/aromatic complexes: Dissecting facial selectivity in CH/π stacking interactions. J. Am. Chem. Soc. 2013, 135, 3347–3350. [Google Scholar] [CrossRef]
- Asensio, J.L.; Ardá, A.; Cañada, F.J.; Jiménez-Barbero, J. Carbohydrate-aromatic interations. Acc. Chem. Res. 2013, 46, 946–954. [Google Scholar] [CrossRef]
- Sylla, B.; Guégan, J.-P.; Wieruszeski, J.-M.; Nugier-Chauvin, C.; Legentil, L.; Daniellou, R.; Ferrières, V. Probing β-(1→3)-D-glucans interactions with recombinant human receptors using high-resolution NMR studies. Carbohydr. Res. 2011, 346, 1490–1494. [Google Scholar] [CrossRef]
- Dayde, B.; Pierra, C.; Gosselin, G.; Surleraux, D.; Ilagouma, A.T.; Laborde, C.; Volle, J.-N.; Virieux, D.; Pirat, J.-L. Synthesis of unnatural phosphonosugar analogues. Eur. J. Org. Chem. 2014, 2014, 1333–1337. [Google Scholar] [CrossRef]
- Ferry, A.; Guinchard, X.; Retailleau, P.; Crich, D. Synthesis, characterization, and coupling reactions of six-membered cyclic P-chiral ammonium phosphonite-boranes; Reactive H-phosphinate equivalents for the stereoselective synthesis of glycomimetics. J. Am. Chem. Soc. 2012, 134, 12289–12301. [Google Scholar] [CrossRef]
- Clarion, L.; Jacquard, C.; Sainte-Catherine, O.; Loiseau, S.; Filippini, D.; Hirlemann, M.-H.; Volle, J.-N.; Virieux, D.; Lecouvey, M.; Pirat, J.-L.; et al. Oxaphosphinanes: New therapeutic perspectives for glioblastoma. J. Med. Chem. 2012, 55, 2196–2211. [Google Scholar] [CrossRef]
- Volle, J.-N.; Filippini, D.; Krawczy, B.; Kaloyanov, N.; Van der Lee, A.; Maurice, T.; Pirat, J.-L.; Virieux, D. Drug discovery: Phosphinolactone, in vivo bioisostere of the lactol group. Org. Biomol. Chem. 2010, 8, 1438–1444. [Google Scholar] [CrossRef]
- Hernández, J.; Ramos, R.; Sastre, N.; Meza, R.; Hommer, H.; Salas, M.; Gordillo, B. Conformational analysis of six-membered ring dioxaphosphinanes. Part 1: Anancomeric thiophosphates. Tetrahedron 2004, 60, 10927–10941. [Google Scholar] [CrossRef]
- Tsunoda, H.; Sasaki, S.; Furuya, T.; Ogawa, S. Synthesis of methyl 5a’-carbamaltoses linked by imino, ether and sulfide bridges and unsaturated derivatives thereof. Liebigs Ann. 1996, 159–165. [Google Scholar] [CrossRef]
- Junge, B.; Heiker, F.-R.; Kurz, J.; Muller, L.; Schmidt, D.D.; Wunsche, C. Untersuchungen zur Struktur des α-D-glucosidaseinhibitors Acarbose. Carbohydr. Res. 1984, 128, 235–268. [Google Scholar] [CrossRef]
- Noguchi, S.; Takemoto, S.; Kidokoro, S.; Yamamoto, K.; Hashimoto, M. Syntheses of cellotriose and cellotetraose analogues as transition state mimics for mechanistic studies of cellulases. Bioorg. Med. Chem. 2011, 19, 3812–3830. [Google Scholar] [CrossRef]
- Kerins, L.; Byrne, S.; Gabba, A.; Murphy, P.V. Anomer preferences for glucuronic and galacturonic acid and derivatives and influence of electron-withdrawing substituents. J. Org. Chem. 2018, 83, 7714–7729. [Google Scholar] [CrossRef]
- Sager, C.P.; Eriş, D.; Smieško, M.; Hevey, R.; Ernst, B. What contributes to an effective mannose recognition domain? Beilstein J. Org. Chem. 2017, 13, 2584–2595. [Google Scholar] [CrossRef]
- Cabani, S.; Gianni, P.; Mollica, V.; Lepori, L. Group contributions to the thermodynamic properties of non-ionic organic solutes in dilute aqueous solution. J. Solution Chem. 1981, 10, 563–595. [Google Scholar] [CrossRef]
- Ge, J.-T.; Li, Y.-Y.; Tian, J.; Liao, R.-Z.; Dong, H. Synthesis of deoxyglycosides by desulfurization under UV light. J. Org. Chem. 2017, 82, 7008–7014. [Google Scholar] [CrossRef]
- Danac, R.; Ball, L.; Gurr, S.J.; Fairbanks, A.J. Synthesis of UDP-glucose derivatives modified at the 3-OH as potential chain terminators of β-glucan biosynthesis. Carbohydr. Res. 2008, 343, 1012–1022. [Google Scholar] [CrossRef]
- Wands, A.M.; Cervin, J.; Huang, H.; Zhang, Y.; Youn, G.; Brautigam, C.A.; Dzebo, M.M.; Björklund, P.; Wallenius, V.; Bright, D.K.; et al. Fucosylated molecules competitively interfere with cholera toxin binding to host cells. ACS Infect. Dis. 2018, 4, 758–770. [Google Scholar] [CrossRef]
- Bondi, A. van der Waals volumes and radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Sadurní, A.; Kehr, G.; Ahlqvist, M.; Wernevik, J.; Sjögren, H.P.; Kankkonen, C.; Knerr, L.; Gilmour, R. Fluorine-directed glycosylation enables the stereocontrolled synthesis of selective SGLT2 inhibitors for type II diabetes. Chem. Eur. J. 2018, 24, 2832–2836. [Google Scholar] [CrossRef]
- Denavit, V.; Lainé, D.; Bouzriba, C.; Shanina, E.; Gillon, É.; Fortin, S.; Rademacher, C.; Imberty, A.; Giguère, D. Stereoselective synthesis of fluroinated galactopyranosides as potential molecular probes for galactophilic proteins: Assessment of monofluorogalactoside-LecA interactions. Chem. Eur. J. 2019, 25, 4478–4490. [Google Scholar] [CrossRef]
- Karban, J.; Horník, Š.; Šťastná, L.Č.; Sýkora, J. A convenient route to peracetylated 3-deoxy-3-fluoro analogues of D-glucosamine and D-galactosamine from a Černý epoxide. Synlett 2014, 25, 1253–1256. [Google Scholar] [CrossRef]
- Kasuya, M.C.; Ito, A.; Hatanaka, K. Simple and convenient synthesis of a fluorinated GM4 analogue. J. Fluor. Chem. 2007, 128, 562–565. [Google Scholar] [CrossRef]
- Nishimura, S.-I.; Hato, M.; Hyugaji, S.; Feng, F.; Amano, M. Glycomics for drug discovery: Metabolic perturbation in androgen-independent prostate cancer cells induced by unnatural hexosamine mimics. Angew. Chem. Int. Ed. 2012, 51, 3386–3390. [Google Scholar] [CrossRef]
- Sprinz, C.; Zanon, M.; Altmayer, S.; Watte, G.; Irion, K.; Marchiori, E.; Hochhegger, B. Effects of blood glucose level on 18F fluorodeoxyglucose (18F-FDG) uptake for PET/CT in normal organs: An analysis on 5623 patients. Sci. Rep. 2018, 8, 2126. [Google Scholar] [CrossRef]
- Maschauer, S.; Haubner, R.; Kuwert, T.; Prante, O. 18F-Glyco-RGD peptides for PET imaging of integrin expression: Efficient radiosynthesis by click chemistry and modulation of biodistribution by glycosylation. Mol. Pharm. 2014, 11, 505–515. [Google Scholar] [CrossRef]
- Namavari, M.; Gowrishankar, G.; Srinivasan, A.; Gambhir, S.S.; Haywood, T.; Beinat, C. A novel synthesis of 6′′-[18F]-fluoromaltotriose as a PET tracer for imaging bacterial infection. J. Label. Compd. Radiopharm. 2017, 61, 408–414. [Google Scholar] [CrossRef]
- Bouchie, A. SGLT2 inhibitors enter crowded diabetes space. Nat. Biotechnol. 2013, 31, 469–470. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Norton, L.; Abdul-Ghani, M. Renal, metabolic and cardiovascular considerations of SGLT2 inhibition. Nat. Rev. Nephrol. 2017, 13. [Google Scholar] [CrossRef]
- Kanai, Y.; Lee, W.-S.; You, G.; Brown, D.; Hediger, M.A. The human kidney low affinity Na+/glucose cotransporter SGLT2. J. Clin. Investig. 1994, 93, 397–404. [Google Scholar] [CrossRef]
- Kasahara, M.; Maeda, M.; Hayashi, S.; Mori, Y.; Abe, T. A missense mutation in the Na+/glucose cotransporter gene SGLT1 in a patient with congenital glucose-galactose malabsorption: Normal trafficking but inactivation of the mutant protein. Biochim. Biophys. Acta 2001, 1536, 141–147. [Google Scholar] [CrossRef]
- Turk, E.; Zabel, B.; Mundlos, S.; Dyer, J.; Wright, E.M. Glucose/galactose malabsorption caused by a defect in the Na+/glucose cotransporter. Nature 1991, 350, 354–356. [Google Scholar] [CrossRef]
- Baumann, A.; Marchner, S.; Daum, M.; Hoffmann-Röder, A. Synthesis of fluorinated Leishmania cap trisaccharides for diagnostic tool and vaccine development. Eur. J. Org. Chem. 2018, 2018, 3803–3815. [Google Scholar] [CrossRef]
- Wagner, S.; Mersch, C.; Hoffmann-Röder, A. Fluorinated glycosyl amino acids for mucin-like glycopeptide antigen analogues. Chem. Eur. J. 2010, 16, 7319–7330. [Google Scholar] [CrossRef]
- Muldard, L.A.; Kováč, P.; Glaudemans, C.P.J. Synthesis of specifically monofluorinated ligands related to the O-polysaccharide of Shigella dysenteriae type 1. Carbohydr. Res. 1994, 259, 21–34. [Google Scholar] [CrossRef]
- Oberbillig, T.; Mersch, C.; Wagner, S.; Hoffmann-Röder, A. Antibody recognition of fluorinated MUC1 glycopeptide antigens. Chem. Commun. 2012, 48, 1487–1489. [Google Scholar] [CrossRef]
- Yang, F.; Zheng, X.-J.; Huo, C.-X.; Wang, Y.; Zhang, Y.; Ye, X.-S. Enhancement of the immunogenicity of synthetic carbohydrate vaccines by chemical modifications of STn antigen. ACS Chem. Biol. 2011, 6, 252–259. [Google Scholar] [CrossRef]
- Kieser, T.J.; Santschi, N.; Nowack, L.; Kehr, G.; Kuhlmann, T.; Albrecht, S.; Gilmour, R. Single site fluorination of the GM4 ganglioside epitope upregulates oligodendrocyte differentiation. ACS Chem. Neurosci. 2018, 9, 1159–1165. [Google Scholar] [CrossRef]
- Fernández, P.; Jiménez-Barbero, J.; Martín-Lomas, M. Syntheses of all the possible monomethyl ethers and several deoxyhalo analogues of methyl β-lactoside as ligands for the Ricinus communis lectins. Carbohydr. Res. 1994, 254, 61–79. [Google Scholar] [CrossRef]
- Willén, D.; Bengtsson, D.; Clementson, S.; Tykesson, E.; Manner, S.; Ellervik, U. Synthesis of double-modified xyloside analogues for probing the β4GalT7 active site. J. Org. Chem. 2018, 83, 1259–1277. [Google Scholar] [CrossRef]
- Idris, I.; Donnelly, R. Sodium-glucose co-transporter-2 inhibitors: An emerging new class of oral antidiabetic drug. Diabetes Obes. Metab. 2009, 11, 79–88. [Google Scholar] [CrossRef]
- Verspohl, E.J. Novel pharmacological approaches to the treatment of type 2 diabetes. Pharmacol. Rev. 2012, 64, 188–237. [Google Scholar] [CrossRef]
- Cao, X.; Zhang, W.; Yan, X.; Huang, Z.; Zhang, Z.; Wang, P.; Shen, J. Modification on the O-glucoside of Sergliflozin-A: A new strategy for SGLT2 inhibitor design. Bioorg. Med. Chem. Lett. 2016, 26, 2170–2173. [Google Scholar] [CrossRef]
- Busca, P.; Piller, V.; Piller, F.; Martin, O.R. Synthesis and biological evaluation of new UDP-GalNAc analogues for the study of polypeptide-α-GalNAc-transferases. Bioorg. Med. Chem. Lett. 2003, 13, 1853–1856. [Google Scholar] [CrossRef]
- Lin, C.-I.; McCarty, R.M.; Liu, H. The biosynthesis of nitrogen-, sulfur-, and high-carbon chain-containing sugars. Chem. Soc. Rev. 2013, 42, 4377–4407. [Google Scholar] [CrossRef]
- Berkov-Zrihen, Y.; Herzog, I.M.; Feldman, M.; Sonn-Segev, A.; Roichman, Y.; Fridman, M. Di-alkylated paromomycin derivatives: Targeting the membranes of Gram positive pathogens that cause skin infections. Bioorg. Med. Chem. 2013, 21, 3624–3631. [Google Scholar] [CrossRef]
- Martinez, J.; Nguyen, L.D.; Hinderlich, S.; Zimmer, R.; Tauberger, E.; Reutter, W.; Saenger, W.; Fan, H.; Moniot, S. Crystal structures of N-acetylmannosamine kinase provide insights into enzyme activity and inhibition. J. Biol. Chem. 2012, 287, 13656–13665. [Google Scholar] [CrossRef]
- Chen, G.C.; Banks, C.H.; Irgolic, K.J.; Zingaro, R.A. Syntheses of 1- and 6-S- and 1- and 6-Se-derivatives of 2-amino-2-deoxy-α/β-D-glucopyranose. J. Chem. Soc. Perkin 1 1980, 10, 2287–2293. [Google Scholar] [CrossRef]
- Liakatos, A.; Kiefel, M.J.; Fleming, F.; Coulson, B.; von Itzstein, M. The synthesis and biological evaluation of lactose-based sialylmimetics as inhibitors of rotaviral infection. Bioorg. Med. Chem. 2006, 14, 739–757. [Google Scholar] [CrossRef]
- Vargas, J.P.; Pinto, L.M.; Savegnago, L.; Lüdtke, D.S. Synthesis of alkylseleno-carbohydrates and evaluation of their antioxidant properties. J. Braz. Chem. Soc. 2015, 26, 810–815. [Google Scholar] [CrossRef]
- Pavashe, P.; Elamparuthi, E.; Hettrich, C.; Möller, H.M.; Linker, T. Synthesis of 2-thiocarbohydrates and their binding to concanavalin A. J. Org. Chem. 2016, 81, 8595–8603. [Google Scholar] [CrossRef]
- Wu, B.; Ge, J.; Ren, B.; Pei, Z.; Dong, H. Synthesis and binding affinity analysis of positional thiol analogs of mannopyranose for the elucidation of sulfur in different position. Tetrahedron 2015, 71, 4023–4030. [Google Scholar] [CrossRef]
- Nieto-Garcia, O.; Wratil, P.R.; Nguyen, L.D.; Böhrsch, V.; Hinderlich, S.; Reutter, W.; Hackenberger, C.P.R. Inhibition of the key enzyme of sialic acid biosynthesis by C6-Se modified N-acetylmannosamine analogs. Chem. Sci. 2016, 7, 3928–3933. [Google Scholar] [CrossRef]
- Blume, A.; Chen, H.; Reutter, W.; Schmidt, R.R.; Hinderlich, S. 2′,3′-Dialdehydo-UDP-N-acetylglucosamine inhibits UDP-N-acetylglucosamine 2-epimerase, the key enzyme of sialic acid biosynthesis. FEBS Lett. 2002, 521, 127–132. [Google Scholar] [CrossRef]
- Al-Rawi, S.; Hinderlich, S.; Reutter, W.; Giannis, A. Synthesis and biochemical properties of reversible inhibitors of UDP-N-acetylglucosamine 2-epimerase. Angew. Chem. Int. Ed. 2004, 43, 4366–4370. [Google Scholar] [CrossRef]
- Matsushita, T.; Sati, G.C.; Kondasinghe, N.; Pirrone, M.G.; Kato, T.; Waduge, P.; Kumar, H.S.; Sanchon, A.C.; Dobosz-Bartoszek, M.; Shcherbakov, D.; et al. Design, multigram synthesis, and in vitro and in vivo evaluation of propylamycin: A semisynthetic 4,5-deoxystreptamine class aminoglycoside for the treatment of drug-resistant Enterobacteriaceae and other Gram-negative pathogens. J. Am. Chem. Soc. 2019, 141, 5051–5061. [Google Scholar] [CrossRef]
- Duscha, S.; Boukari, H.; Shcherbakov, D.; Salian, S.; Silva, S.; Kendall, A.; Kato, T.; Akbergenov, R.; Perez-Fernandez, D.; Bernet, B.; et al. Identification and evaluation of improved 4′-O-(alkyl) 4,5-disubstituted 2-deoxystreptamines as next-generation aminoglycoside antibiotics. mBio 2014, 5, e01827-14. [Google Scholar] [CrossRef]
- Büll, C.; Boltje, T.J.; van Dinther, E.A.W.; Peters, T.; de Graaf, A.M.A.; Leusen, J.H.W.; Kreutz, M.; Figdor, C.G.; den Brok, M.H.; Adema, G.J. Targeted delivery of a sialic acid-blocking glycomimetic to cancer cells inhibits metastatic spread. ACS Nano 2015, 9, 733–745. [Google Scholar] [CrossRef]
- Büll, C.; Boltje, T.J.; Wassink, M.; de Graaf, A.M.A.; van Delft, F.L.; den Brok, M.H.; Adema, G.J. Targeting aberrant sialylation in cancer cells using a fluorinated sialic acid analog impairs adhesion, migration, and in vivo tumor growth. Mol. Cancer Ther. 2016, 12, 1935–1946. [Google Scholar] [CrossRef]
- Zhu, J.-S.; McCormick, N.E.; Timmons, S.C.; Jakeman, D.L. Synthesis of α-deoxymono and difluorohexopyranosyl 1-phosphates and kinetic evaluation with thymidylyl- and guanidylyltransferases. J. Org. Chem. 2016, 81, 8816–8825. [Google Scholar] [CrossRef]
- Hanzawa, Y.; Uda, J.; Kobayashi, Y.; Ishido, Y.; Taguchi, T.; Shiro, M. Trifluoromethylation of chiral aldehyde and synthesis of 6-deoxy-6,6,6-trifluorohexoses. Chem. Pharm. Bull. 1991, 39, 2459–2461. [Google Scholar] [CrossRef][Green Version]
- Achmatowicz, M.M.; Allen, J.G.; Bio, M.M.; Bartberger, M.D.; Borths, C.J.; Colyer, J.T.; Crockett, R.D.; Hwang, T.-L.; Koek, J.N.; Osgood, S.A.; et al. Telescoped process to manufacture 6,6,6-trifluorofucose via diastereoselective transfer hydrogenation: Scalable access to an inhibitor of fucosylation utilized in monoclonal antibody production. J. Org. Chem. 2016, 81, 4736–4743. [Google Scholar] [CrossRef]
- Allen, J.G.; Mujacic, M.; Frohn, M.J.; Pickrell, A.J.; Kodama, P.; Bagal, D.; San Miguel, T.; Sickmier, E.A.; Osgood, S.; Swietlow, A.; et al. Facile modulation of antibody fucosylation with small molecule fucostatin inhibitors and cocrystal structure with GDP-mannose 4,6-dehydratase. ACS Chem. Biol. 2016, 11, 2734–2743. [Google Scholar] [CrossRef]
- Okeley, N.M.; Alley, S.C.; Anderson, M.E.; Boursalian, T.E.; Burke, P.J.; Emmerton, K.M.; Jeffrey, S.C.; Klussman, K.; Law, C.-L.; Sussman, D.; et al. Development of orally active inhibitors of protein and cellular fucosylation. Proc. Natl. Acad. Sci. USA 2013, 110, 5404–5409. [Google Scholar] [CrossRef]
- Li, J.; Hsu, H.-C.; Ding, Y.; Li, H.; Wu, Q.; Yang, P.; Luo, B.; Rowse, A.L.; Spalding, D.M.; Bridges, S.L., Jr.; et al. Inhibition of fucosylation reshapes inflammatory macrophages and suppresses type II collagen-induced arthritis. Arthritis Rheumatol. 2014, 66, 2368–2379. [Google Scholar] [CrossRef]
- Belcher, J.D.; Chen, C.; Nguyen, J.; Abdulla, F.; Nguyen, P.; Nguyen, M.; Okeley, N.M.; Benjamin, D.R.; Senter, P.D.; Vercellotti, G.M. The fucosylation inhibitor, 2-fluorofucose, inhibits vaso-occlusion, leukocyte-endothelium interactions and NF-κB activation in transgenic sickle mice. PLoS ONE 2015, 10, e0117772. [Google Scholar] [CrossRef]
- Soriano del Amo, D.; Wang, W.; Besanceney, C.; Zheng, T.; He, Y.; Gerwe, B.; Seidel, R.D., III; Wu, P. Chemoenzymatic synthesis of the sialyl Lewis X glycan and its derivatives. Carbohydr. Res. 2010, 345, 1107–1113. [Google Scholar] [CrossRef][Green Version]
- Wang, W.; Hu, T.; Frantom, P.A.; Zheng, T.; Gerwe, B.; Soriano del Amo, D.; Garret, S.; Seidel, R.D., III; Wu, P. Chemoenzymatic synthesis of GDP-L-fucose and the Lewis X glycan derivatives. Proc. Natl. Acad. Sci. USA 2009, 106, 16096–16101. [Google Scholar] [CrossRef]
- Ichikawa, Y.; Lin, Y.-C.; Dumas, D.P.; Shen, G.-J.; Garcia-Junceda, E.; Williams, M.A.; Bayer, R.; Ketcham, C.; Walker, L.E.; Paulson, J.C.; et al. Chemical-enzymatic synthesis and conformational analysis of sialyl Lewis x and derivatives. J. Am. Chem. Soc. 1992, 114, 9283–9298. [Google Scholar] [CrossRef]
- Lee, H.-Y.; Chen, C.-Y.; Tsai, T.-I.; Li, S.-T.; Lin, K.-H.; Cheng, Y.-Y.; Ren, C.-T.; Cheng, T.-J.R.; Wu, C.-Y.; Wong, C.-H. Immunogenicity study of Globo H analogues with modification at the reducing or nonreducing end of the tumor antigen. J. Am. Chem. Soc. 2014, 136, 16844–16853. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, Y.; Wang, Z.; Wang, Z.; Huang, L. The synthesis of a 2-deoxy-2-acetonyl sugar from its corresponding natural saccharide. J. Chem. Res. 2012, 36, 244–246. [Google Scholar] [CrossRef]
- Hang, H.C.; Bertozzi, C.R. Ketone isosteres of 2-N-acetamidosugars as substrates for metabolic cell surface engineering. J. Am. Chem. Soc. 2001, 123, 1242–1243. [Google Scholar] [CrossRef]
- Cai, L.; Guan, W.; Chen, W.; Wang, P.G. Chemoenzymatic synthesis of uridine 5′-diphospho-2-acetonyl-2-deoxy-α-D-glucose as C2-carbon isostere of UDP-GlcNAc. J. Org. Chem. 2010, 75, 3492–3494. [Google Scholar] [CrossRef]
- Rawat, M.; Newton, G.L.; Ko, M.; Martinez, G.J.; Fahey, R.C.; Av-Gay, Y. Mycothiol-deficient Mycobacterium smegmatis mutants are hypersensitive to alkylating agents, free radicals, and antibiotics. Antimicrob. Agents Chemother. 2002, 46, 3348–3355. [Google Scholar] [CrossRef]
- Gammon, D.W.; Hunter, R.; Steenkamp, D.J.; Mudzunga, T.T. Synthesis of 2-deoxy-2-C-alkylglucosides of myo-inositol as possible inhibitors of a N-deacetylase enzyme in the biosynthesis of mycothiol. Bioorg. Med. Chem. 2003, 13, 2045–2049. [Google Scholar] [CrossRef]
- Abdelwahab, N.Z.; Crossman, A.T.; Sullivan, L.; Ferguson, M.A.J.; Urbaniak, M.D. Inhibitors incorporating zinc-binding groups target the GlcNAc-PI de-N-acetylase in Trypanosoma brucei, the causative agent of African sleeping sickness. Chem. Biol. Drug Des. 2012, 79, 270–278. [Google Scholar] [CrossRef]
- Mehlert, A.; Zitzmann, N.; Richardson, J.M.; Treumann, A.; Ferguson, M.A.J. The glycosylation of the variant surface glycoproteins and procyclic acidic repetitive proteins of Trypanosoma brucei. Mol. Biochem. Parasitol. 1998, 91, 145–152. [Google Scholar] [CrossRef]
- Urbaniak, M.D.; Crossman, A.; Chang, T.; Smith, T.K.; van Aalten, D.M.F.; Ferguson, M.A.J. The N-acetyl-D-glucosaminylphosphatidylinositol de-N-acetylase of glycosylphosphatidylinositol biosynthesis is a zinc metalloenzyme. J. Biol. Chem. 2005, 280, 22831–22838. [Google Scholar] [CrossRef]
- Abdelwahab, N.Z.; Urbaniak, M.D.; Ferguson, M.A.J.; Crossman, A.T. Synthesis of potential metal-binding group compounds to examine the zinc dependency of the GPI de-N-acetylase metalloenzyme in Trypanosoma brucei. Carbohydr. Res. 2011, 346, 708–714. [Google Scholar] [CrossRef][Green Version]
- DiFrancesco, B.R.; Morrison, Z.A.; Nitz, M. Monosaccharide inhibitors targeting carbohydrate esterase family 4 de-N-acetylases. Bioorg. Med. Chem. 2018, 26, 5631–5643. [Google Scholar] [CrossRef]
- Crous, W.; Naidoo, K.J. Conformational and electrostatic analysis of SN1 donor analogue glycomimetic inhibitors of ST3Gal-I mammalian sialyltransferase. Bioorg. Med. Chem. 2016, 24, 4998–5005. [Google Scholar] [CrossRef]
- Izumi, M.; Wada, K.; Yuasa, H.; Hashimoto, H. Synthesis of bisubstrate and donor analogues of sialyltransferase and their inhibitory activities. J. Org. Chem. 2005, 70, 8817–8824. [Google Scholar] [CrossRef]
- Büll, C.; Heise, T.; Adema, G.J.; Boltje, T.J. Sialic acid mimetics to target the sialic acid-Siglec axis. Trends Biochem. Sci. 2016, 41, 519–531. [Google Scholar] [CrossRef]
- Bradley, S.J.; Fazli, A.; Kiefel, M.J.; von Itzstein, M. Synthesis of novel sialylmimetics as biological probes. Bioorg. Med. Chem. Lett. 2001, 11, 1587–1590. [Google Scholar] [CrossRef]
- Fazli, A.; Bradley, S.J.; Kiefel, M.J.; Jolly, C.; Holmes, I.H.; von Itzstein, M. Synthesis and biological evaluation of sialylmimetics as rotavirus inhibitors. J. Med. Chem. 2001, 44, 3292–3301. [Google Scholar] [CrossRef]
- Hoos, R.; Huixin, J.; Vasella, A.; Weiss, P. Synthesis and enzymatic evaluation of substrates and inhibitors of β-glucuronidases. Helv. Chim. Acta 1996, 79, 1757–1784. [Google Scholar] [CrossRef]
- Mochizuki, T.; Iwano, Y.; Shiozaki, M.; Kurakata, S.; Kanai, S.; Nishijima, M. Lipid A-type pyrancarboxylic acid derivatives, their synthesis and their biological activities. Tetrahedron 2000, 56, 7691–7703. [Google Scholar] [CrossRef]
- Downey, A.M.; Cairo, C.W. α-Bromophosphonate analogs of glucose-6-phosphate are inhibitors of glucose-6-phosphatase. Carbohydr. Res. 2013, 381. [Google Scholar] [CrossRef]
- Kim-Muller, J.Y.; Accili, D. Selective insulin sensitizers. Science 2011, 331, 1529–1531. [Google Scholar] [CrossRef]
- Ashmore, J.; Hastings, A.B.; Nesbett, F.B. The effect of diabetes and fasting on liver glucose-6-phosphatase. Proc. Natl. Acad. Sci. USA 1954, 40, 673–678. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
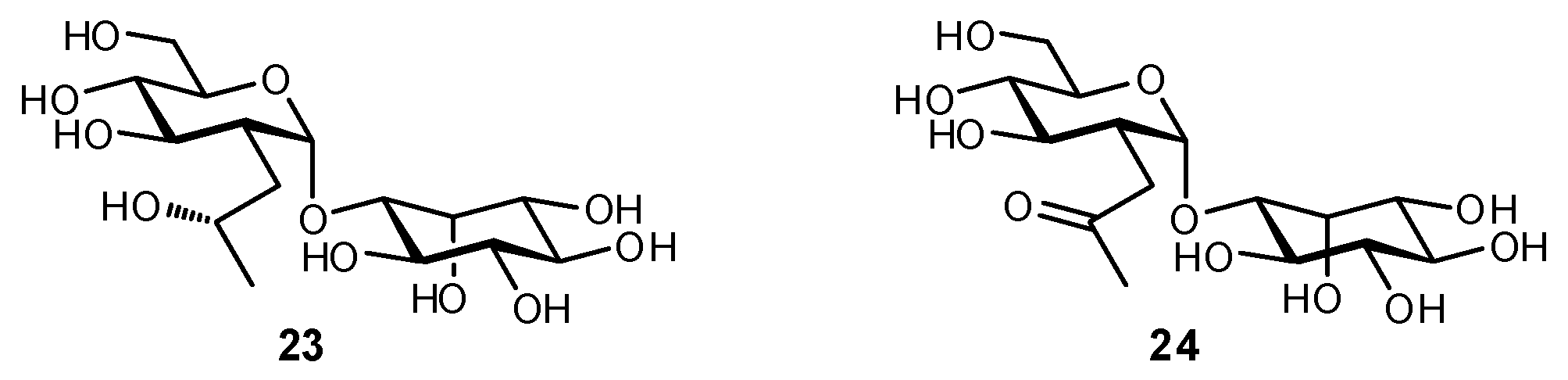{kind=link}
{kind=link}
{kind=link}
| Functional Group | Bioisosteric Replacement | Rationale | Disadvantages |
|---|---|---|---|
| O-Glycosidic linkage | N-, C-, S-, Se-Glycosides | O-Glycosides can be susceptible to chemical/enzymatic hydrolysis in vivo | Enhanced flexibility (larger atom, loss of anomeric effect) |
| Endocyclic O atom | Imino-, thio-, carbasugars, phostones, phostines | Enhance stability; Reduce polar surface area; Iminosugars can mimic charged oxocarbenium transition state | Changes in pyranoside conformation; Loss of anomeric effect |
| OH | Deoxygenation | Reduce polar surface area; Increase hydrophobic contacts with protein | Potentially disrupts critical ligand-protein interactions; Disrupts ligand pre-organization |
| OH | Deoxyfluorination | Similar polarity and size; H-bond acceptor ability; Reduce polar surface area; Destabilize oxocarbenium transition state | Removes H-bond donor ability |
| OH | Methyl etherification | Reduce polar surface area | Removes H-bond donor ability; Potential steric incompatibilities |
| OH | SH/SeH substitution | Reduce polar surface area (enhanced atom polarizability); Enhance π-interactions | Larger atoms; Longer bonds/altered bond angles; Weaker H-bond donors |
| H | Fluorination | Similar size and hydrophobicity; Chemically inert; Destabilize oxocarbenium transition state | Alters electron-density in neighboring substituents |
| NHAc | C-, N-derivatives | Enhance metal chelation; Introduce novel functionalities for bioconjugation (e.g., ketone) | Potentially introduces steric incompatibilities or charged substituents |
| CO2− | Amide, sulfonate, phosphonate | Reduce polar surface area; Enhance charged protein interactions | Disrupts critical carboxylate–protein interactions |
| Original Group | Potential Replacements |
|---|---|
 |  |
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hevey, R. Bioisosteres of Carbohydrate Functional Groups in Glycomimetic Design. Biomimetics 2019, 4, 53. https://doi.org/10.3390/biomimetics4030053
Hevey R. Bioisosteres of Carbohydrate Functional Groups in Glycomimetic Design. Biomimetics. 2019; 4(3):53. https://doi.org/10.3390/biomimetics4030053
Chicago/Turabian StyleHevey, Rachel. 2019. "Bioisosteres of Carbohydrate Functional Groups in Glycomimetic Design" Biomimetics 4, no. 3: 53. https://doi.org/10.3390/biomimetics4030053
APA StyleHevey, R. (2019). Bioisosteres of Carbohydrate Functional Groups in Glycomimetic Design. Biomimetics, 4(3), 53. https://doi.org/10.3390/biomimetics4030053