Fabrication of Human Keratinocyte Cell Clusters for Skin Graft Applications by Templating Water-in-Water Pickering Emulsions

 , , , ,
, , , ,  and
and
Abstract

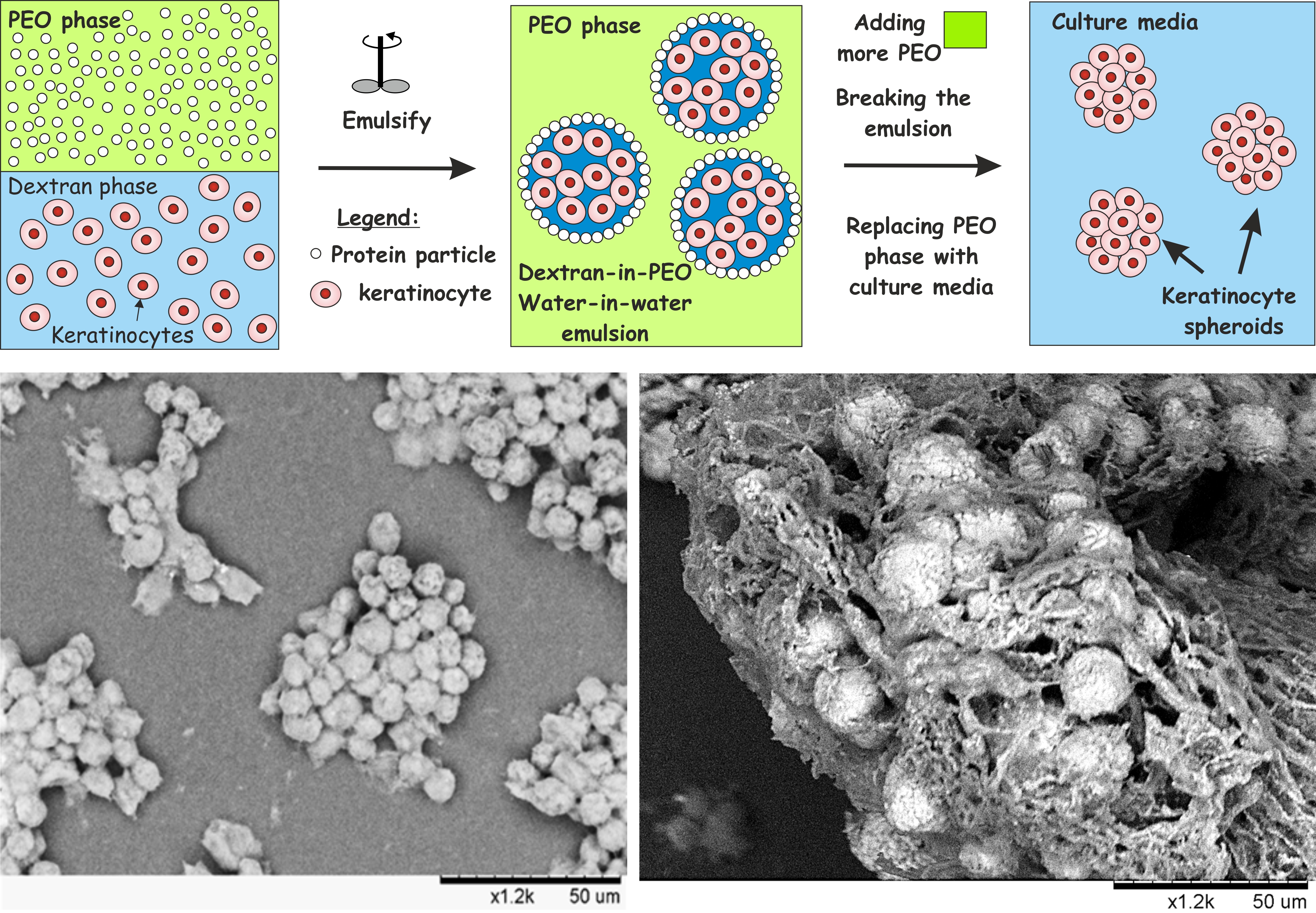{kind=link}
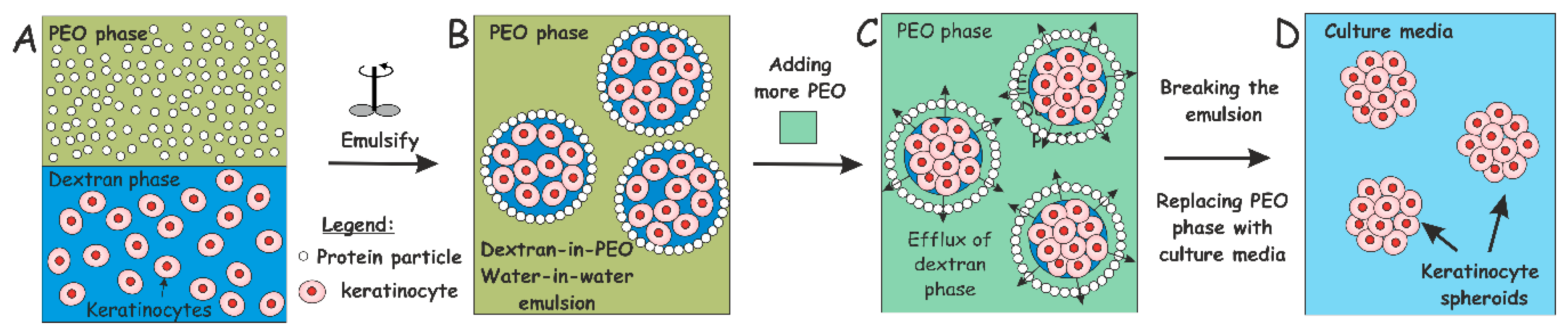{kind=link}
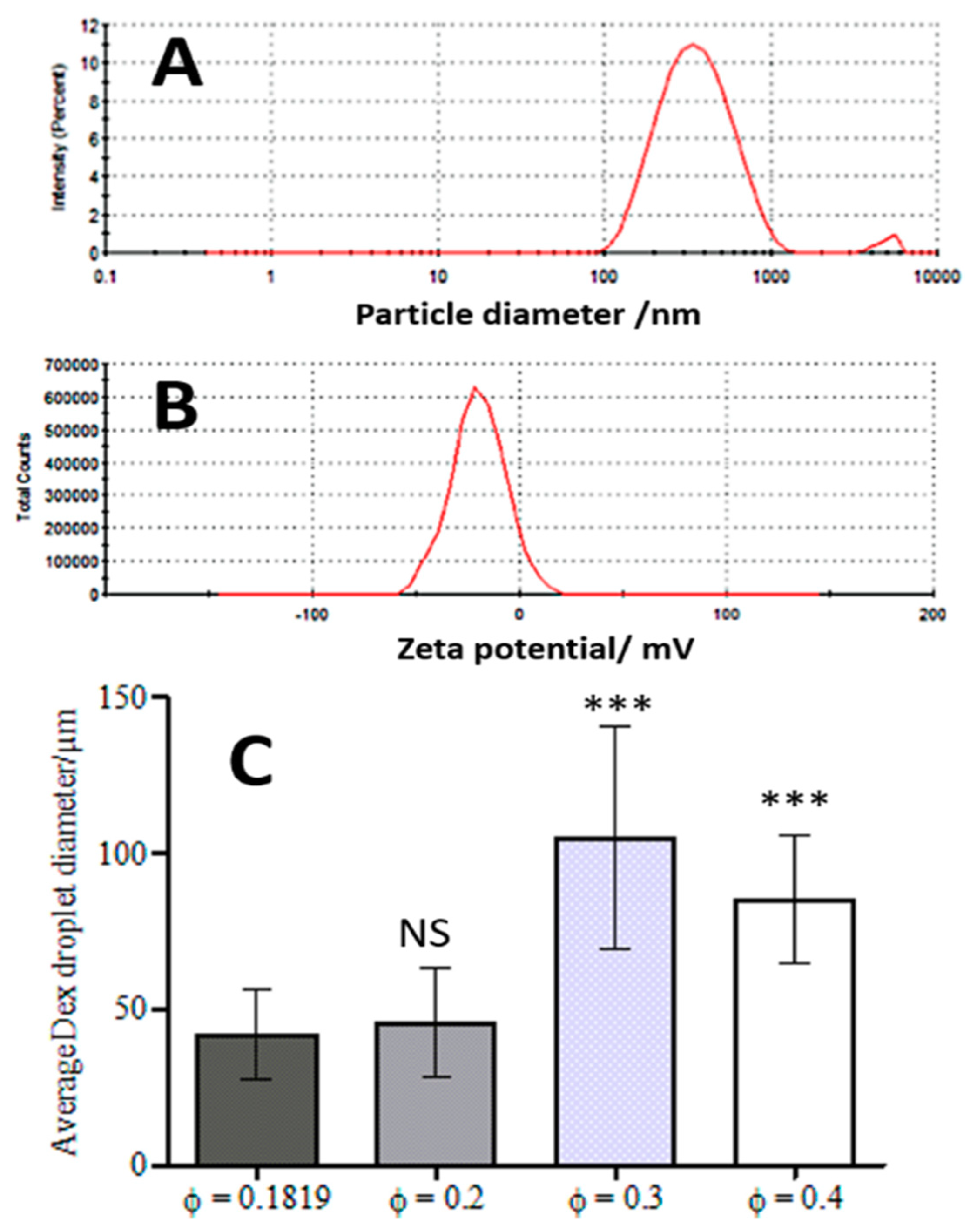{kind=link}
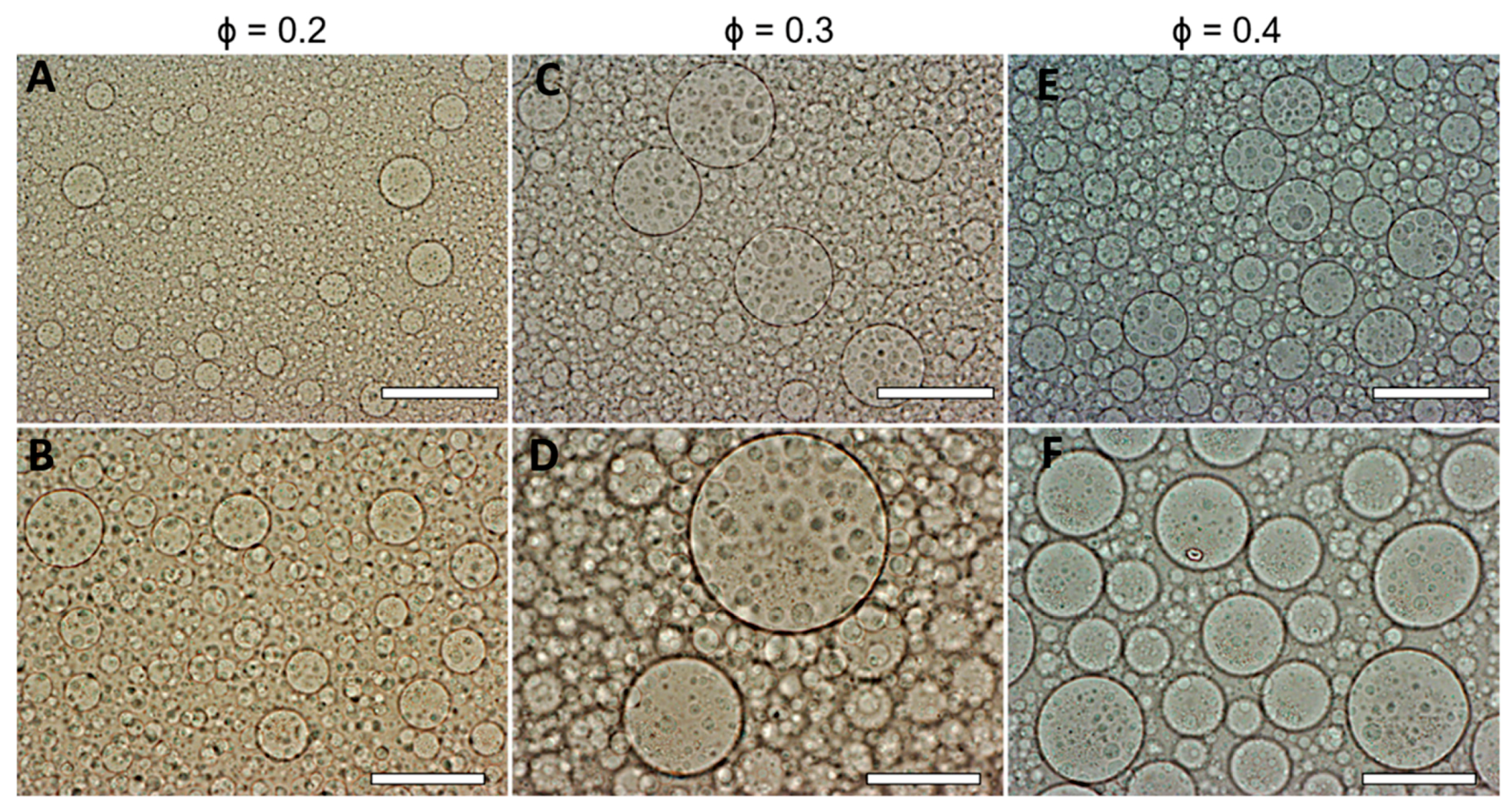{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. 2D HaCaT Cell Culture
2.3. Preparation of the Whey Protein (WP) Particles
2.4. Production of w/w Pickering Emulsions, Cell Encapsulation and Clusteroid Isolation
2.5. Cell Viability Assay
2.6. 3D Keratinocyte Clusteroids Culture
2.7. Fabrication of HaCaT Cell Clusteroids
2.8. Preparation of Model HaCaT Clusteroids in Alginate Hydrogels Formulations
2.9. SEM Imaging of HaCaT Clusteroids
2.10. Statistical Analysis
3. Results
3.1. WP Particle Characterisation
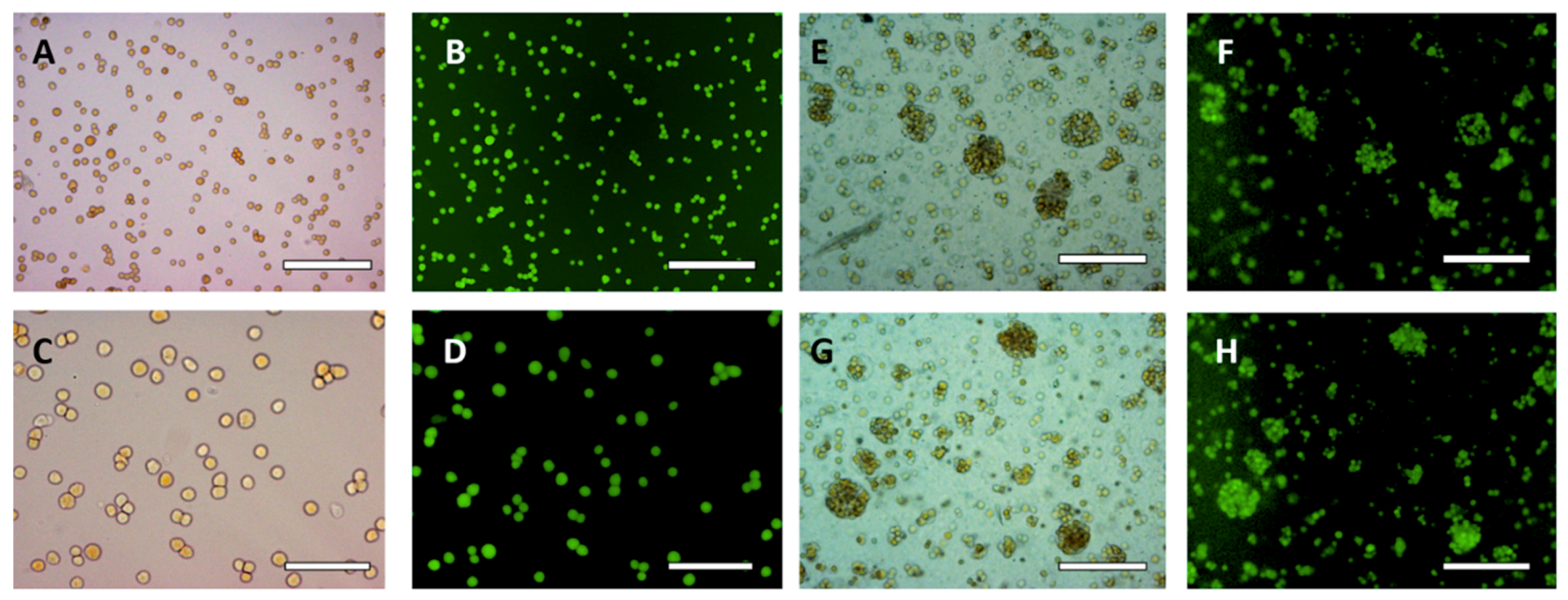
3.2. HaCaT Cell Encapsulation in w/w Emulsions
3.3. Effect of the DEX Phase Volume Fraction
3.4. Effect of the w/w Pickering Emulsion Homogenization
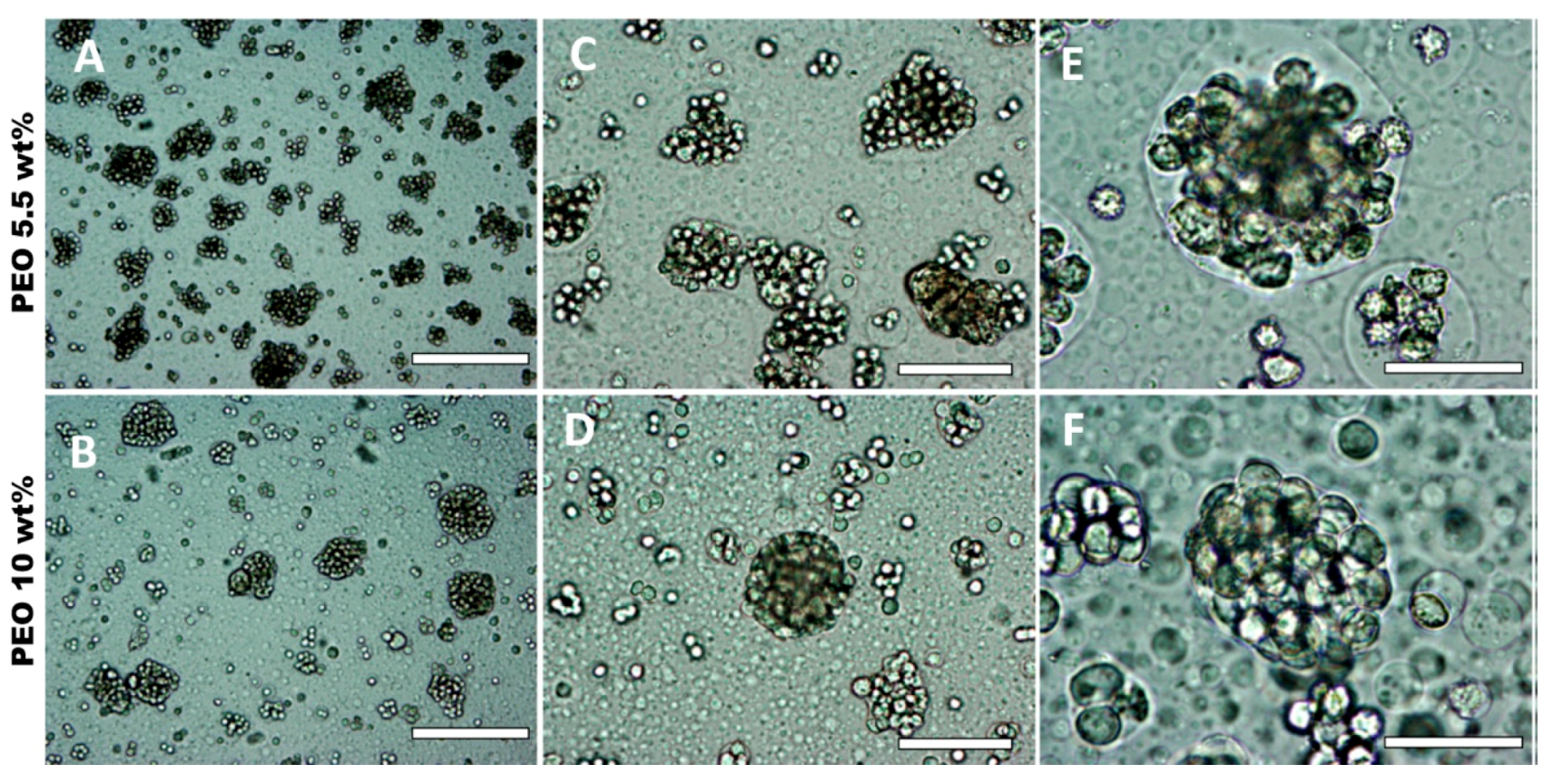
3.5. Effect of HaCaT Cell Volume Fraction and PEO Concentration
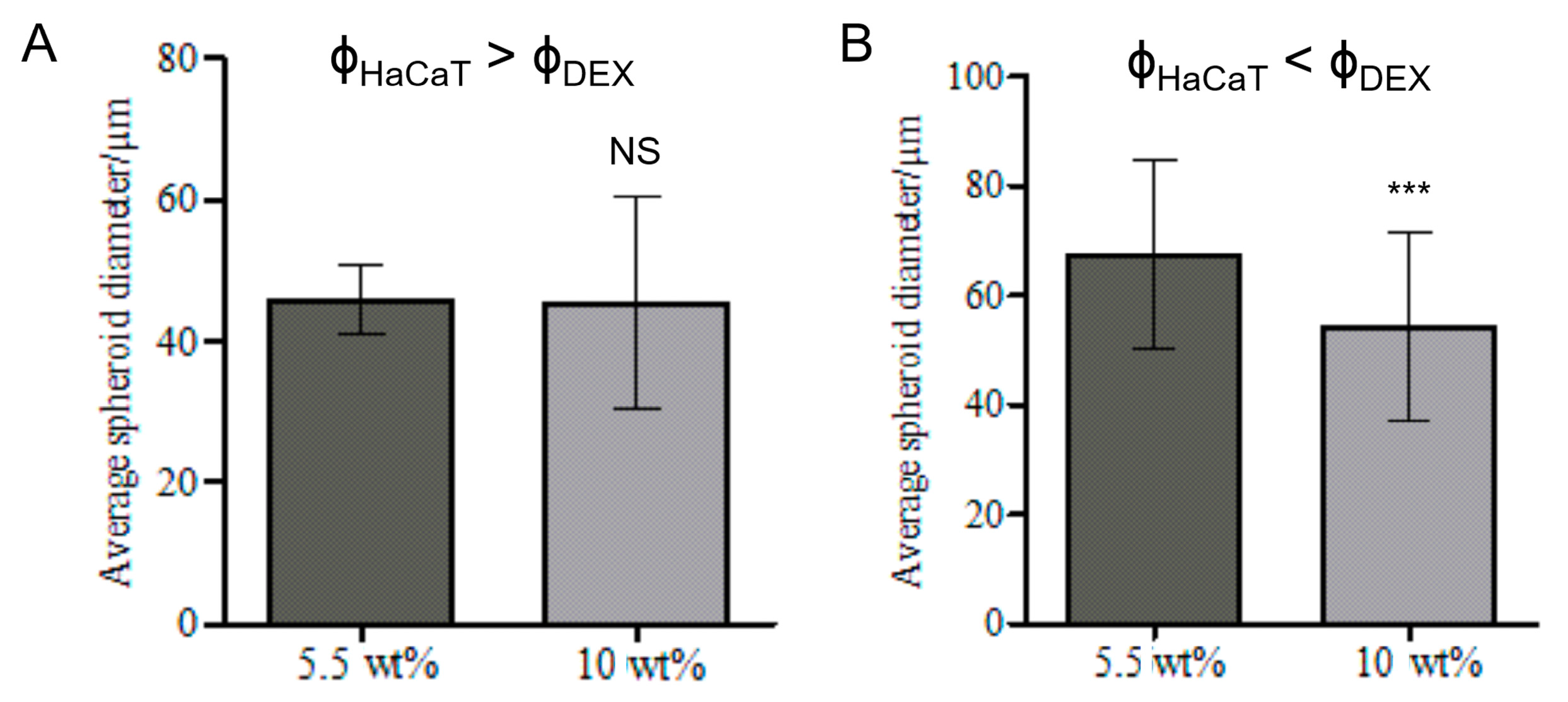
3.6. Effect of the Volume Fractions of DEX Phase and HaCaT Cells
3.7. Hydrogel-HaCaT Spheroid Formulations
3.7.1. Effect of the Cell Spheroid Density
3.7.2. Effect of the Breaking of the w/w Emulsion
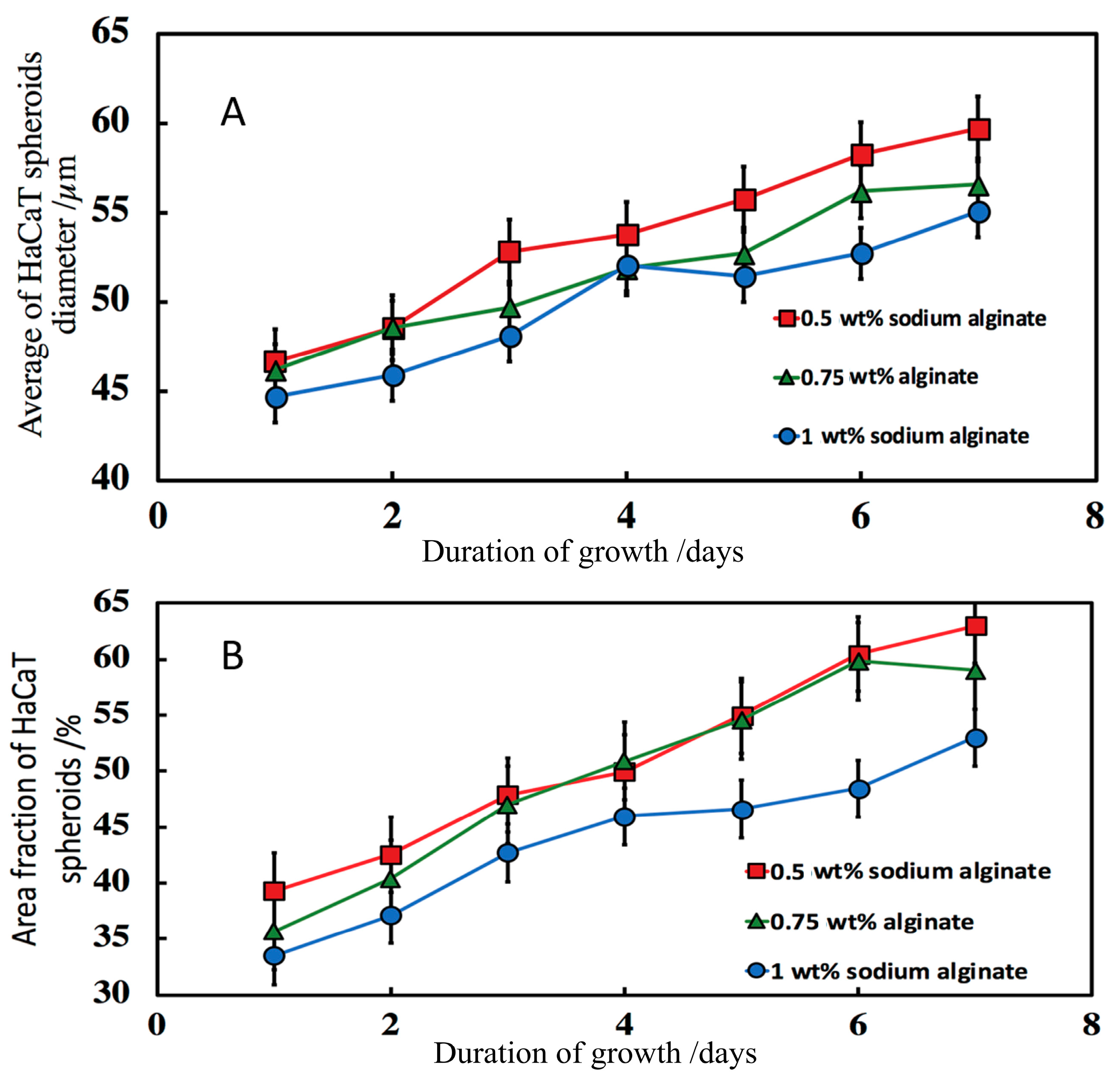
3.7.3. Effect of the Sodium Alginate Concentration and the Calcium Chloride Solution Incubation Time
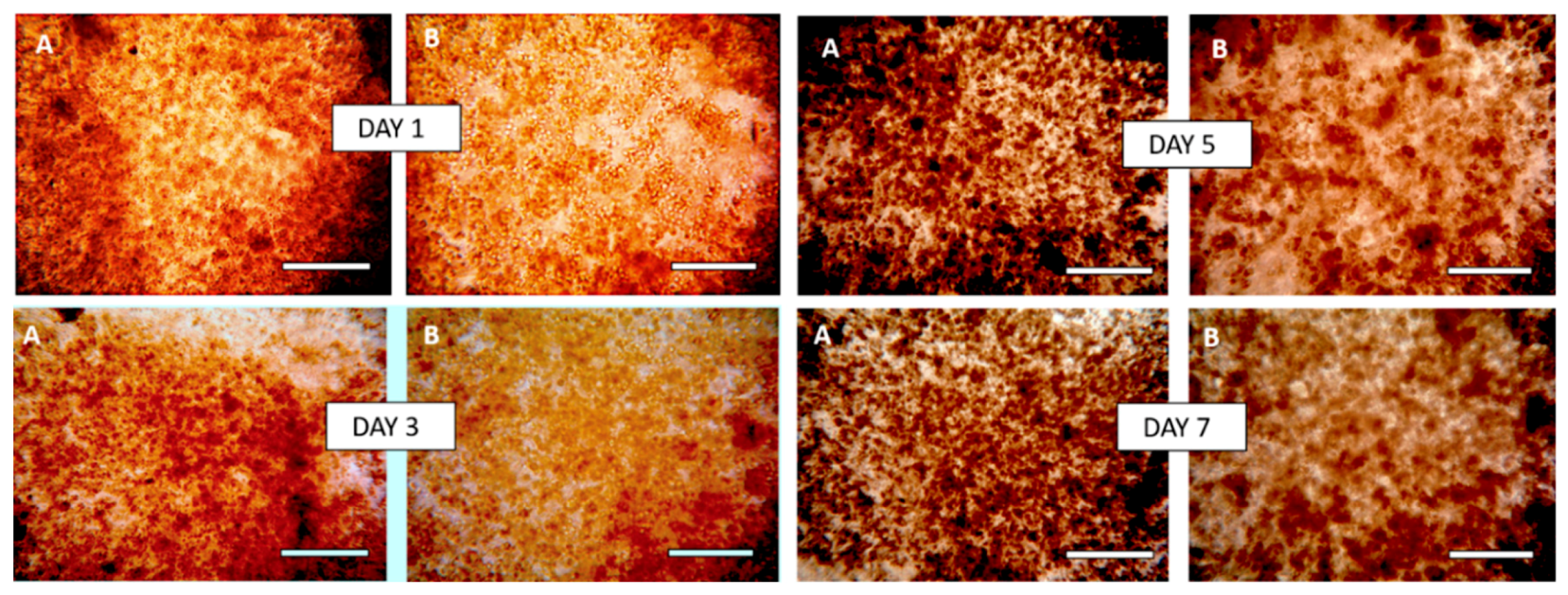
3.8. Morphology of Composite Alginate Films with HaCaT Clusteroids
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ji, S.; Guvendiren, M. Recent Advances in Bioink Design for 3D Bioprinting of Tissues and Organs. Front. Bioeng. Biotechnol. 2017, 5, 23. [Google Scholar] [CrossRef] [PubMed]
- Rustad, K.C.; Sorkin, M.; Levi, B. Strategies for organ level tissue engineering. Organogenesis 2010, 6, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Malda, J.; Viser, J.; Melchels, F.P. 25th Anniversary Article: Engineering Hydrogels for Biofabrication. Adv. Mater. 2013, 25, 5011–5028. [Google Scholar] [CrossRef] [PubMed]
- Edmondson, R.; Broglie, J.J.; Addock, A.F.; Yang, L. Three-dimensional cell culture systems and their applications in drug discovery and cell-based biosensors. Assay Drug Dev. Technol. 2014, 12, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Kular, J.K.; Basu, S.; Sharma, R.I. The extracellular matrix: Structure, composition, age-related differences, tools for analysis and applications for tissue engineering. J. Tissue Eng. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Frantz, C.; Stewart, K.M.; Weaver, V.M. The extracellular matrix at a glance. J. Cell Sci. 2010, 123, 4195–4200. [Google Scholar] [CrossRef] [PubMed]
- Larson, B. 3D Cell Culture: A Review of Current Technique. Available online: https://www.biotek.com/resources/white-papers/3d-cell-culture-a-review-of-currenttechniques/ (accessed on 20 April 2018).
- Fang, Y.; Eglen, R.M. Three-Dimensional Cell Cultures in Drug Discovery and Development. SLAS Discov. 2017, 22, 456–472. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, Y.; Takeuchi, S. Three-dimensional cell culture based on microfluidic techniques to mimic living tissues. Biomater. Sci. 2013, 1, 257–264. [Google Scholar] [CrossRef]
- Park, K.M.; Shin, Y.M.; Kim, K. Tissue Engineering and Regenerative Medicine 2017: A Year in Review. Tissue Eng. Part B Rev. 2018, 24, 327–344. [Google Scholar] [CrossRef]
- Hardwick, R.N.; Viergever, C.; Nguyen, D.G. 3D bioengineered tissues: From advancements in in vitro safety to new horizons in disease modeling. Clin. Pharmacol. Ther. 2017, 101, 453–457. [Google Scholar] [CrossRef]
- Tung, Y.C.; Hsiao, A.Y.; Allen, S.G.; Torisawa, Y.S.; Ho, M.; Takayama, S. High-throughput 3D spheroid culture and drug testing using a 384 hanging drop array. Analyst 2011, 136, 473–478. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.G.; Ortmann, D.; Hancock, M.J.; Bae, H.; Khademhosseini, A. A Hollow Sphere Soft Lithography Approach for Long-Term Hanging Drop Methods. Tissue Eng. Part C Methods 2010, 16, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.Z.; Chou, L.F.; Chien, C.C.; Chang, H.Y. Dynamic analysis of hepatoma spheroid formation: Roles of E-cadherin and β1-integrin. Cell Tissue Res. 2006, 324, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Kelm, J.M.; Timmins, N.E.; Brown, C.J.M.; Fussenegger, M.; Nielsen, L.K. Method for generation of homogeneous multicellular tumor spheroids applicable to a wide variety of cell types. Biotechnol. Bioeng. 2003, 83, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Kelm, J.M.; Fussenegger, M. Microscale tissue engineering using gravity-enforced cell assembly. Trends Biotechnol. 2004, 22, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Kato-Negishi, M.; Tsuda, Y.; Onoe, H.; Takeuchi, S. A neurospheroid network-stamping method for neural transplantation to the brain. Biomaterials 2010, 31, 8939–8945. [Google Scholar] [CrossRef] [PubMed]
- Ota, H.; Yamamoto, R.; Deguchi, K.; Tanaka, Y.; Kazoe, Y.; Sato, Y.; Miki, N. Three-dimensional spheroid-forming lab-on-a-chip using micro-rotational flow. Sens. Actuators B Chem. 2010, 147, 359–365. [Google Scholar] [CrossRef]
- Akiyama, Y.; Morishima, K. Long-term and room temperature operable bioactuator powered by insect dorsal vessel tissue. Lab Chip 2009, 9, 140–144. [Google Scholar] [CrossRef] [PubMed]
- Fakhrullin, R.F.; Brandy, M.L.; Cayre, O.J.; Velev, O.D.; Paunov, V.N. Live celloidosome structures based on the assembly of individual cells by colloid interactions. Phys. Chem. Chem. Phys. 2010, 12, 11912–11922. [Google Scholar] [CrossRef]
- Brandy, M.L.; Cayre, O.J.; Fakhrullin, R.F.; Velev, O.D.; Paunov, V.N. Directed assembly of yeast cells into living yeastosomes by microbubble templating. Soft Matter 2010, 6, 3494–3498. [Google Scholar] [CrossRef]
- Gasperini, L.; Mano, J.F.; Reis, R.L. Natural polymers for the microencapsulation of cells. J. R. Soc. Interface 2014, 11, 20140817. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, B.T.; Nicolai, T.; Benayahia, L. Stabilization of water-in-water emulsions by addition of protein particles. Langmuir 2013, 29, 10658–10664. [Google Scholar] [CrossRef] [PubMed]
- Ganley, W.; Ryan, P.; Van Duijneveldt, J. Stabilisation of water-in-water emulsions by montmorillonite platelets. J. Colloid Interface Sci. 2017, 505, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, E. Particle-based stabilization of water-in-water emulsions containing mixed biopolymers. Trends Food Sci. Technol. 2019, 83, 31–40. [Google Scholar] [CrossRef]
- Singh, P.; Medronho, B.; Miguel, M.G.; Esquena, J. On the encapsulation and viability of probiotic bacteria in edible carboxymethyl cellulose-gelatin water-in-water emulsions. Food Hydrocoll. 2018, 75, 41–50. [Google Scholar] [CrossRef]
- Atefi, E.; Joshi, R.; Mann, J.A.; Tavana, H. Interfacial Tension Effect on Cell Partition in Aqueous Two-Phase Systems. ACS Appl. Mater. Interfaces 2015, 7, 21305–21314. [Google Scholar] [CrossRef] [PubMed]
- Atefi, E.; Lemmo, S.; Fyffe, D.; Luker, G.D.; Tavana, H. High Throughput, Polymeric Aqueous Two-Phase Printing of Tumor Spheroids. Adv. Funct. Mater. 2014, 24, 6509–6515. [Google Scholar] [CrossRef]
- Lemmo, S.; Atefi, E.; Luker, G.D.; Tavana, H. Optimization of Aqueous Biphasic Tumor Spheroid Microtechnology for Anti-cancer Drug Testing in 3D Culture. Cell. Mol. Bioeng. 2014, 7, 344–354. [Google Scholar] [CrossRef]
- Singh, S.; Tavana, H. Collagen Partition in Polymeric Aqueous Two-Phase Systems for Tissue Engineering. Front. Chem. 2018, 6, 379. [Google Scholar] [CrossRef]
- Das, A.A.K.; Filby, B.W.; Geddes, D.A.; Legrand, D.; Paunov, V.N. High throughput fabrication of cell spheroids by templating water-in-water Pickering emulsions. Mater. Horiz. 2017, 4, 1196–1200. [Google Scholar] [CrossRef]
- Ng, W.L.; Yeong, W.Y.; Naing, M.W. Cellular Approaches to Tissue-Engineering of Skin: A Review. J. Tissue Sci. Eng. 2015, 6, 1000150. [Google Scholar] [CrossRef]
- Vig, K.; Chaudhari, A.; Tripathi, S. Advances in Skin Regeneration Using Tissue Engineering. Int. J. Mol. Sci. 2017, 18, 789. [Google Scholar] [CrossRef] [PubMed]
- Varaprasad, K.; Raghavendra, G.M.; Jayaramudu, T. A mini review on hydrogels classification and recent developments in miscellaneous applications. Mater. Sci. Eng. 2017, 79, 958–971. [Google Scholar] [CrossRef]
- Santana, B.P.; Nedel, F.; Piva, E.; de Carvalho, R.V.; Demarco, F.F.; Carreño, N.L.V. Preparation, Modification, and Characterization of Alginate Hydrogel with Nano-/Microfibers: A New Perspective for Tissue Engineering. BioMed. Res. Int. 2013, 2013, 307602. [Google Scholar] [CrossRef]
- Jang, J.; Seol, Y.J.; Kim, H.J. Effects of alginate hydrogel cross-linking density on mechanical and biological behaviors for tissue engineering. J. Mech. Behav. Biomed. Mater. 2014, 37, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Adams, G.; Buttery, L. Alginate Encapsulation Technology Supports Embryonic Stem Cell. J. Biotechnol. 2009, 144, 304–312. [Google Scholar] [CrossRef]
- Smits, J.P.H.; Niehues, H.; Rikken, G. Immortalized N/TERT keratinocytes as an alternative cell source in 3D human epidermal models. Sci. Rep. 2017, 7, 11838. [Google Scholar] [CrossRef]
- Boukamp, P.; Petrussevska, R.T.; Breitkreutz, D. Normal Keratinization in a Spontaneously Immortalized Aneuploid Human Keratinocyte Cell Line. J. Cell Biol. 1988, 106, 761–771. [Google Scholar] [CrossRef] [PubMed]
- Chrzanowski, T.H.; Crotty, R.D.; Hubbard, J.G.; Welch, R.P. Applicability of the fluorescein diacetate method of detecting active bacteria in freshwater. Microb. Ecol. 1984, 10, 179–185. [Google Scholar] [CrossRef] [PubMed]
- McClements, D.J.; Jafari, S.M. Improving emulsion formation, stability and performance using mixed emulsifiers: A review. Adv. Colloid Interface Sci. 2018, 251, 55–79. [Google Scholar] [CrossRef]
- Breeuwer, P.; Drocourt, J.; Rombouts, F.M.; Abee, T. A Novel Method for Continuous Determination of the Intracellular pH in Bacteria with the Internally Conjugated Fluorescent Probe 5 (and 6-)-Carboxyfluorescein Succinimidyl Ester. Appl. Environ. Microbiol. 1996, 62, 178–183. [Google Scholar] [PubMed]
- Yang, C.H.; Wang, M.X.; Haider, H. Strengthening alginate/polyacrylamide hydrogels using various multivalent cations. ACS Appl. Mater. Interfaces 2013, 5, 10418–10422. [Google Scholar] [CrossRef] [PubMed]
- Laschke, M.W.; Menger, M.D. Life is 3D: Boosting Spheroid Function for Tissue Engineering. Trends Biotechnol. 2017, 35, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Brohem, C.A.; da Silva Cardeal, L.B.; Tiago, M. Artificial skin in perspective: Concepts and applications. Pigment Cell Melanoma Res. 2013, 24, 35–50. [Google Scholar] [CrossRef] [PubMed]
- Wikramanayake, T.C.; Stojadinovic, O.; Tomic-Canic, M. Epidermal Differentiation in Barrier Maintenance and Wound Healing. Adv. Wound Care 2014, 3, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Mironov, V.; Visconti, R.P.; Kasyanov, V.; Forgacs, G.; Drake, C.J.; Markwald, R.R. Organ printing: Tissue spheroids as building blocks. Biomaterials 2009, 30, 2164–2174. [Google Scholar] [CrossRef] [PubMed]
- Kitala, D.; Kawecki, M.; Klama-Baryla, A.; Labus, W.; Kraut, M.; Glik, J.; Ryszkiel, I.; Kawecki, M.P.; Nowak, M. Allogeneic vs. Autologous Skin Grafts in the Therapy of Patients with Burn Injuries: A Restrospective, Open-label Clinical Study with Pair Matching. Adv. Clin. Exp. Med. 2016, 25, 923–929. [Google Scholar] [CrossRef]
- Murphy, P.S.; Evans, G.R.D. Advances in Wound Healing: A Review of Current Wound Healing Products. Plast. Surg. Int. 2012, 2012, 190436. [Google Scholar] [CrossRef]










© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Celik, S.B.G.; Dominici, S.R.; Filby, B.W.; Das, A.A.K.; Madden, L.A.; Paunov, V.N. Fabrication of Human Keratinocyte Cell Clusters for Skin Graft Applications by Templating Water-in-Water Pickering Emulsions. Biomimetics 2019, 4, 50. https://doi.org/10.3390/biomimetics4030050
Celik SBG, Dominici SR, Filby BW, Das AAK, Madden LA, Paunov VN. Fabrication of Human Keratinocyte Cell Clusters for Skin Graft Applications by Templating Water-in-Water Pickering Emulsions. Biomimetics. 2019; 4(3):50. https://doi.org/10.3390/biomimetics4030050
Chicago/Turabian StyleCelik, Sevde B. G., Sébastien R. Dominici, Benjamin W. Filby, Anupam A. K. Das, Leigh A. Madden, and Vesselin N. Paunov. 2019. "Fabrication of Human Keratinocyte Cell Clusters for Skin Graft Applications by Templating Water-in-Water Pickering Emulsions" Biomimetics 4, no. 3: 50. https://doi.org/10.3390/biomimetics4030050
APA StyleCelik, S. B. G., Dominici, S. R., Filby, B. W., Das, A. A. K., Madden, L. A., & Paunov, V. N. (2019). Fabrication of Human Keratinocyte Cell Clusters for Skin Graft Applications by Templating Water-in-Water Pickering Emulsions. Biomimetics, 4(3), 50. https://doi.org/10.3390/biomimetics4030050