Synthesis and Characterization of Acetic Acid-Doped Polyaniline and Polyaniline–Chitosan Composite

 , and
, and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of PAni Powders
2.3. Synthesis of PAni–Cs Composite
2.4. Characterization of PAni Powder and PAni–Cs Composites
2.5. Cell Viability Assay
2.5.1. Preparation of the Lymphocyte Culture
2.5.2. Cell Viability Assay of Lymphocytes on PAni–Cs Composites
Preparation of Test Samples
Treatment of Lymphocyte Culture
Cell Count
Statistical Analysis
3. Results
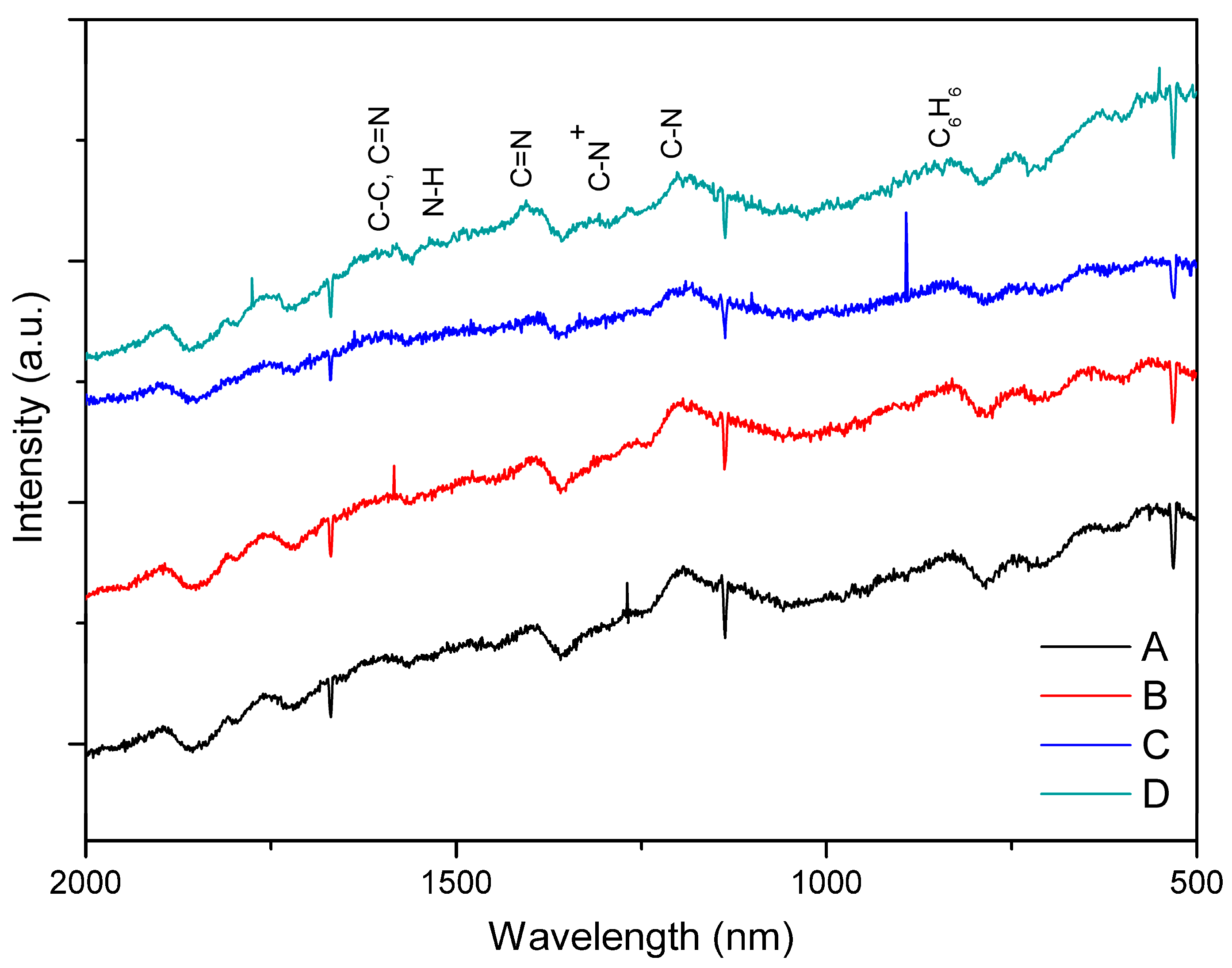
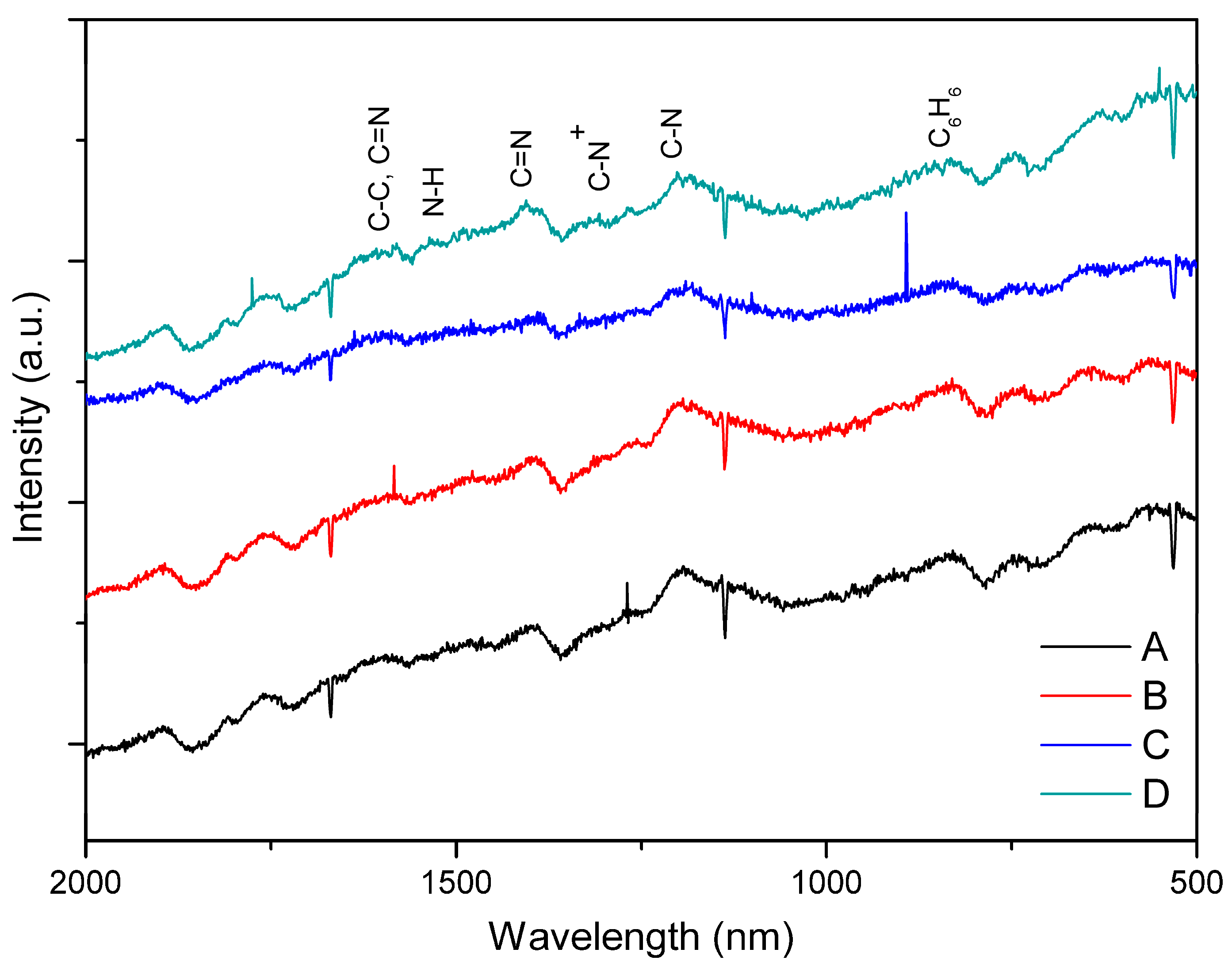
3.1. PAni Sample Analysis
3.2. Surface Morphology and Composition of PAni–Cs Films
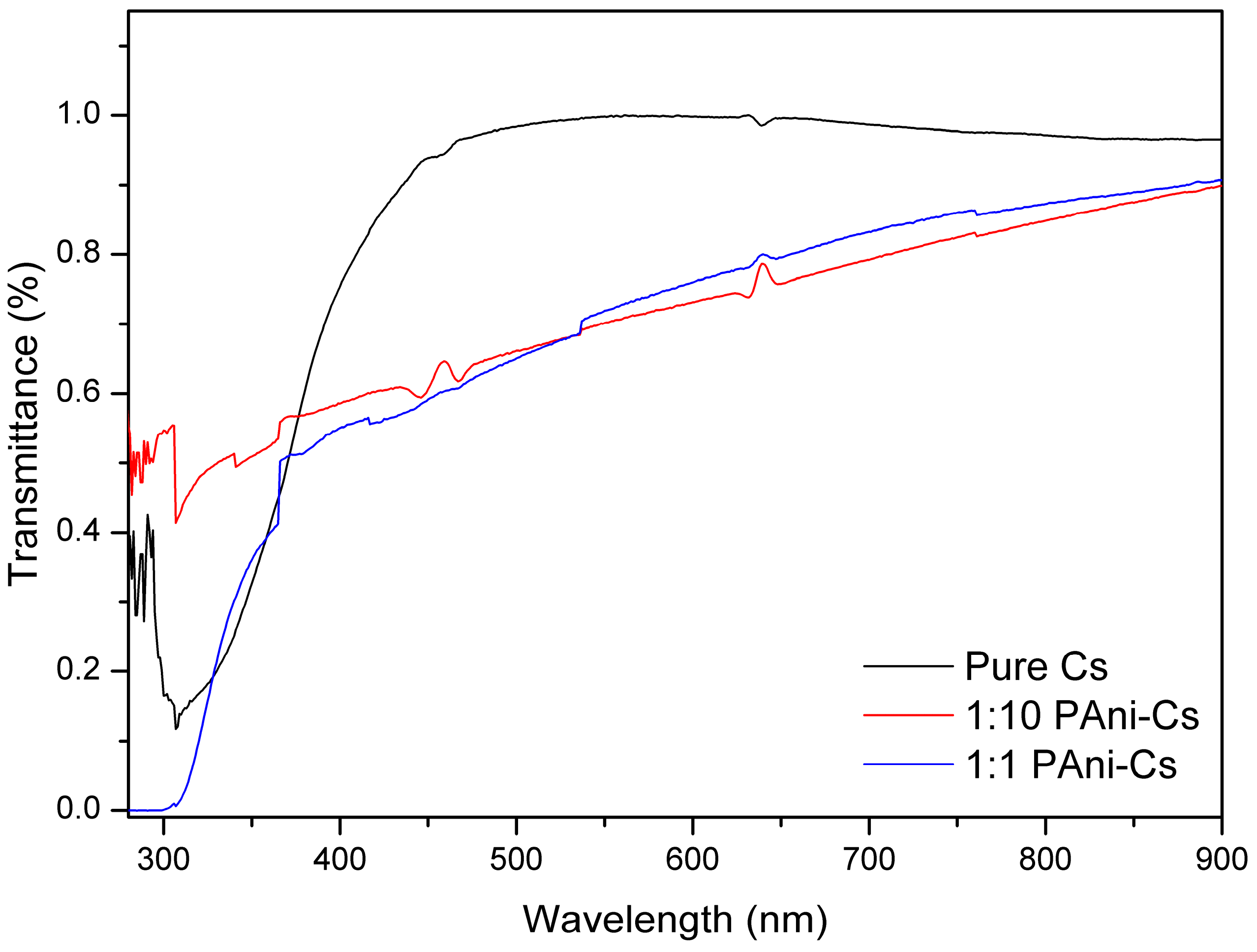
3.3. UV–Vis Spectroscopic Study of PAni–Cs Films
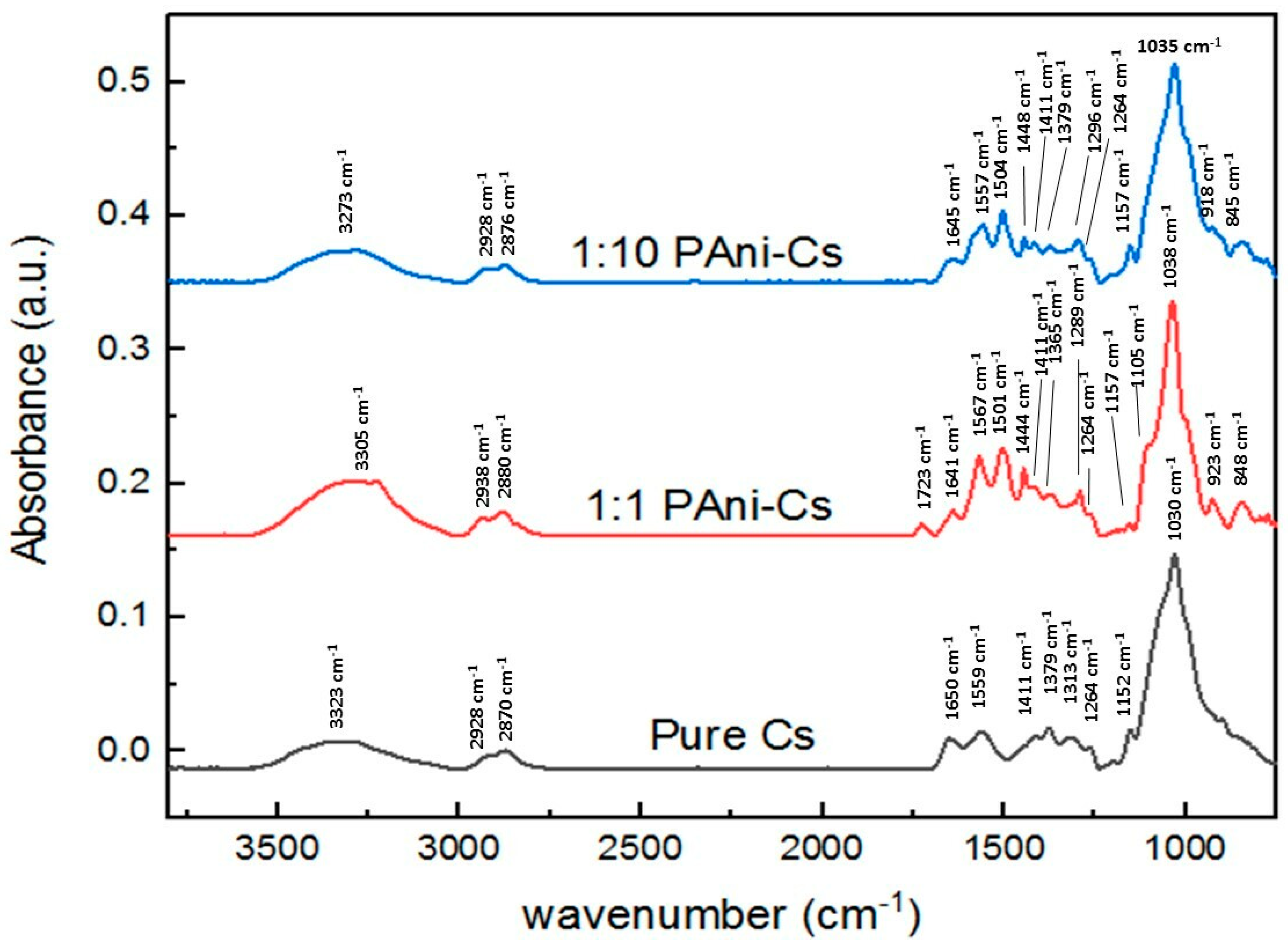
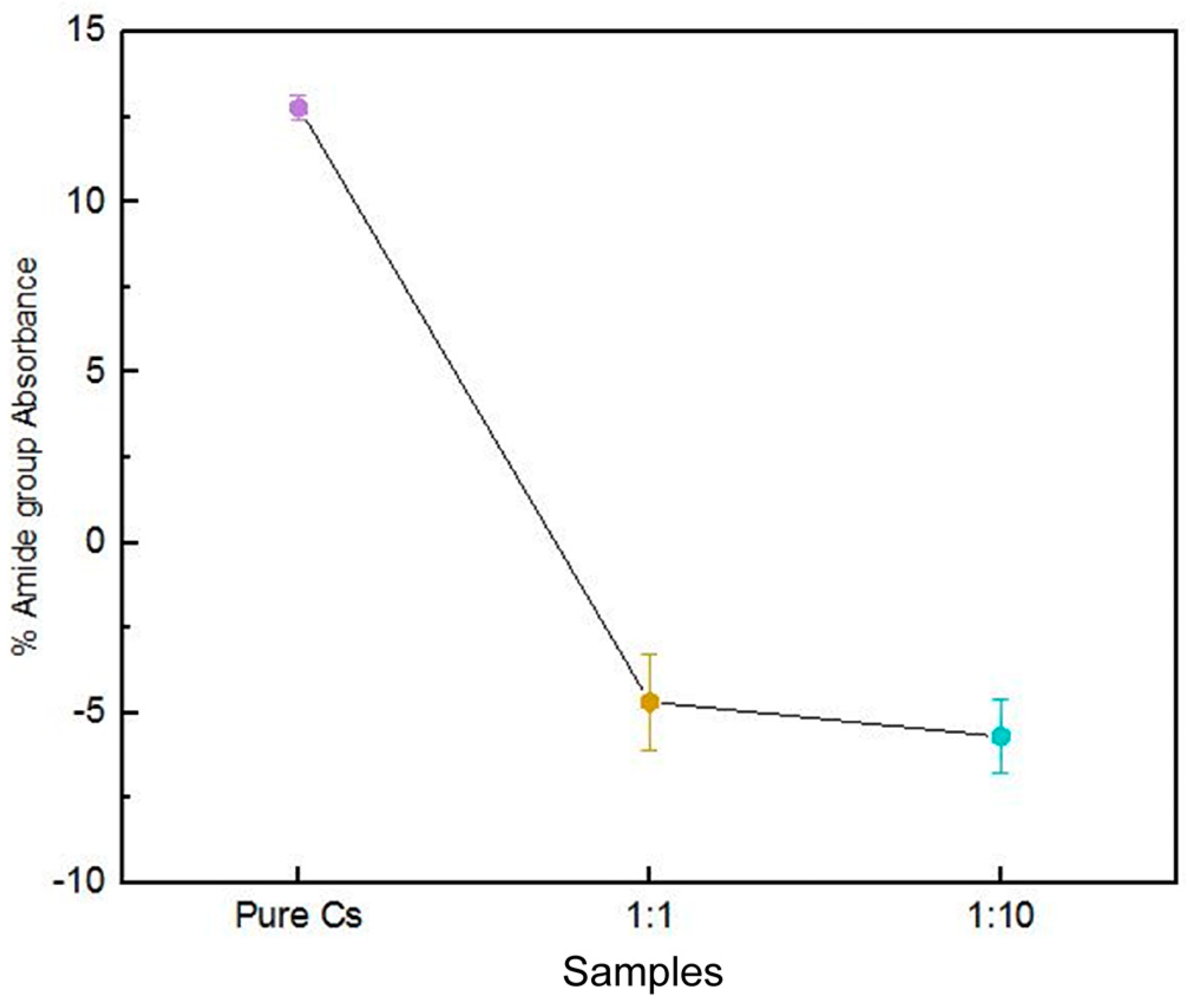
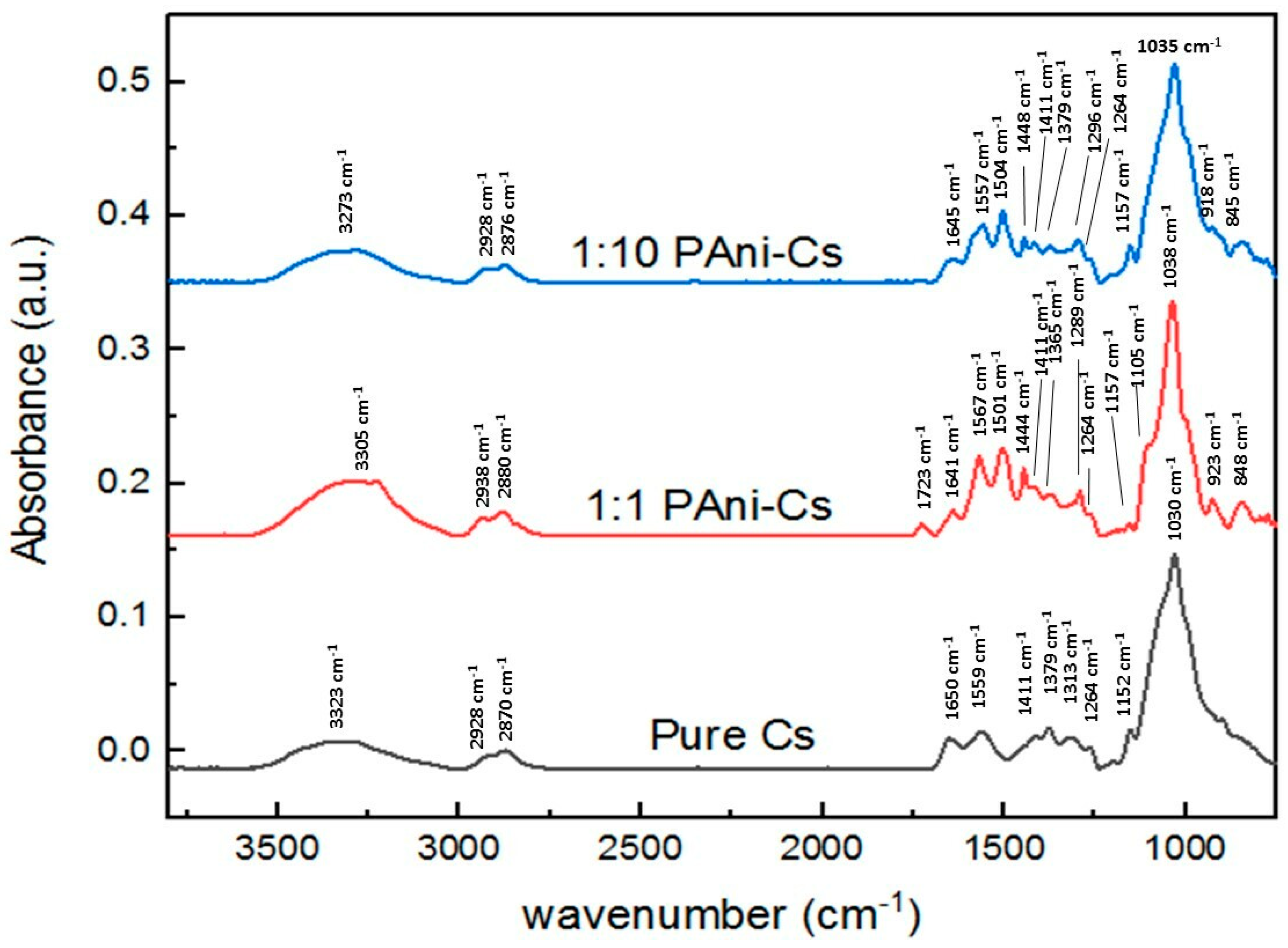
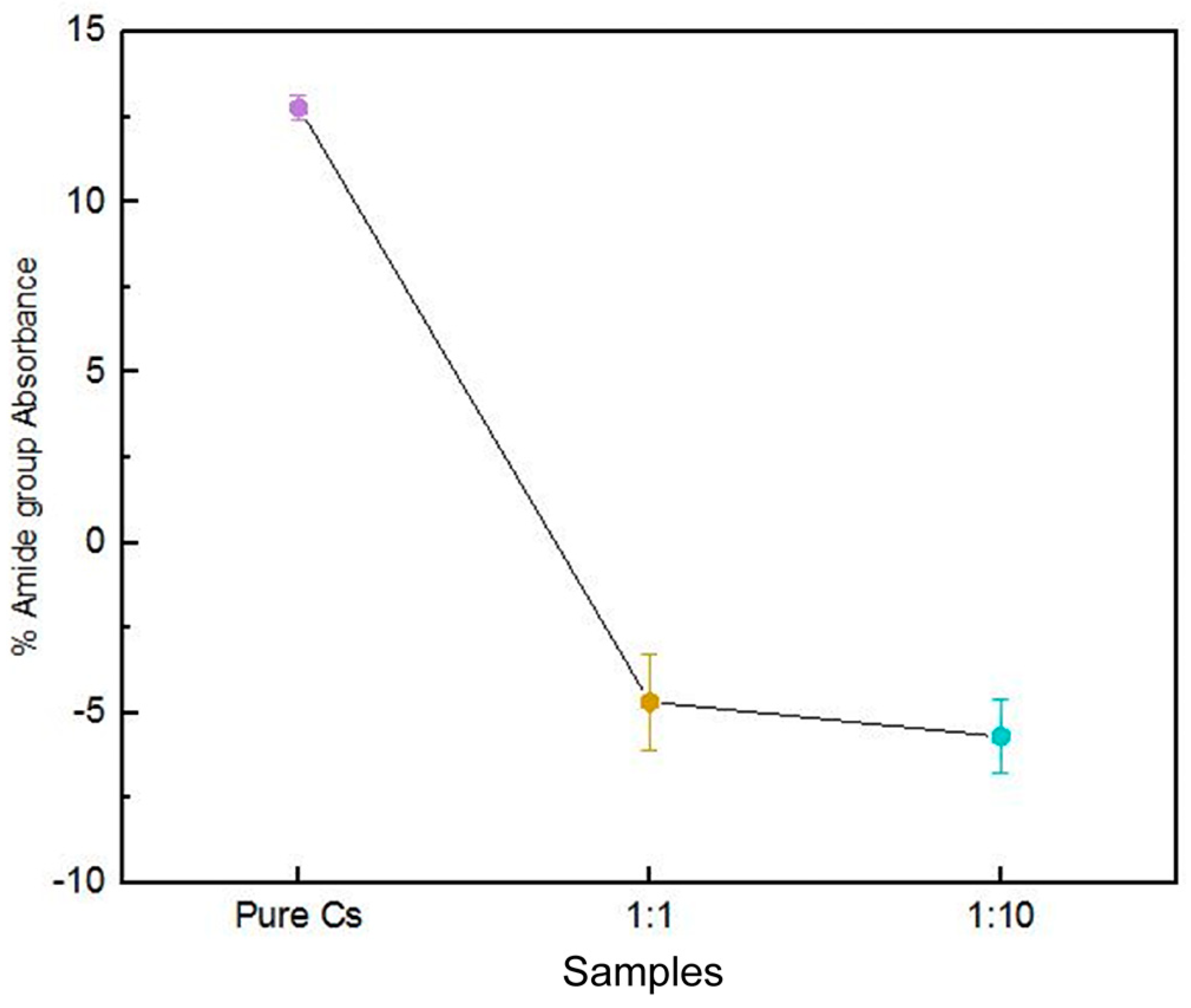
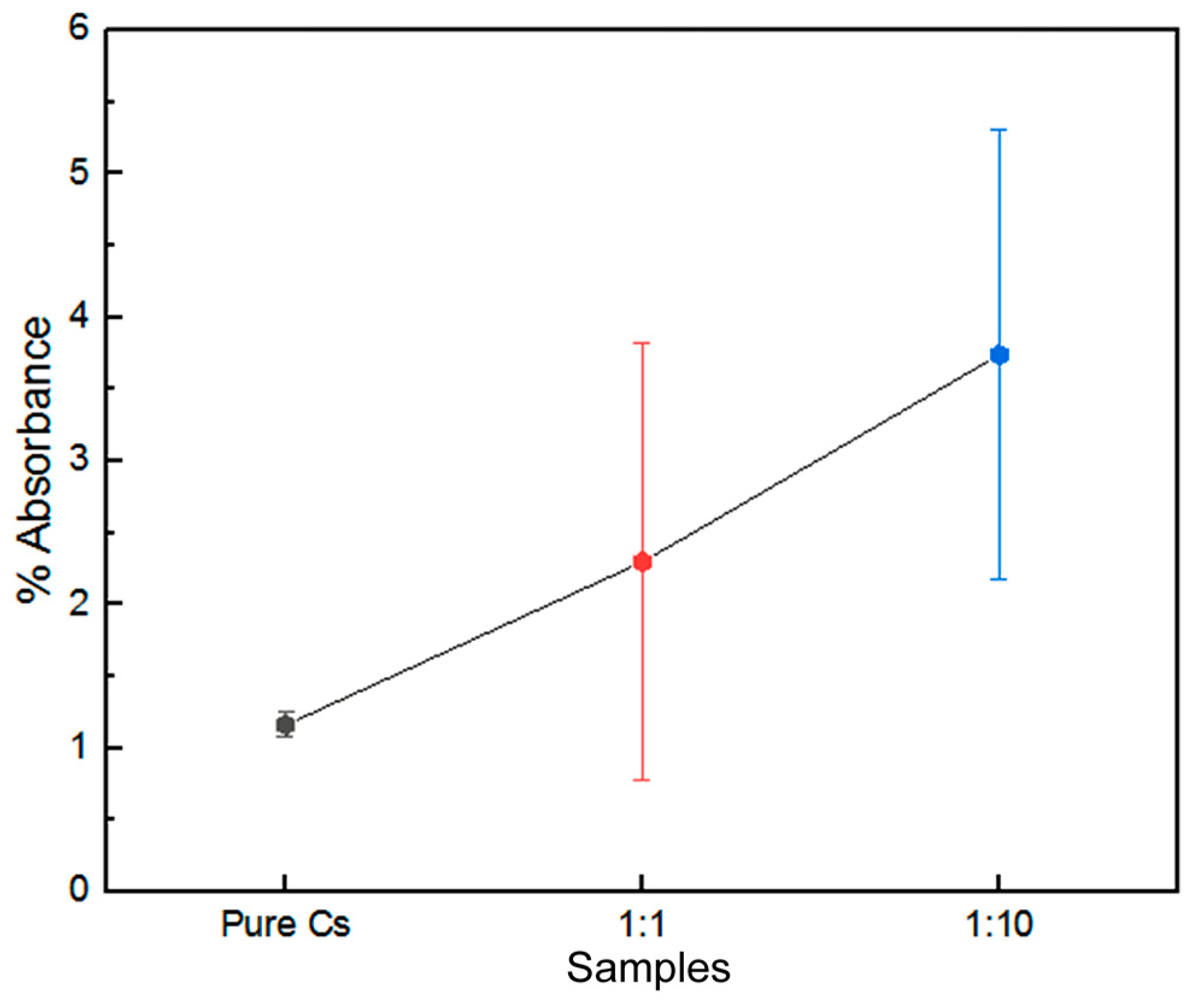
3.4. SR-FTIR Spectral Analyses of PAni–Cs Composites
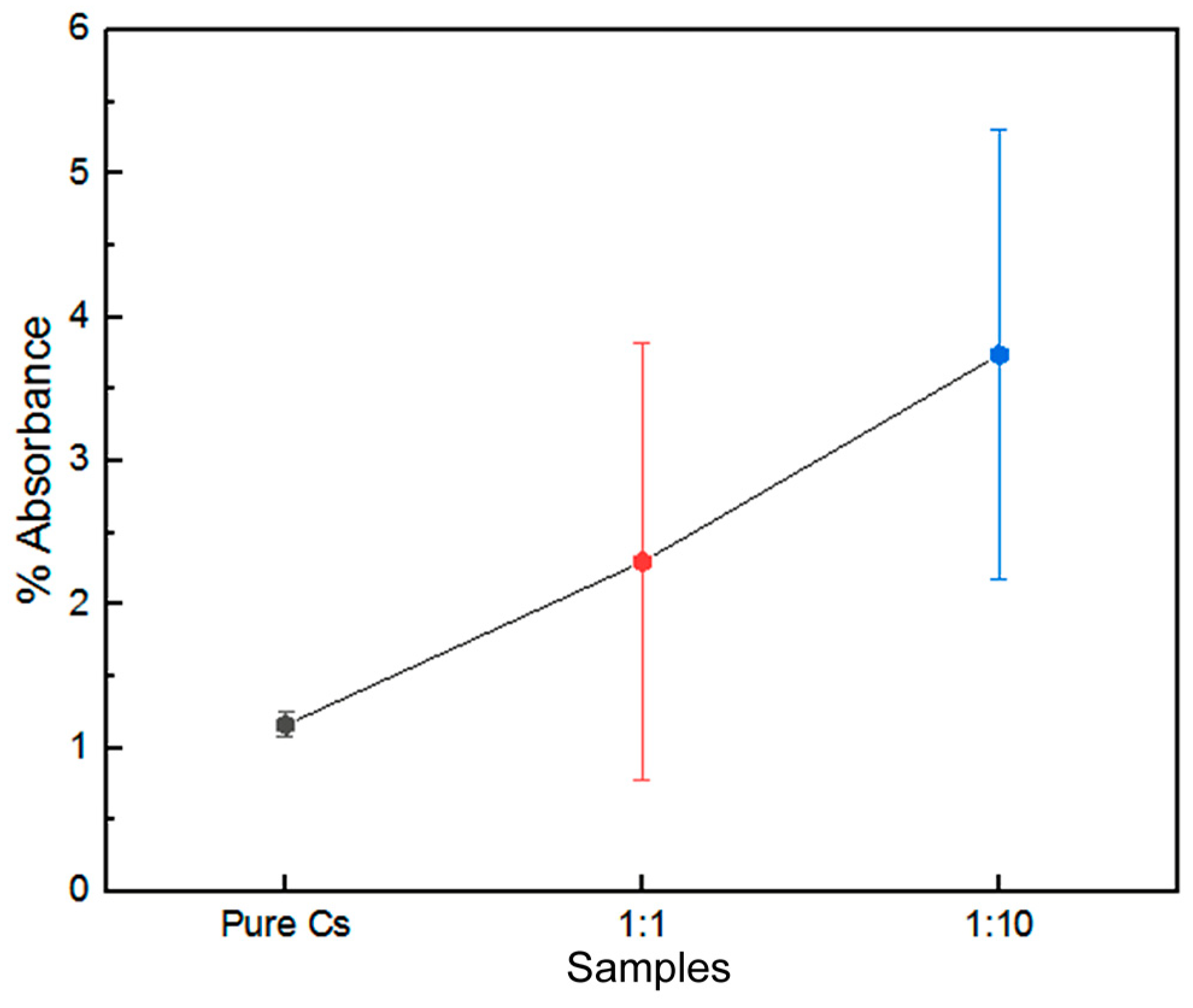
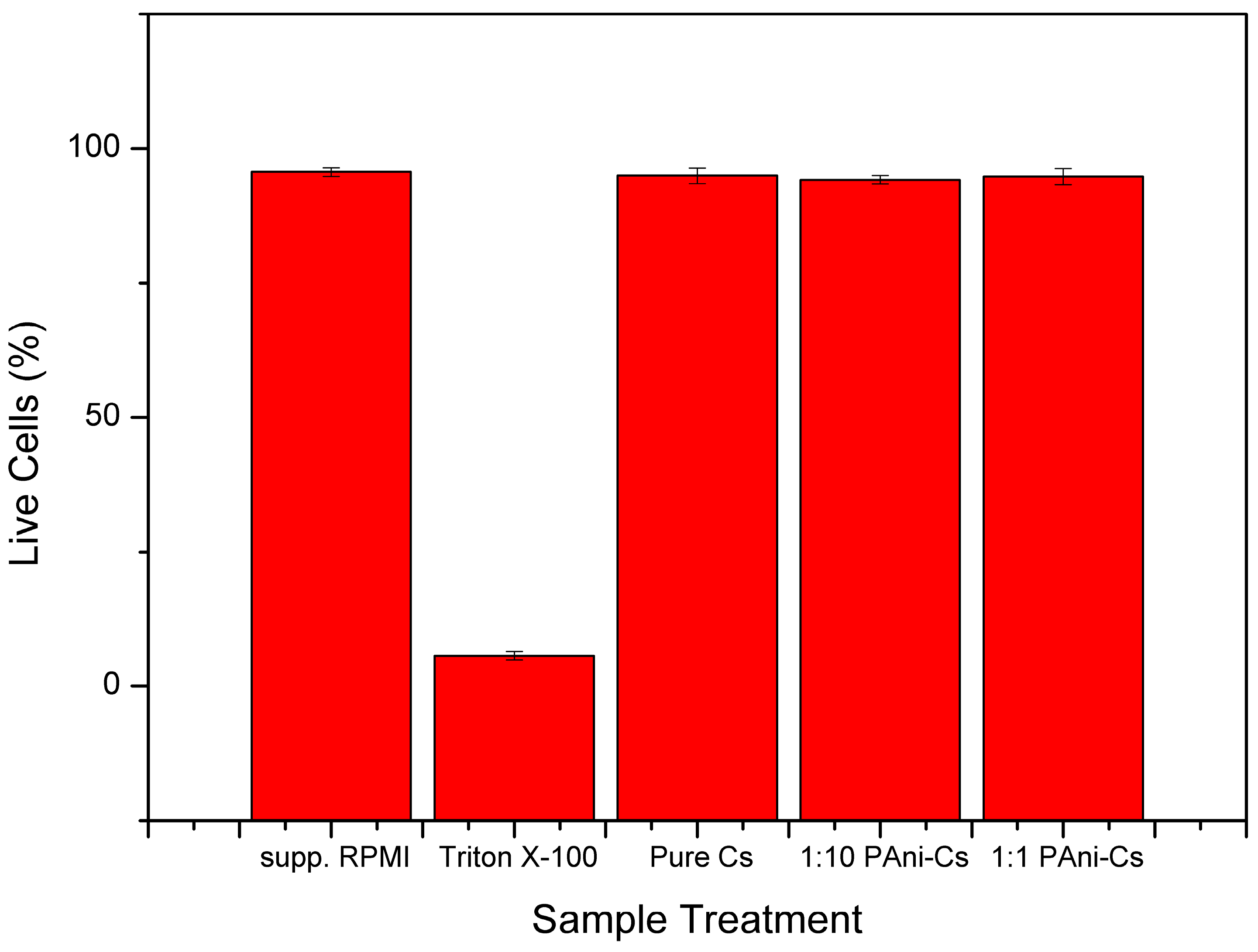
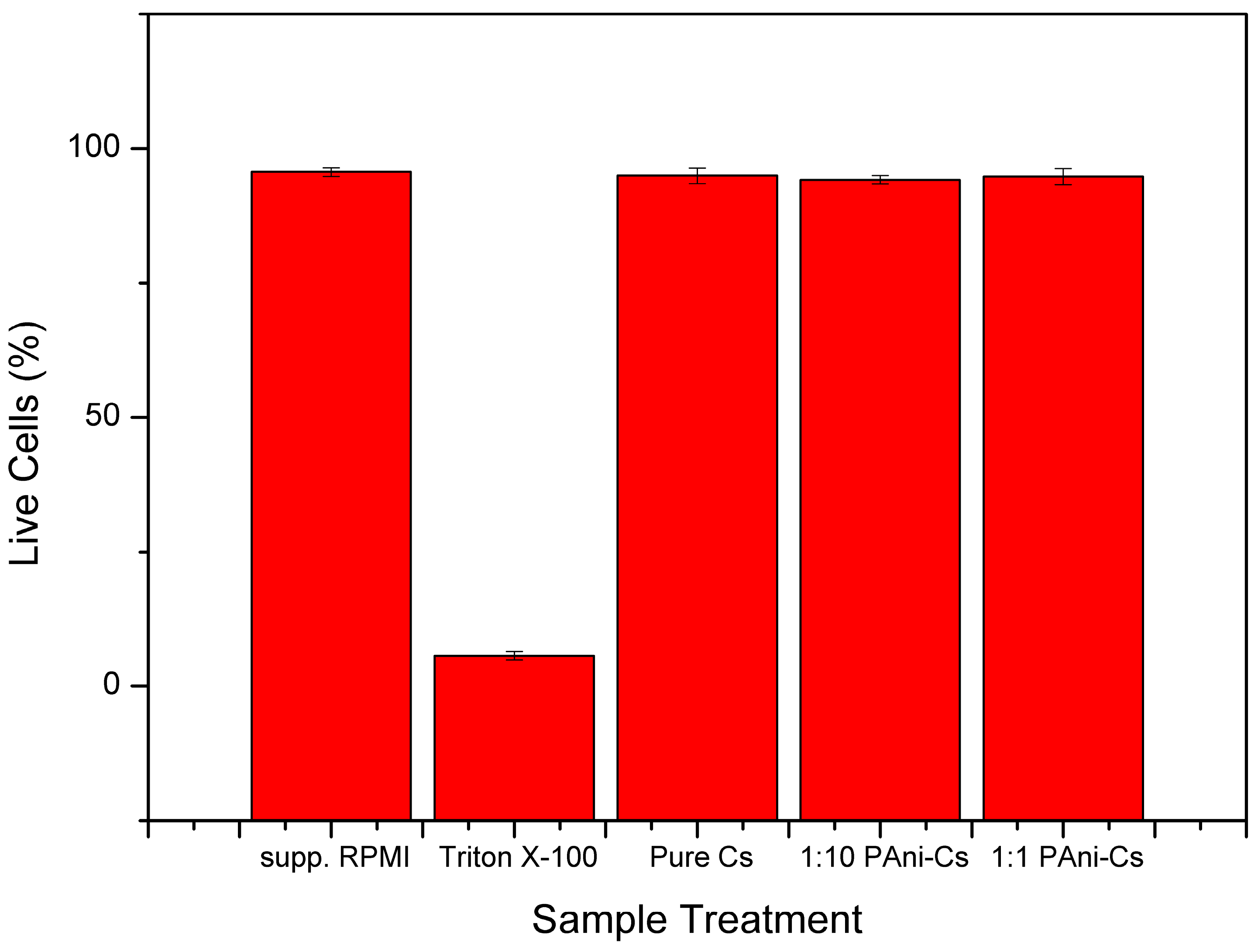
3.5. Cell Viability Assessment
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Taaca, K.L.M.; Vasquez, M.R., Jr. Hemocompatibility and cytocompatibility of pristine and plasma-treated silver-zeolite-chitosan composites. Appl. Surf. Sci. 2018, 432, 324–331. [Google Scholar] [CrossRef]
- He, W.; Benson, R. Handbook of Polymer Applications in Medicine and Medical Devices, 1st ed.; William Andrew: Oxford, UK, 2013; pp. 55–76. ISBN 9780323228053. [Google Scholar]
- Pornanong, A. Introduction to biomaterials for wound healing. Wound Heal. Biomater. 2016, 2, 3–38. [Google Scholar] [CrossRef]
- O’Brien, F.J. Biomaterials and scaffolds for tissue engineering. Mater. Today 2011, 14, 88–95. [Google Scholar] [CrossRef]
- Taaca, K.L.M.; Vasquez, M.R., Jr. Fabrication of Ag-exchanged zeolite/chitosan composites and effects of plasma treatment. Microporous Mesoporous Mater. 2017, 241, 383–391. [Google Scholar] [CrossRef]
- Qi, W.; Zhang, X.; Wang, H. Self-assembled polymer nanocomposites for biomedical application. Curr. Opin. Colloid Interface Sci. 2018, 35, 36–41. [Google Scholar] [CrossRef]
- Tozar, A.; Karahan, I.H. A comprehensive study on electrophoretic deposition of a novel type of collagen and hexagonal boron nitride reinforced hydroxyapatite/chitosan biocomposite coating. Appl. Surf. Sci. 2018, 452, 322–336. [Google Scholar] [CrossRef]
- Kayan, D.B.; Kocak, D.; Ilhan, M. The activity of PAni-Chitosan composite film decorated with Pt nanoparticles for electrocatalytic hydrogen generation. Int. J. Hydrogen Energy 2016, 41, 10522–10529. [Google Scholar] [CrossRef]
- Soni, B.; Mahmoud, B.; Chang, S.; El-Giar, E.M.; Hassan, E.B. Physicochemical, antimicrobial and antioxidant properties of chitosan/TEMPO biocomposite packaging films. Food Packag. Shelf Life 2018, 17, 73–79. [Google Scholar] [CrossRef]
- Rathinam, K.; Singh, S.P.; Arnusch, C.J.; Kasher, R. An environmentally-friendly chitosan-lysozyme biocomposite for the T effective removal of dyes and heavy metals from aqueous solutions. Carbohydr. Polym. 2018, 199, 506–515. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Oves, M.; Almeelbi, T.; Al-Makishah, N.H.; Barakat, M.A. Hybrid chitosan/polyaniline-polypyrrole biomaterial for enhanced adsorption and antimicrobial activity. J. Colloid Interface Sci. 2017, 490, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Sedaghat, S. Synthesis and characterization of new biocompatible copolymer: Chitosan-graft-polyaniline. Int. Nano Lett. 2014, 4, 99. [Google Scholar] [CrossRef]
- Janaki, V.; Oh, B.T.; Shanthi, K.; Lee, K.J.; Ramasamy, A.K.; Kamala-Kannan, S. Polyaniline/chitosan composite: An eco-friendly polymer for enhanced removal of dyes from aqueous solution. Synth. Met. 2012, 162, 974–980. [Google Scholar] [CrossRef]
- Kasparkova, V.; Humpolicek, P.; Capáková, Z.; Bober, P.; Stejskal, J.; Trchová, M.; Rejmontová, P.; Junkar, I.; Lehocký, M.; Mozetič, M. Cell-compatible conducting polyaniline films prepared in colloidal dispersion mode. Colloids Surf. B Biointerfaces 2017, 157, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Kašpárková, V.; Humpolíček, P.; Stejskal, J.; Kopecká, J.; Kuceková, Z.; Moučka, R. Conductivity impurity profile, and cytotoxicity of solvent-extractedpolyaniline. Polym. Adv. Technol. 2016, 27, 156–161. [Google Scholar] [CrossRef]
- Li, M.; Guo, Y.; Wei, Y.; MacDiarmid, A.G.; Lelkes, P.I. Electrospinning polyaniline-contained gelatin nanofibers for tissue engineering applications. Biomaterials 2006, 27, 2705–2715. [Google Scholar] [CrossRef] [PubMed]
- Yavuz, A.G.; Uygun, A.; Bhethanabotla, V. Substituted polyaniline/chitosan composites: Synthesis and characterization. Carbohydr. Polym. 2009, 75, 448–453. [Google Scholar] [CrossRef]
- Yavuz, A.G.; Uygun, A.; Bhethanabotla, V. Preparation of substituted polyaniline/chitosan composites by in situ electropolymerization and their application to glucose sensing. Carbohydr. Polym. 2010, 81, 712–719. [Google Scholar] [CrossRef]
- Thanpitcha, T.; Sirivat, A.; Jamieson, A.M.; Rujiravanit, R. Preparation and characterization of polyaniline/chitosan blend film. Carbohydr. Polym. 2006, 64, 560–568. [Google Scholar] [CrossRef]
- Defensor, M.J.; Salvador, A.A. Development of Cost Effective Confocal Micro-Raman System. In Proceedings of the 28th Samahang Pisika ng Pilipinas Physics Congress (SPP 2010), Antipolo City, Philippines, 25–27 October 2010. [Google Scholar]
- Dunkhunthod, B.; Thumanu, K.; Eumkeb, G. Application of FTIR microspectroscopy for monitoring and discrimination of the anti-adipogenesis activity of baicalein in 3T3-L1 adipocytes. Vib. Spectrosc. 2017, 89, 92–101. [Google Scholar] [CrossRef]
- Helthcare, G.E. Ficoll-Paque PLUS Instructions; GE Healthcare Bio-Sciences AB: Uppsala, Sweden, 2007. [Google Scholar]
- Sedenkova, I.; Trchova, M.; Stejskal, J. Thermal degradation of polyaniline films prepared in solutions of strong and weak acids and in water–FTIR and Raman spectroscopic studies. Polym. Degrad. Stab. 2008, 93, 2147–2157. [Google Scholar] [CrossRef]
- Grzeszcuk, M.; Grańska, A.; Szostak, R. Raman spectroelectrochemistry of polyaniline synthesized using different electrolytic regimes-multivariate analysis. Int. J. Electrochem. Sci. 2013, 8, 8951–8965. [Google Scholar]
- Lago, M.A.; Rodriguez Bernaldo de Quiros, A.; Sendon, R.; Sanches-Silva, A.; Costa, H.S.; Sánchez-Machado, D.I.; Paseiro Losada, P. Compilation of analytical methods to characterize and determine chitosan, and main applications of the polymer in food active packaging. CyTA-J. Food. 2011, 9, 319–328. [Google Scholar] [CrossRef]
- Khairkar, S.R.; Raut, A.R. Synthesis of chitosan-graft-polyaniline-based composites. Am. J. Mater. Sci. Eng. 2014, 2, 62–67. [Google Scholar] [CrossRef]
- Abbasian, M.; Jaymand, M.; Niroomand, P.; Farnoudian-Habibi, A.; Karaj-Abad, S.G. Grafting of aniline derivatives onto chitosan and their applications for removal of reactive dyes from industrial effluents. Int. J. Biol. Macromol. 2017, 95, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Bandara, S.; Carnegie, C.; Johnson, C.; Akindoju, F.; Williams, E.; Swaby, J.M.; Oki, A.; Carson, L.E. Synthesis and characterization of zinc/chitosan-folic acid complex. Heliyon 2018, 4, e00737. [Google Scholar] [CrossRef] [PubMed]
- Tumacder, D.v.C. Synthesis and Characteristics of Polyaniline (PAni) Produced via Two-Electrode Electrochemical Technique. Undergraduate Thesis, MSU-IIT, Iligan, Philippines, 2012. [Google Scholar]
- Bhadra, S.; Khastgir, D.; Singha, N.K.; Lee, J.H. Progress in preparation, processing and applications of polyaniline. Prog. Polym. Sci. 2009, 34, 783–810. [Google Scholar] [CrossRef]
- Hu, Z.; Hong, P.; Liao, M.; Kong, S.; Huang, N.; Ou, C.; Li, S. Preparation and characterization of chitosan–agarose composite films. Materials 2016, 9, 816. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Aniline to Acid Dopant Concentration Ratio |
|---|---|
| A | 5.48 M aniline:0.1 M HCl |
| B | 5.48 M aniline:0.1 M CH3COOH |
| C | 5.48 M aniline:0.01 M HCl |
| D | 5.48 M aniline:0.01 M CH3COOH |
| % Weight | Pure Cs | 1:10 PAni–Cs | 1:1 PAni–Cs |
|---|---|---|---|
| C | 37.505 | 59.249 | 53.056 |
| O | 41.419 | 40.751 | 46.944 |
| Wavenumber (cm−1) | Vibration Modes | ||
|---|---|---|---|
| Pure Cs | 1:1 PAni–Cs | 1:10 PAni–Cs | |
| 3323 | 3305 | 3273 | ν(NH2) in primary amines ν(OH) in pyranose ring |
| 2928 | 2938 | 2924 | ν(CH2) in CH2OH group |
| 2870 | 2880 | 2876 | ν(C–H) in pyranose ring |
| 1650–1559 | 1723–1641 | 1645 | ν(C=O) in amide I band δ(N–H) of amide II |
| - | 1567 | 1557 | ν(C=N) of quinoid ring |
| - | 1501 | 1504 | ν(C=C) of benzenoid ring |
| - | 1444 | 1448 | ν(C=C) of aromatic ring ν(N=N) in PAni structure |
| 1411 | 1411 | 1411 | ν- vibrations in amide I, II, and III |
| 1379 | 1365 | 1379 | δ(C–H) in methyl group of amide |
| 1313 | 1289 | 1296 | δ(C–H) in chitosan ring structure ν(C–N) of benzenoid ring |
| 1264 | 1264 | 1264 | δ(N–H) in amide group |
| 1152 | 1157 | 1157 | ν(C–O) in glycosidic linkage |
| - | 1105 (shoulder) | - | ν(C–O) in glycosidic linkage |
| 1030 | 1038 | 1035 | ν(C–O) in secondary OH group |
| - | 923 | 918 | vibrations in pyranose ring |
| - | 848 | 845 | δ(C–H) of benzenoid ring |
| Descriptive Statistics | Test Statistics | ||||||
|---|---|---|---|---|---|---|---|
| N | Mean | SD | Variance | Chi-Square | df | p-value | |
| Pure Cs | 3 | 94.93333 | 1.50111 | 2.25333 | 4.50667 | 2 | 0.2101 |
| 1:1 | 3 | 94.76667 | 1.51438 | 2.29333 | 4.58667 | 2 | 0.20186 |
| 1:10 | 3 | 94.16667 | 0.75719 | 0.57333 | 1.14667 | 2 | 0.87271 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pasela, B.R.; Castillo, A.P.; Simon, R.; Pulido, M.T.; Mana-ay, H.; Abiquibil, M.R.; Montecillo, R.; Thumanu, K.; Tumacder, D.v.; Taaca, K.L. Synthesis and Characterization of Acetic Acid-Doped Polyaniline and Polyaniline–Chitosan Composite. Biomimetics 2019, 4, 15. https://doi.org/10.3390/biomimetics4010015
Pasela BR, Castillo AP, Simon R, Pulido MT, Mana-ay H, Abiquibil MR, Montecillo R, Thumanu K, Tumacder Dv, Taaca KL. Synthesis and Characterization of Acetic Acid-Doped Polyaniline and Polyaniline–Chitosan Composite. Biomimetics. 2019; 4(1):15. https://doi.org/10.3390/biomimetics4010015
Chicago/Turabian StylePasela, Bianca Rae, Acelle Pearl Castillo, Rhenish Simon, Maria Teresa Pulido, Haidee Mana-ay, Ma. Roxan Abiquibil, Rhys Montecillo, Kanjana Thumanu, Doebner von Tumacder, and Kathrina Lois Taaca. 2019. "Synthesis and Characterization of Acetic Acid-Doped Polyaniline and Polyaniline–Chitosan Composite" Biomimetics 4, no. 1: 15. https://doi.org/10.3390/biomimetics4010015
APA StylePasela, B. R., Castillo, A. P., Simon, R., Pulido, M. T., Mana-ay, H., Abiquibil, M. R., Montecillo, R., Thumanu, K., Tumacder, D. v., & Taaca, K. L. (2019). Synthesis and Characterization of Acetic Acid-Doped Polyaniline and Polyaniline–Chitosan Composite. Biomimetics, 4(1), 15. https://doi.org/10.3390/biomimetics4010015