
Numerical Parametric Investigation of Nonaqueous Vanadium Redox Flow Batteries

Abstract
:1. Introduction
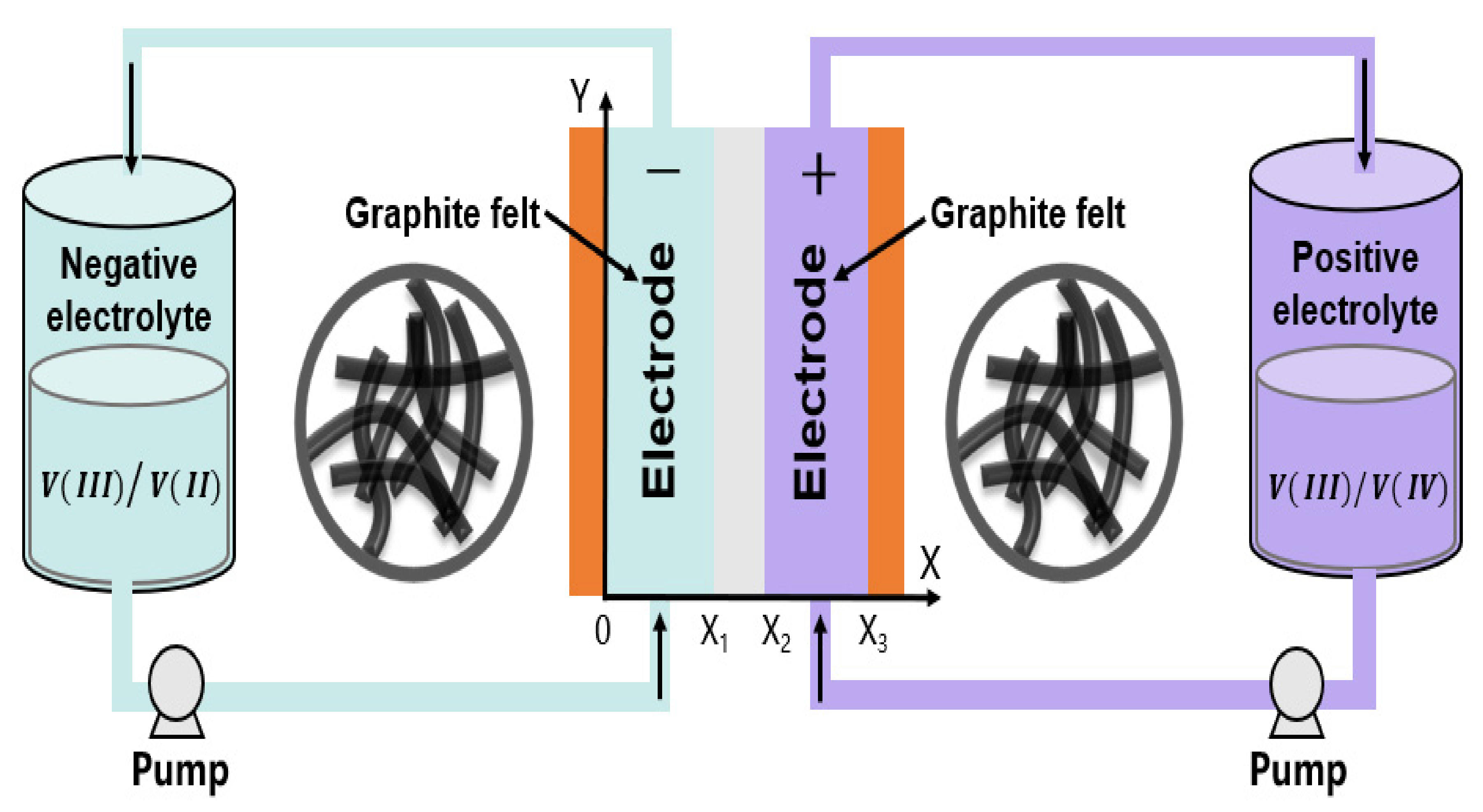
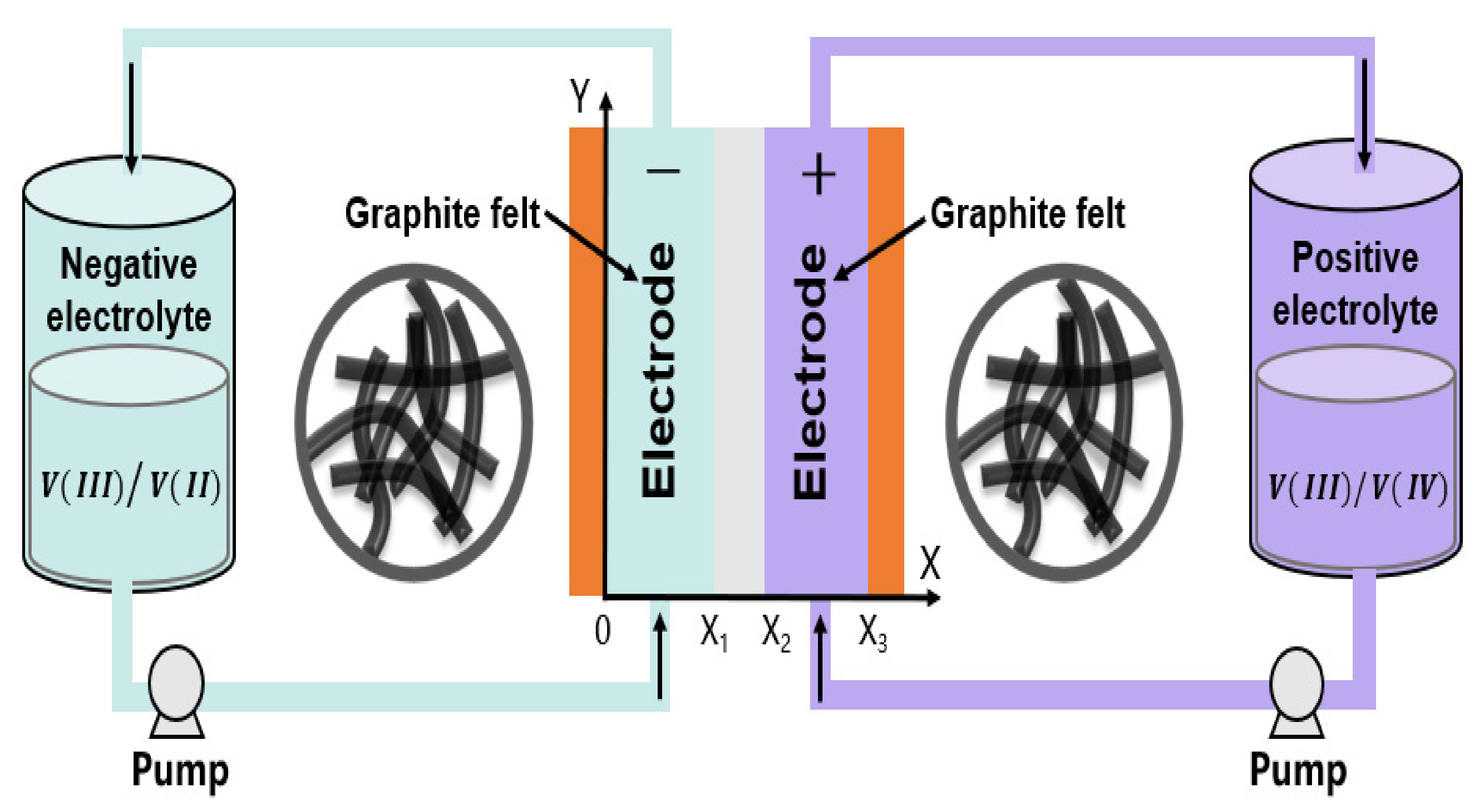
2. Model Formulation
- (1)
- Variations of redox-active species, solid/liquid potentials, pressure, and velocity in the direction perpendicular to the x–y plane were neglected.
- (2)
- The fluid flow through the porous electrodes was considered to be incompressible.
- (3)
- The cell was assumed to be isothermal.
- (4)
- Parasitic reactions were neglected.
- (5)
- The membrane was a cation exchange membrane, and tetraethylammonium ions (TEA+) were assumed to be the only charge carrier and transfer in the membrane; other solvent and redox-active species crossover through the membrane was not considered.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Term | Value |
|---|---|
| Electrolyte density | 800 kg m−3 |
| Electrolyte dynamic viscosity | 0.34 Pa s |
| V(II) ion diffusivity, DV2 [53] | 3.93 × 10−10 m2 s−1 |
| V(III) ion diffusivity, DV3 [53] | 3.93 × 10−10 m2 s−1 |
| V(IV) ion diffusivity, DV4 [53] | 3.93 × 10−10 m2 s−1 |
| TEA+ ion diffusivity, DTEA+ [54] | 1.88 × 10−9 m2 s−1 |
| Tetrafluoroborate (BF4-) ion diffusivity, DBF4- [54] | 3.33 × 10−9 m2 s−1 |
| * | 9 |
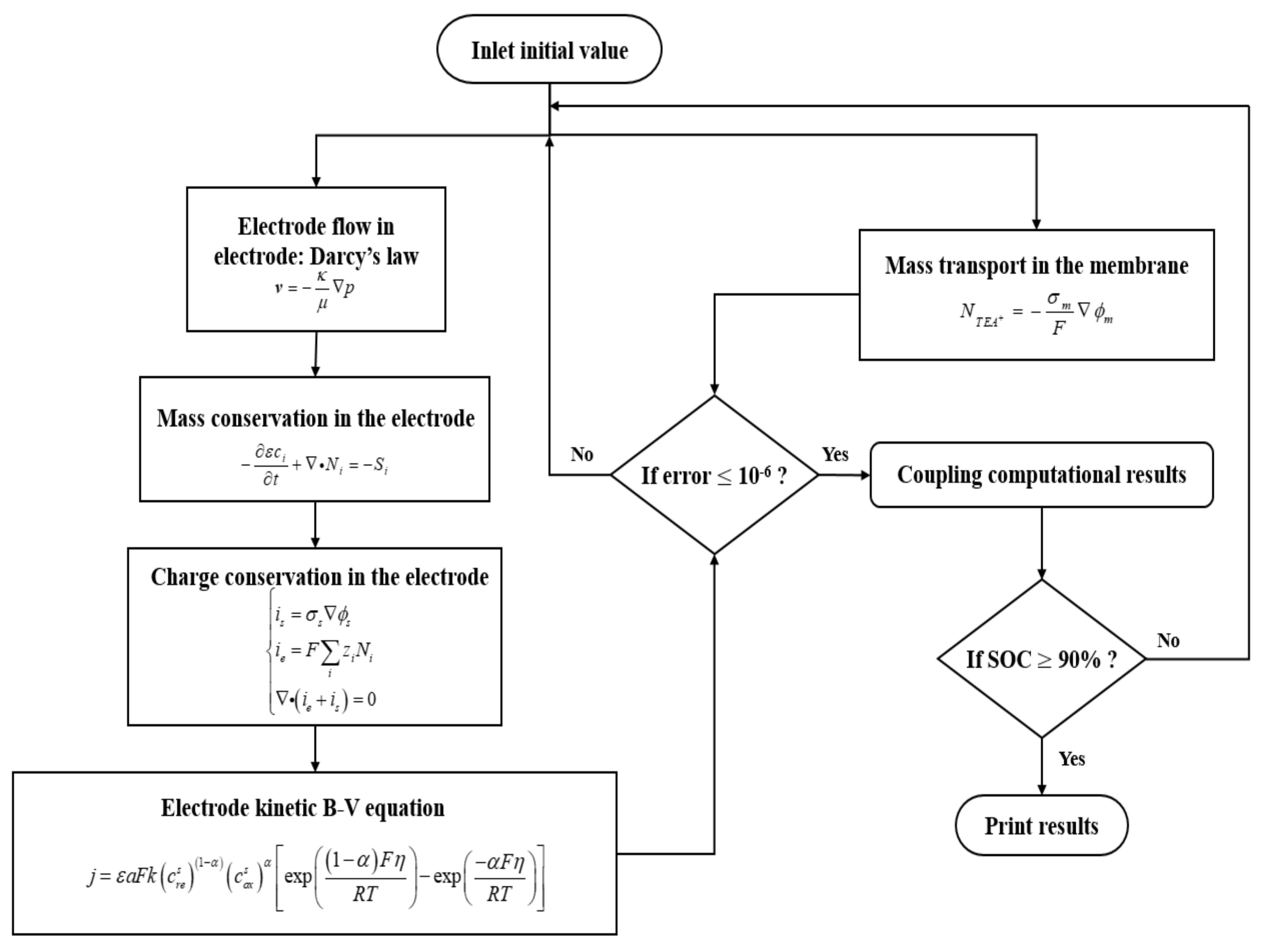
2.1. Governing Equations
2.1.1. Transport Processes in Porous Electrodes
2.1.2. Transport Processes in the Membrane
2.1.3. Electrochemical Reactions in Porous Electrodes
2.2. Boundary Conditions
2.3. Electrolyte Tanks, Inlet Concentrations, and Initial Values
3. Results and Discussion
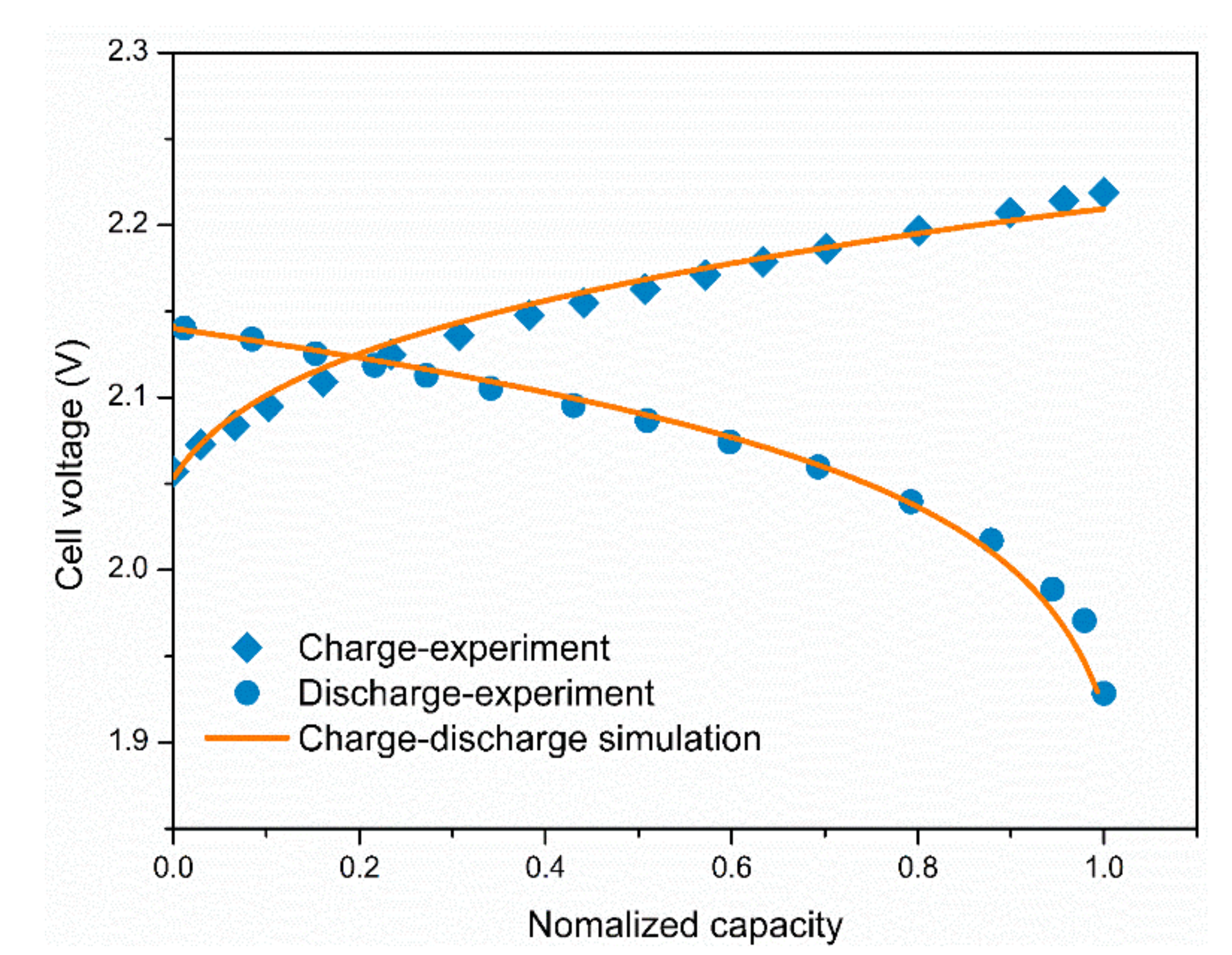
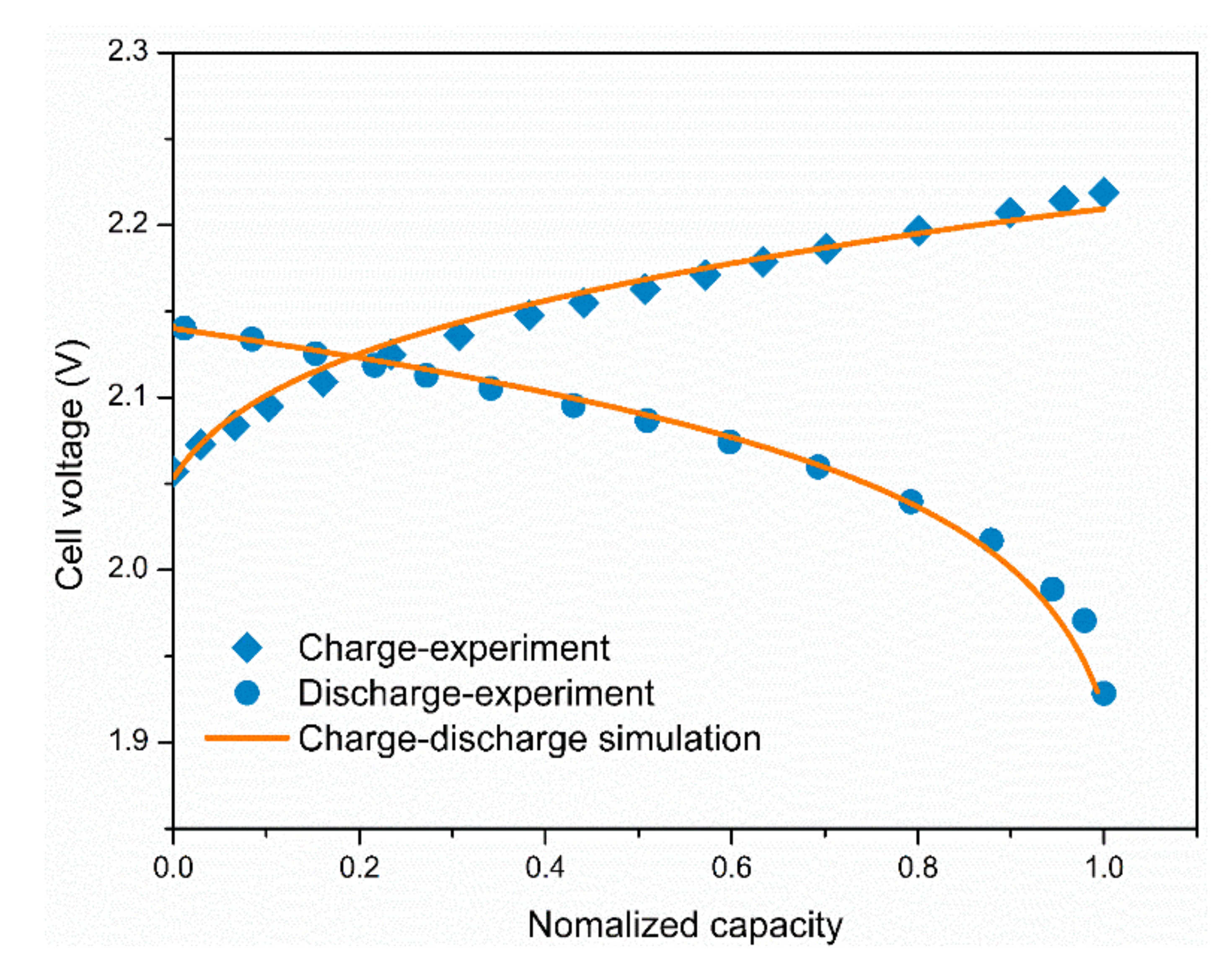
3.1. Model Validation
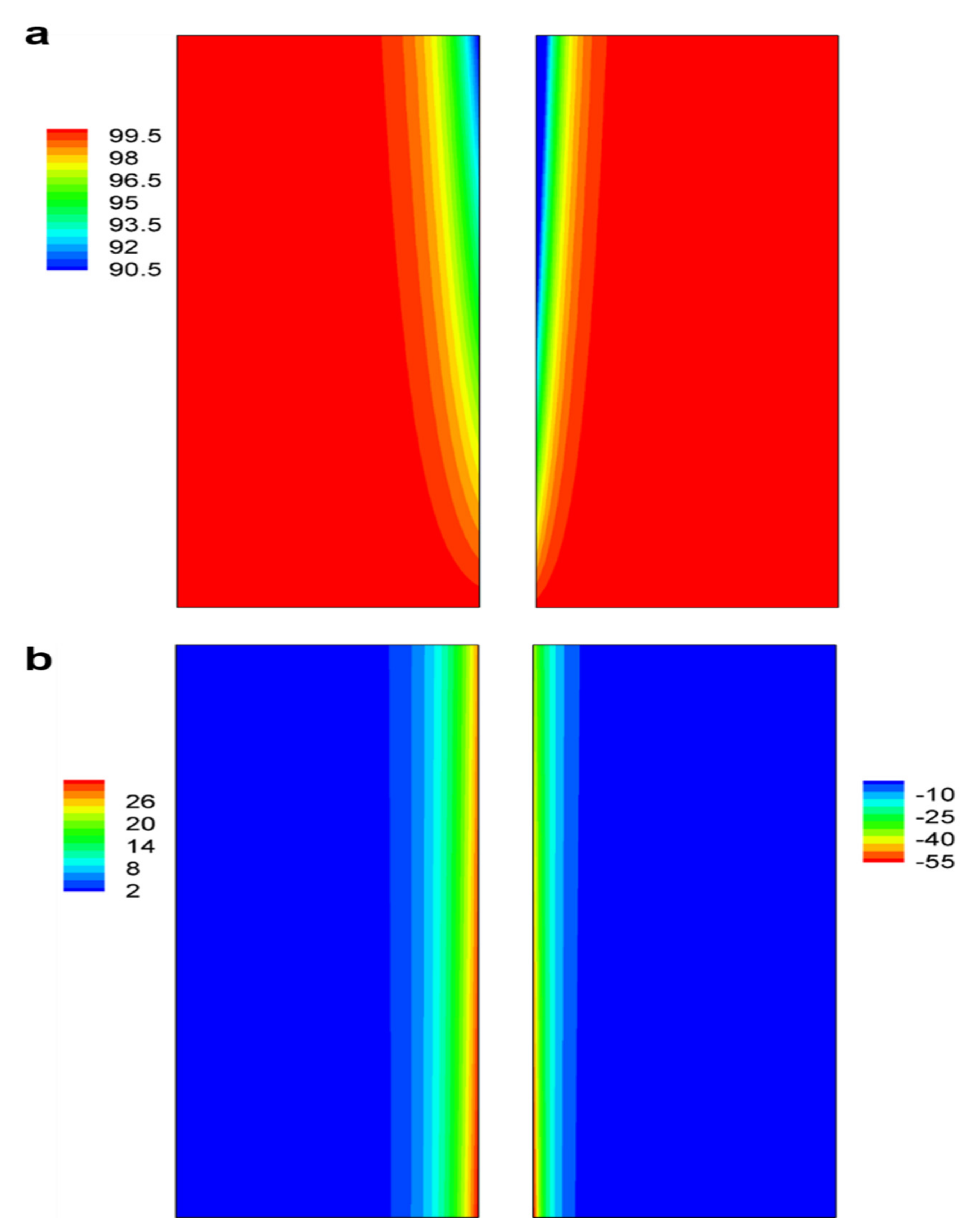
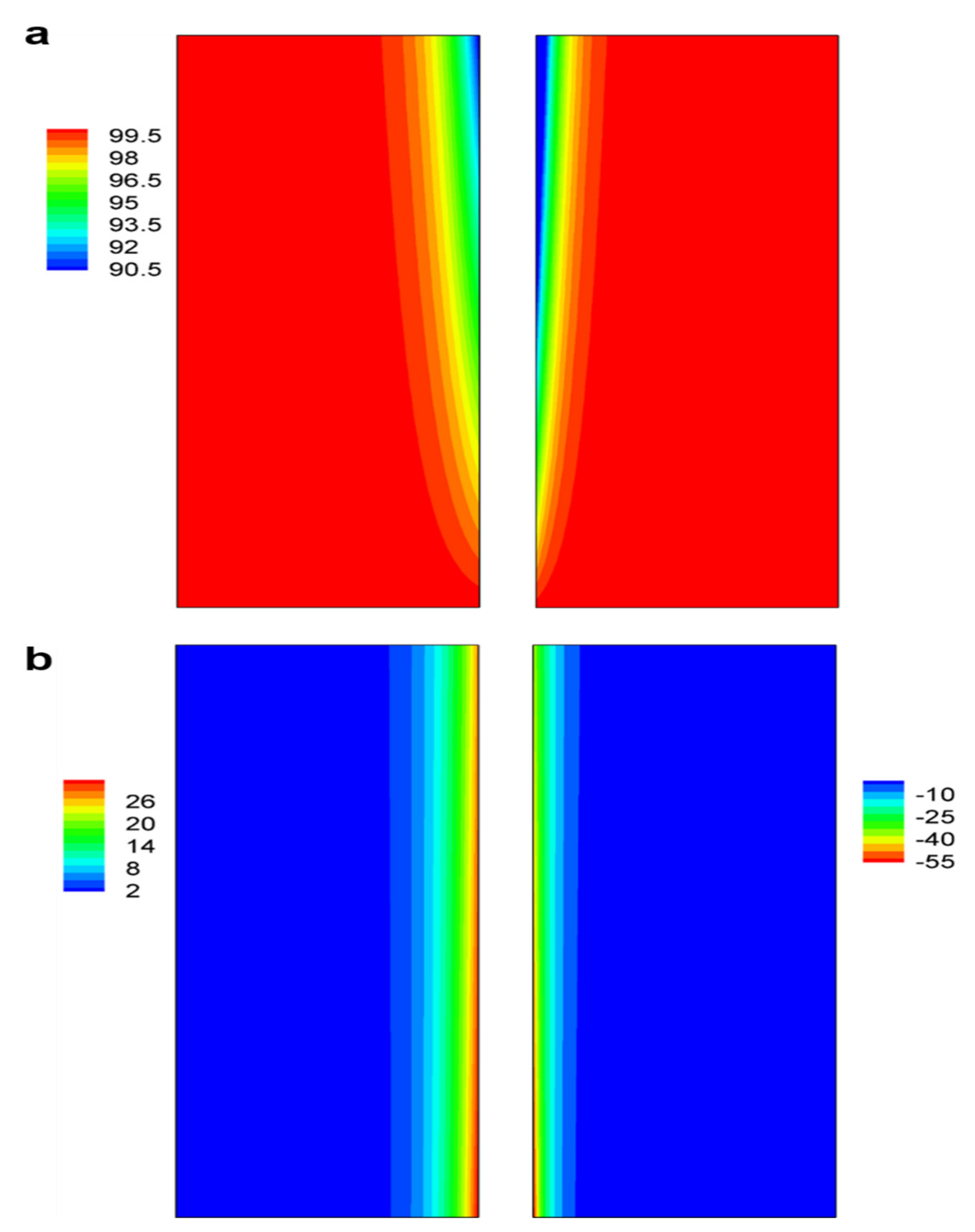
3.2. Concentration and Reaction Current Density Distributions in Electrodes
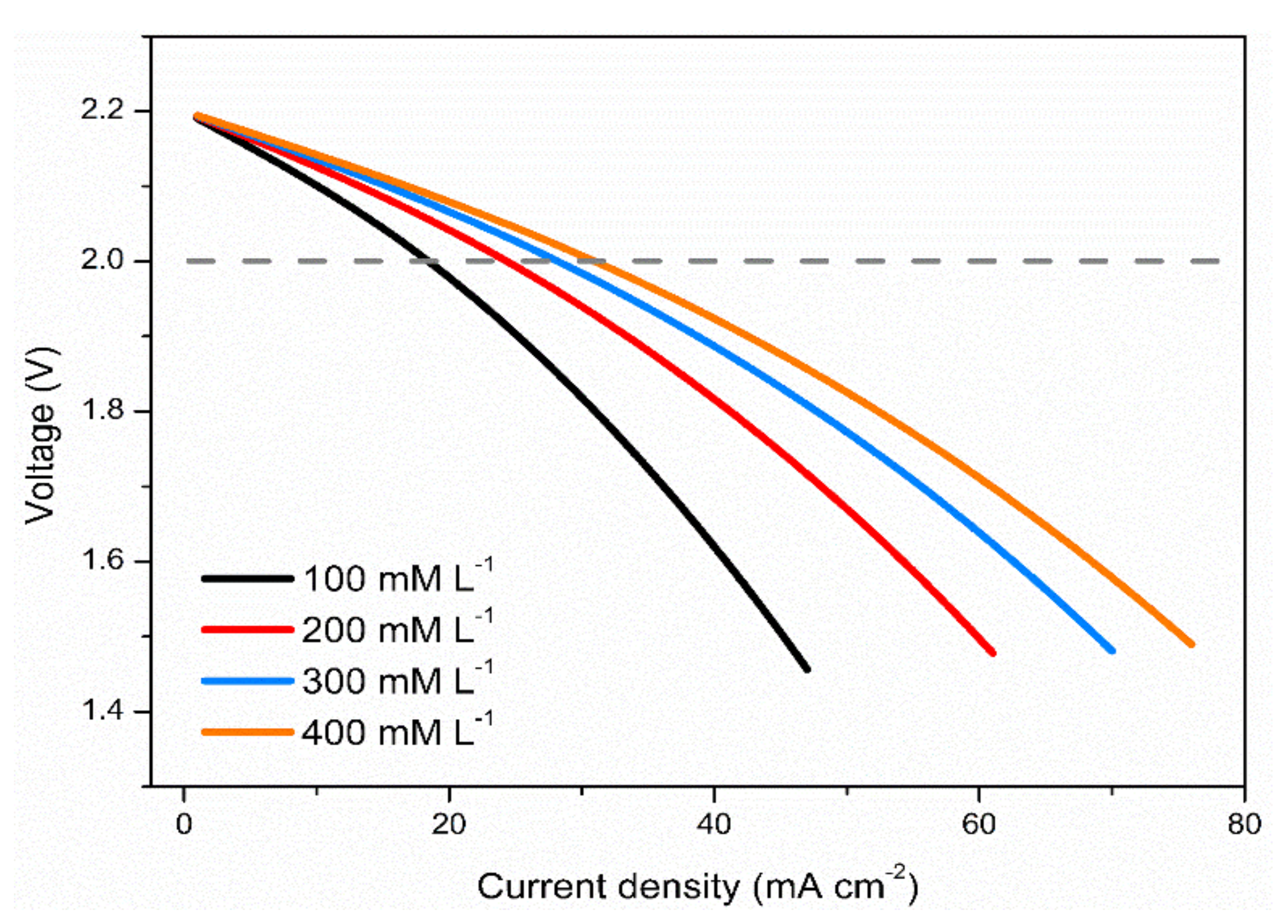
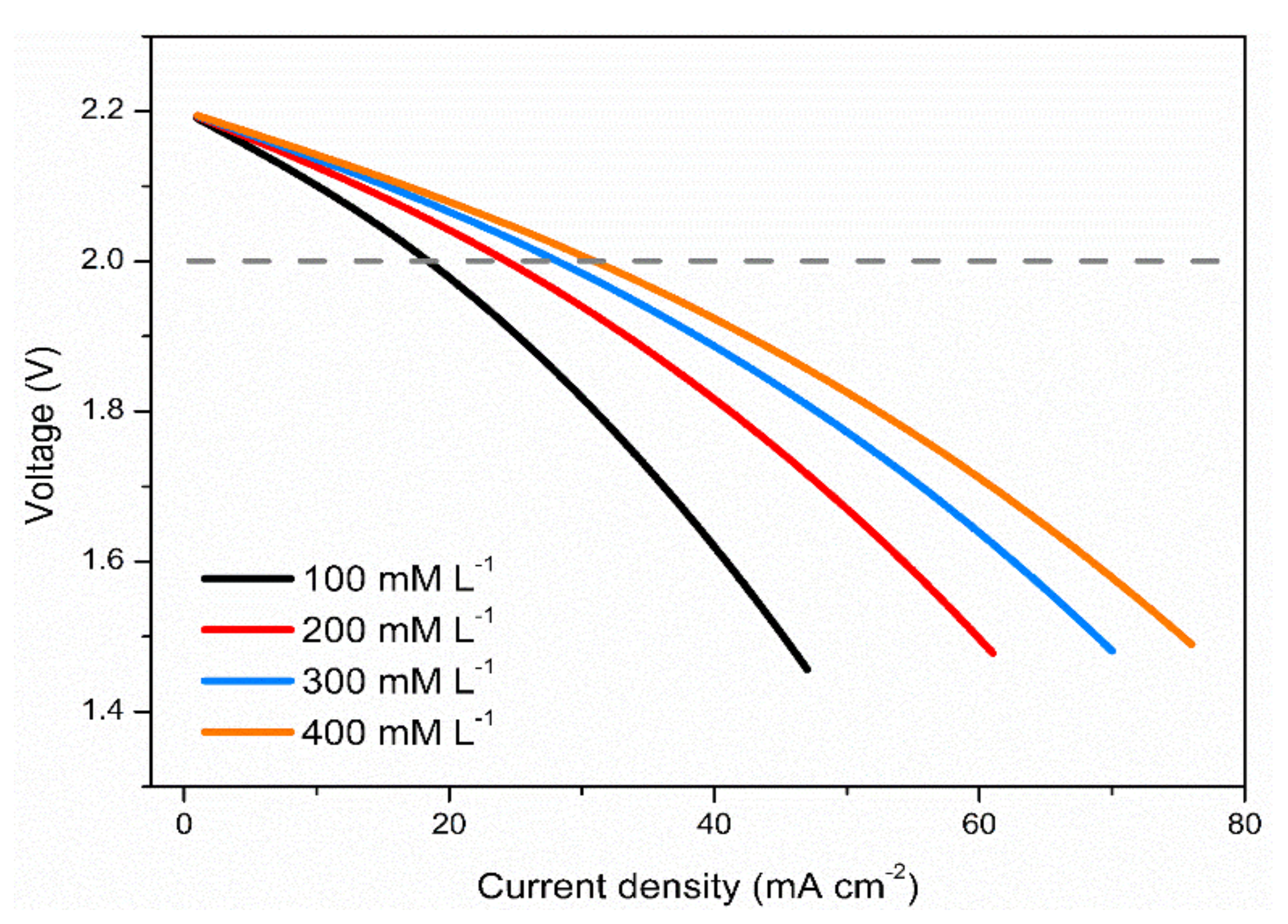
3.3. Effects of Vanadium Salt Concentrations
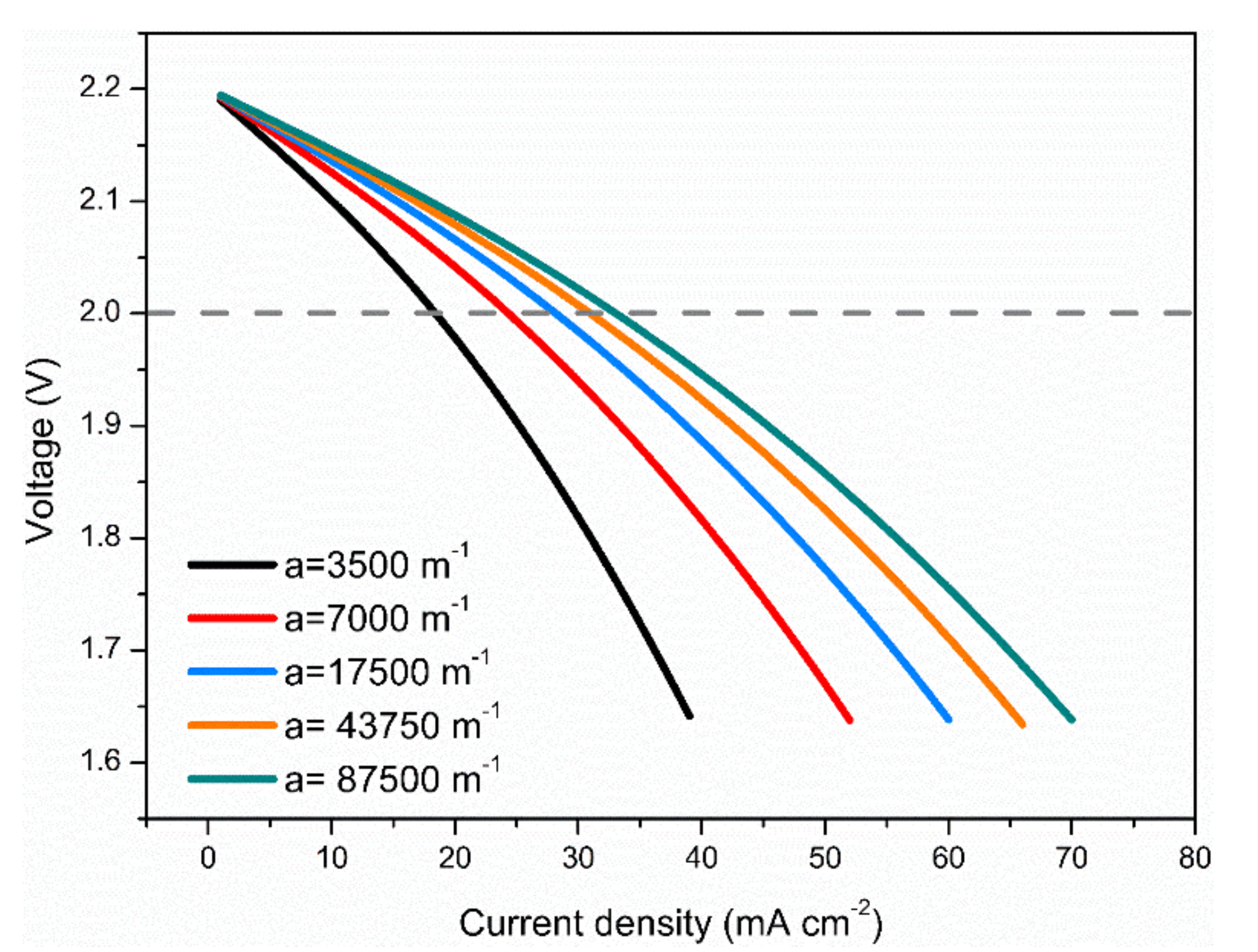
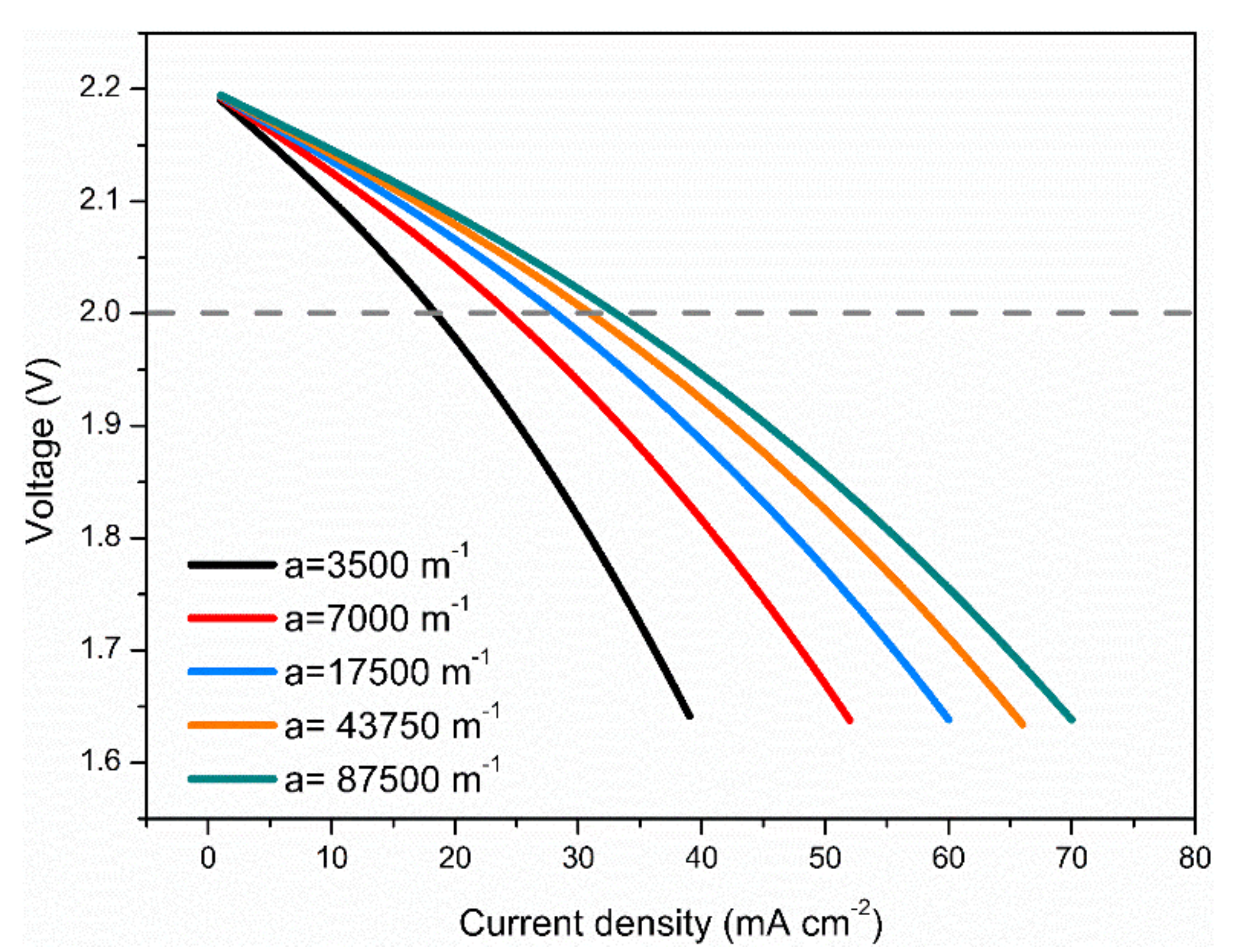
3.4. Effects of Electrode Surface Area
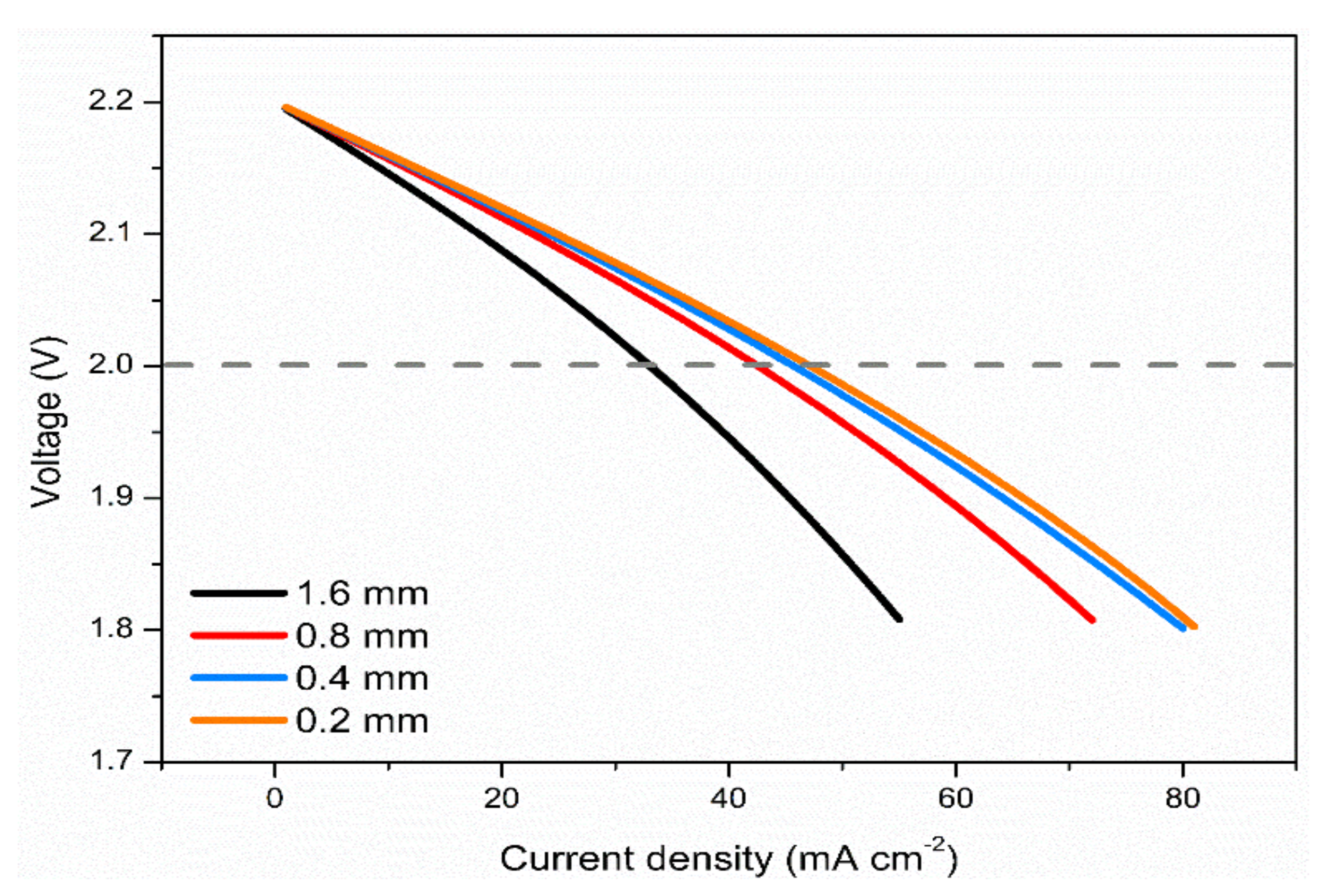
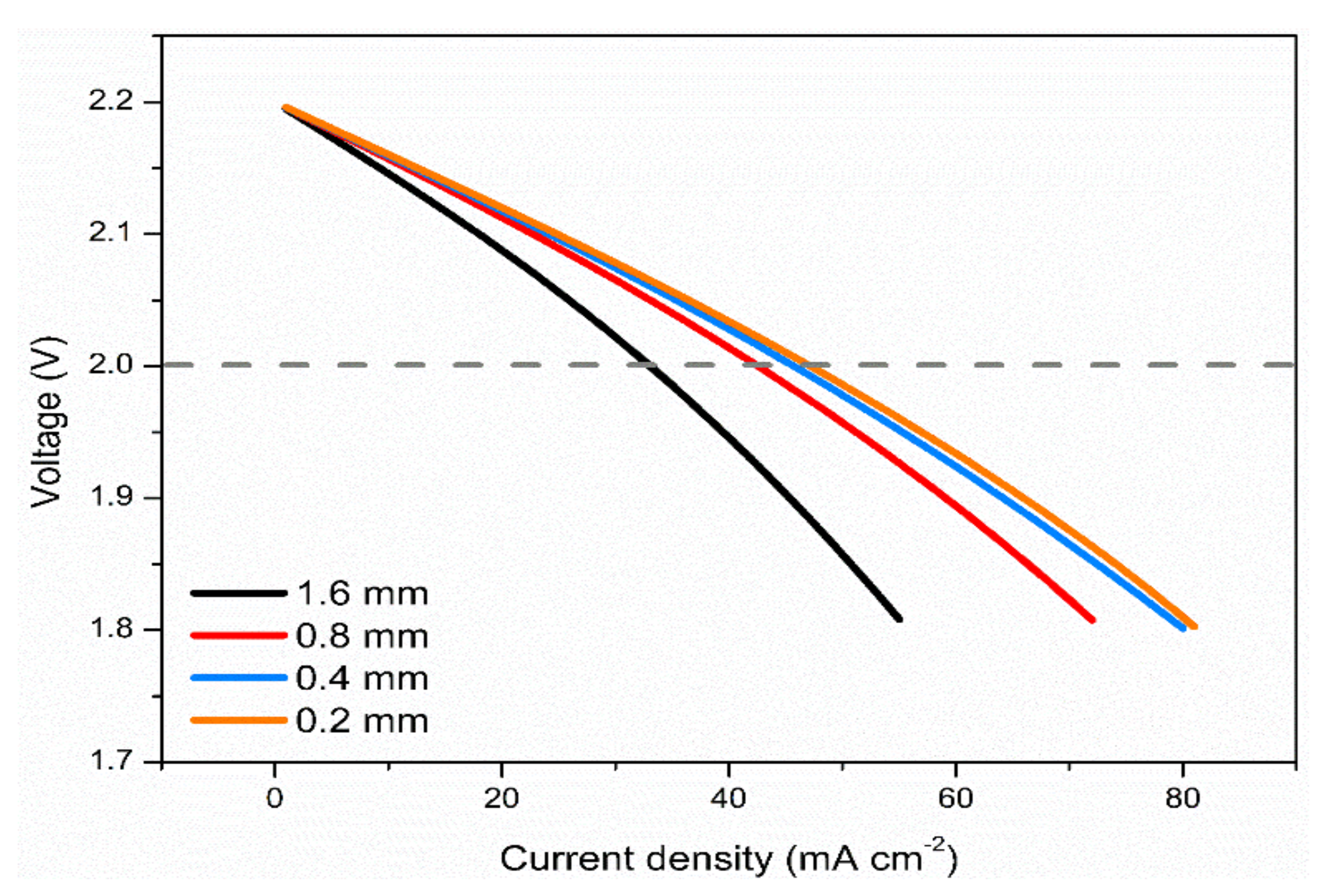
3.5. Effects of Electrode Thickness
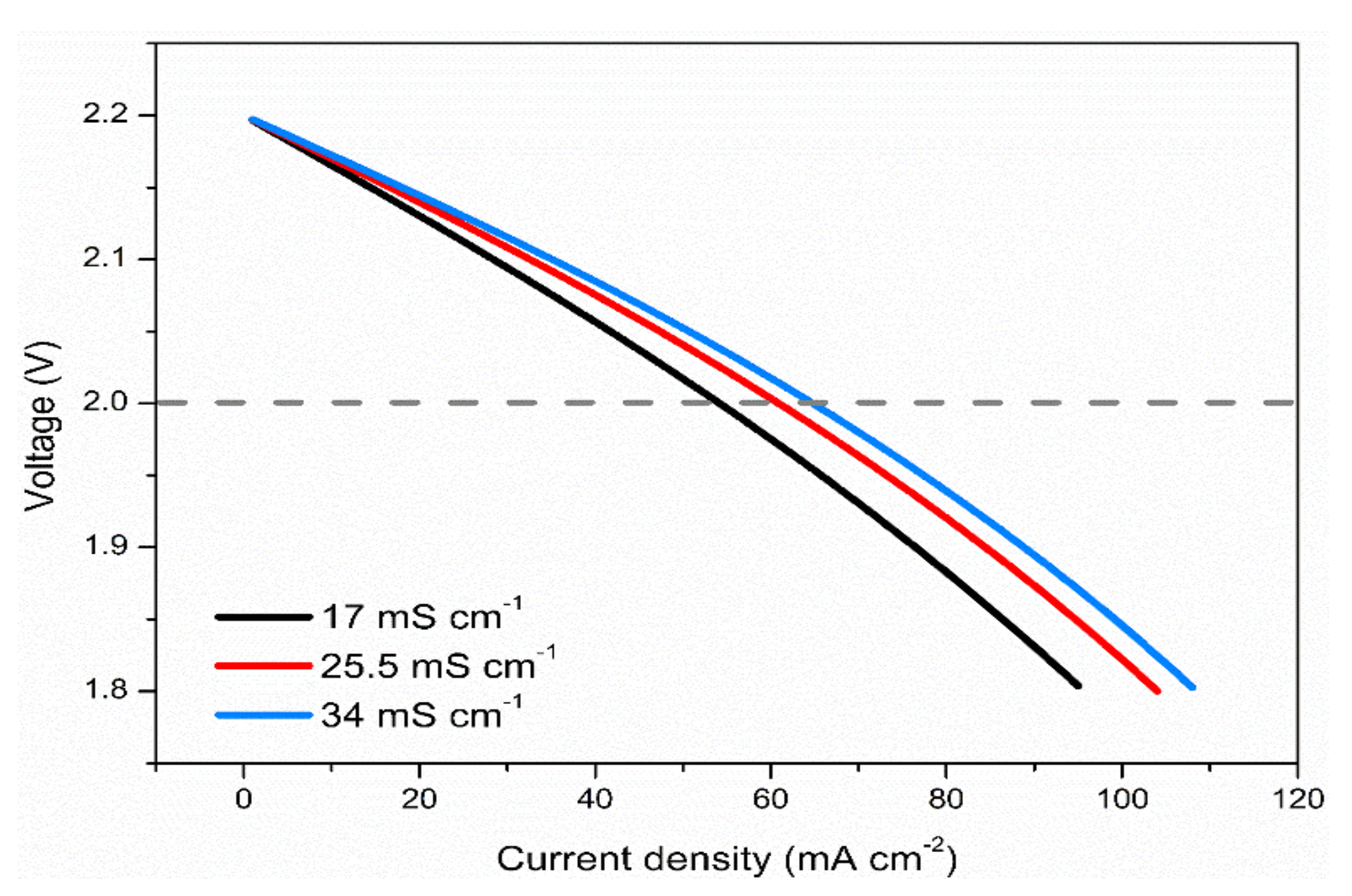
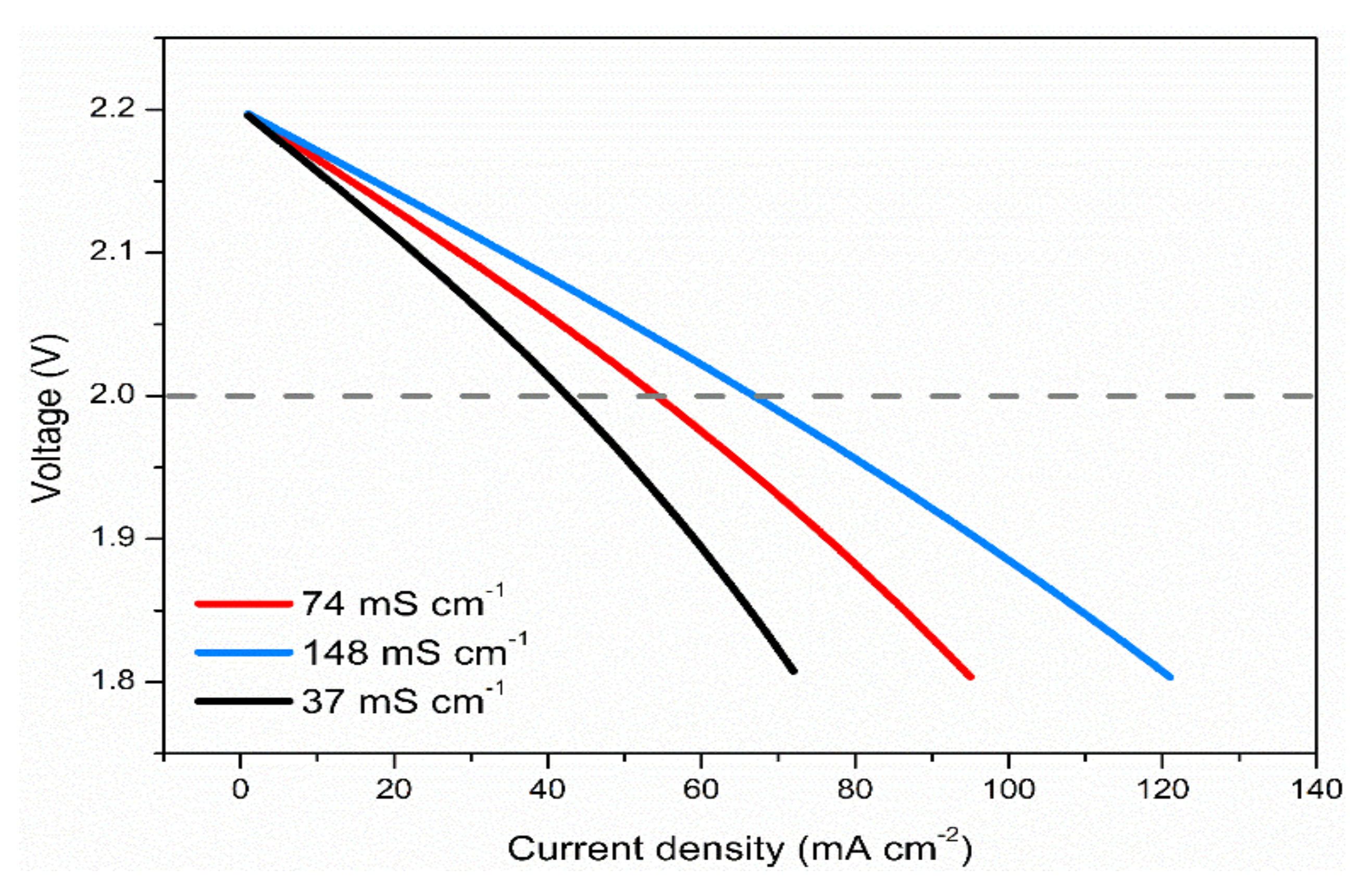
3.6. Effects of Electrolyte Conductivity
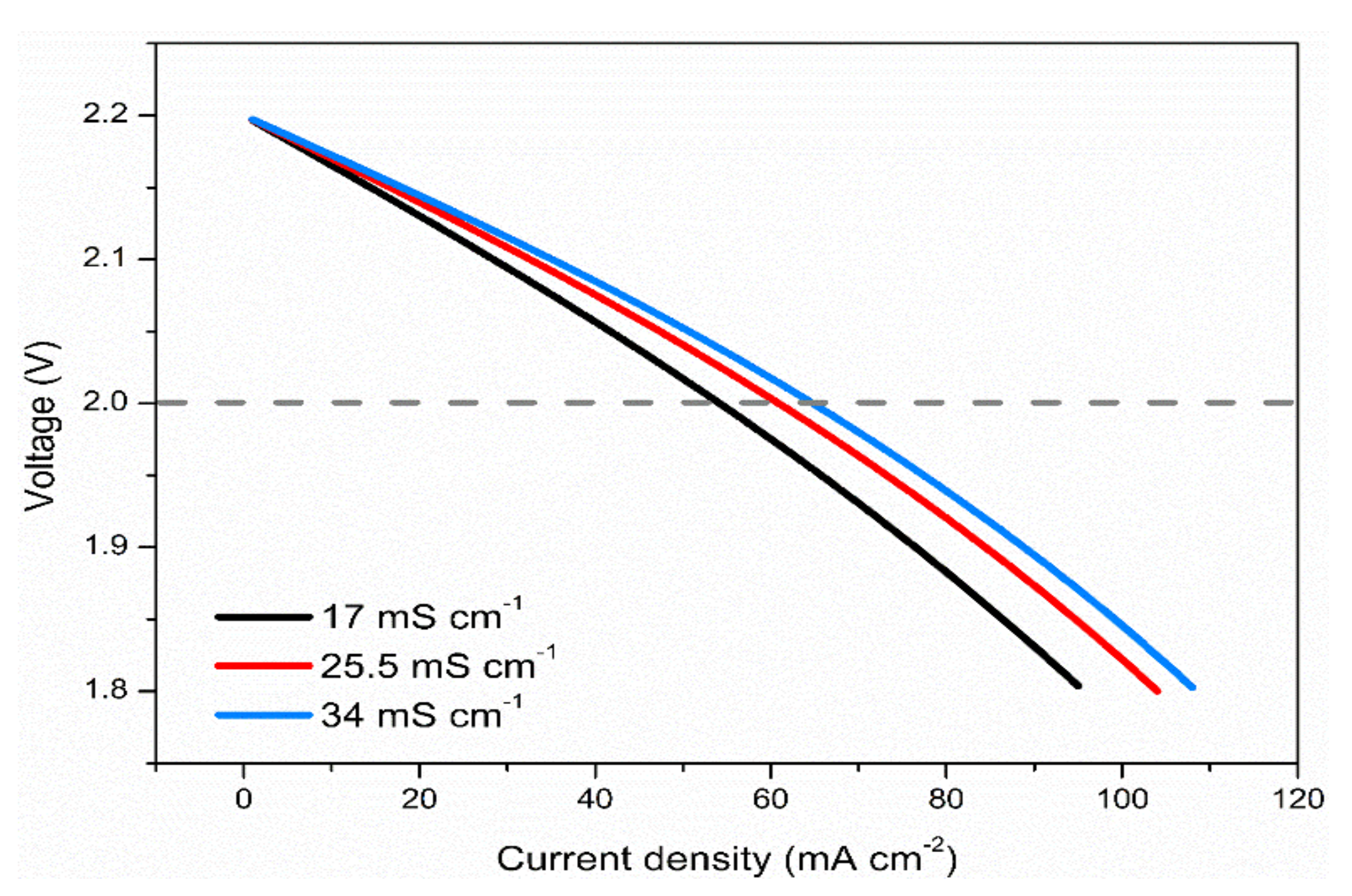
3.7. Effects of Membrane Conductivity
4. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Nomenclature
| a | the specific surface area of the porous electrode, m−1 | |
| c | concentration of species, mol m−3 | |
| d | thickness of the electrode, m | |
| D | diffusivity of the species, m2 s−1 | |
| F | Faraday’s constant, C mol−1 | |
| Hcell | length of the porous electrode | |
| I | operational current density, A m−2 | |
| j | local reaction current density, A m−2 | |
| k+/- | reaction rate constant, m s−1 | |
| km | pore-level mass transport coefficient, s−1 | |
| N | superficial molar flux, mol m2 s−1 | |
| p | pressure, Pa | |
| R | universal gas constant, J mol−1 K−1 | |
| S | source term | |
| t | time, s | |
| T | temperature, K | |
| u | mobility, mol s−1 kg−1 | |
| superficial velocity, m s−1 | ||
| U0 | equilibrium potential, V | |
| standard equilibrium potential, V | ||
| V | tank volume, m−3 | |
| wcell | width of the porous electrode | |
| z | valence | |
| Greek | ||
| ρ | density, kg m−3 | |
| μ | viscosity, kg m−1 s−1 | |
| ε | porosity | |
| κ | hydraulic permeability, m2 | |
| σ | conductivity, S m−1 | |
| potential, V | ||
| η | overpotential, V | |
| α | charge transfer coefficient | |
| ω | volumetric flow rate, m3 s−1 | |
| Superscripts and subscripts | ||
| e | electrolyte | |
| eff | effective | |
| i | species | |
| in | inlet | |
| m | membrane | |
| out | outlet | |
| s | solid | |
| V(II) | V(acac)3− | |
| V(III) | V(acac)3 | |
| V(IV) | V(acac)3+ | |
References
- Sun, C.; Negro, E.; Vezzu, K.; Pagot, G.; Cavinato, G.; Nale, A.; Bang, Y.H.; Noto, V.D. Hybrid inorganic-organic proton-conducting membranes based on SPEEK doped with WO3 nanoparticles for application in vanadium redox flow batteries. Electrochim. Acta 2019, 309, 311–325. [Google Scholar] [CrossRef]
- Weber, A.Z.; Mench, M.M.; Meyers, J.P.; Ross, P.N.; Gostick, J.T.; Liu, Q. Redox flow batteries: A review. J. Appl. Electrochem. 2011, 41, 1137–1164. [Google Scholar] [CrossRef] [Green Version]
- Huskinson, B.; Marshak, M.P.; Suh, C.; Er, S.; Gerhardt, M.R.; Galvin, C.J.; Chen, X.; Aspuru-Guzik, A.; Gordon, R.G.; Aziz, M.J. A metal-free organic–inorganic aqueous flow battery. Nature 2014, 505, 195–198. [Google Scholar] [CrossRef]
- Lin, K.; Chen, Q.; Gerhardt, M.R.; Tong, L.; Kim, S.B.; Eisenach, L.; Valle, A.W.; Hardee, D.; Gordon, R.G.; Aziz, M.J.; et al. Alkaline quinone flow battery. Science 2015, 349, 1529–1532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chalamala, B.R.; Soundappan, T.; Fisher, G.R.; Anstey, M.R.; Viswanathan, V.V.; Perry, M.L. Redox flow batteries: An engineering perspective. Proc. IEEE 2014, 102, 976–999. [Google Scholar] [CrossRef]
- Perry, M.L.; Weber, A.Z. Advanced redox-flow batteries: A perspective. J. Electrochem. Soc. 2015, 163, A5064–A5067. [Google Scholar] [CrossRef]
- Zhang, H.; Sun, C. Cost-effective iron-based aqueous redox flow batteries for large-scale energy storage application: A review. J. Power Sources 2021, 493, 229445. [Google Scholar] [CrossRef]
- Darling, R.M.; Gallagher, K.G.; Kowalski, J.A.; Ha, S.; Brushett, F.R. Pathways to low-cost electrochemical energy storage: A comparison of aqueous and nonaqueous flow batteries. Energy Environ. Sci. 2014, 7, 3459–3477. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Z.; Yin, Y.; Xie, C.; Zhang, H.; Yao, Y.; Li, X. Advanced materials for zinc-based flow battery: Development and challenge. Adv. Mater. 2019, 31, 1902025. [Google Scholar] [CrossRef]
- Kim, D.; Seo, S.; Lee, M.; Park, J.; Moon, S.; Kang, Y.S.; Choi, Y.; Kang, M. Pore-filled anion-exchange membranes for non-aqueous redox flow batteries with dual-metal-complex redox shuttles. J. Membr. Sci. 2014, 454, 44–50. [Google Scholar] [CrossRef]
- Davies, T.J.; Tummino, J.J. High-performance vanadium redox flow batteries with graphite felt electrodes. C—J. Carbon Res. 2018, 4, 8. [Google Scholar] [CrossRef] [Green Version]
- Khataee, A.; Drazevic, E.; Catalano, J.; Bentien, A. Performance optimization of differential pH quinone-bromide redox flow battery. J. Electrochem. Soc. 2018, 165, A3918–A3924. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Sleightholme, A.E.; Shinkle, A.A.; Li, Y.; Thompson, L.T. Non-aqueous vanadium acetylacetonate electrolyte for redox flow batteries. Electrochem. Commun. 2009, 11, 2312–2315. [Google Scholar] [CrossRef]
- Brushett, F.R.; Vaughey, J.T.; Jansen, A.N. An all-organic non-aqueous lithium-ion redox flow battery. Adv. Energy Mater. 2012, 2, 1390–1396. [Google Scholar] [CrossRef]
- Liu, Q.; Shinkle, A.A.; Li, Y.; Monroe, C.W.; Thompson, L.T.; Sleightholme, A.E. Non-aqueous chromium acetylacetonate electrolyte for redox flow batteries. Electrochem. Commun. 2010, 12, 1634–1637. [Google Scholar] [CrossRef]
- Shin, S.; Yun, S.; Moon, S. A review of current developments in non-aqueous redox flow batteries: Characterization of their membranes for design perspective. Rsc Adv. 2013, 3, 9095–9116. [Google Scholar] [CrossRef]
- Hamelet, S.; Tzedakis, T.; Leriche, J.; Sailler, S.; Larcher, D.; Taberna, P.; Simon, P.; Tarascon, J. Non-aqueous Li-based redox flow batteries. J. Electrochem. Soc. 2012, 159, A1360–A1367. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Liu, Q.; Shi, X.; Li, Y. Tetrabutylammonium hexafluorophosphate and 1-ethyl-3-methyl imidazolium hexafluorophosphate ionic liquids as supporting electrolytes for non-aqueous vanadium redox flow batteries. J. Power Sources 2012, 203, 201–205. [Google Scholar] [CrossRef]
- Sleightholme, A.E.; Shinkle, A.A.; Liu, Q.; Li, Y.; Monroe, C.W.; Thompson, L.T. Non-aqueous manganese acetylacetonate electrolyte for redox flow batteries. J. Power Sources 2011, 196, 5742–5745. [Google Scholar] [CrossRef]
- Mun, J.; Lee, M.; Park, J.; Oh, D.; Lee, D.; Doo, S. Non-aqueous redox flow batteries with nickel and iron tris (2, 2′-bipyridine) complex electrolyte. Electrochem. Solid-State Lett. 2012, 15, A80–A82. [Google Scholar] [CrossRef]
- Shinkle, A.A.; Sleightholme, A.E.; Griffith, L.D.; Thompson, L.T.; Monroe, C.W. Degradation mechanisms in the non-aqueous vanadium acetylacetonate redox flow battery. J. Power Sources 2012, 206, 490–496. [Google Scholar] [CrossRef]
- Herr, T.; Noack, J.; Fischer, P.; Tübke, J. 1, 3-Dioxolane, tetrahydrofuran, acetylacetone and dimethyl sulfoxide as solvents for non-aqueous vanadium acetylacetonate redox-flow-batteries. Electrochim. Acta 2013, 113, 127–133. [Google Scholar] [CrossRef]
- Maurya, S.; Shin, S.; Sung, K.; Moon, S. Anion exchange membrane prepared from simultaneous polymerization and quaternization of 4-vinyl pyridine for non-aqueous vanadium redox flow battery applications. J. Power Sources 2014, 255, 325–334. [Google Scholar] [CrossRef]
- Suttil, J.; Kucharyson, J.; Escalante-Garcia, I.; Cabrera, P.; James, B.; Savinell, R.; Sanford, M.; Thompson, L. Metal acetylacetonate complexes for high energy density non-aqueous redox flow batteries. J. Mater. Chem. A 2015, 3, 7929–7938. [Google Scholar] [CrossRef]
- Wei, X.; Xu, W.; Vijayakumar, M.; Cosimbescu, L.; Liu, T.; Sprenkle, V.; Wang, W. TEMPO-based catholyte for high-energy density nonaqueous redox flow batteries. Adv. Mater. 2014, 26, 7649–7653. [Google Scholar] [CrossRef] [PubMed]
- Su, L.; Ferrandon, M.; Kowalski, J.A.; Vaughey, J.T.; Brushett, F.R. Electrolyte development for non-aqueous redox flow batteries using a high-throughput screening platform. J. Electrochem. Soc. 2014, 161, A1905–A1914. [Google Scholar] [CrossRef]
- Herr, T.; Fischer, P.; Tübke, J.; Pinkwart, K.; Elsner, P. Increasing the energy density of the non-aqueous vanadium redox flow battery with the acetonitrile-1, 3-dioxolane–dimethyl sulfoxide solvent mixture. J. Power Sources 2014, 265, 317–324. [Google Scholar]
- Darling, R.; Gallagher, K.; Xie, W.; Su, L.; Brushett, F. Transport Property Requirements for Flow Battery Separators. J. Electrochem. Soc. 2015, 163, A5029–A5040. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.; Negro, E.; Nale, A.; Pagot, G.; Vezzu, K.; Zawodzinski, T.A.; Meda, L.; Gambaro, C.; Noto, V.D. An efficient barrier toward vanadium crossover in redox flow batteries: The bilayer [Nafion/(WO3)x] hybrid inorganic-organic membrane. Electrochim. Acta 2021, 378, 138133. [Google Scholar]
- Yuan, J.; Pan, Z.Z.; Jin, Y.; Qiu, Q.; Zhang, C.; Zhao, Y.; Li, Y. Membranes in non-aqueous redox flow battery: A review. J. Power Sources 2021, 500, 229983. [Google Scholar]
- Milshtein, J.D.; Barton, J.L.; Darling, R.M.; Brushett, F.R. 4-acetamido-2, 2, 6, 6-tetramethylpiperidine-1-oxyl as a model organic redox active compound for nonaqueous flow batteries. J. Power Sources 2016, 327, 151–159. [Google Scholar]
- Escalante-García, I.L.; Wainright, J.S.; Thompson, L.T.; Savinell, R.F. Performance of a non-aqueous vanadium acetylacetonate prototype redox flow battery: Examination of separators and capacity decay. J. Electrochem. Soc. 2014, 162, A363–A372. [Google Scholar]
- Zhou, X.; Zhao, T.; An, L.; Zeng, Y.; Yan, X. A vanadium redox flow battery model incorporating the effect of ion concentrations on ion mobility. Appl. Energy 2015, 158, 157–166. [Google Scholar]
- Li, Y.; Skyllas-Kazacos, M.; Bao, J. A dynamic plug flow reactor model for a vanadium redox flow battery cell. J. Power Sources 2016, 311, 57–67. [Google Scholar]
- Shah, A.; Watt-Smith, M.; Walsh, F. A dynamic performance model for redox-flow batteries involving soluble species. Electrochim. Acta 2008, 53, 8087–8100. [Google Scholar]
- You, D.; Zhang, H.; Chen, J. A simple model for the vanadium redox battery. Electrochim. Acta 2009, 54, 6827–6836. [Google Scholar]
- Al-Fetlawi, H.; Shah, A.; Walsh, F. Non-isothermal modelling of the all-vanadium redox flow battery. Electrochim. Acta 2009, 55, 78–89. [Google Scholar]
- Knehr, K.; Agar, E.; Dennison, C.; Kalidindi, A.; Kumbur, E. A transient vanadium flow battery model incorporating vanadium crossover and water transport through the membrane. J. Electrochem. Soc. 2012, 159, A1446–A1459. [Google Scholar]
- Zhou, X.; Zhao, T.; An, L.; Zeng, Y.; Wei, L. Modeling of ion transport through a porous separator in vanadium redox flow batteries. J. Power Sources 2016, 327, 67–76. [Google Scholar]
- Yang, W.; He, Y.; Li, Y. Performance modeling of a vanadium redox flow battery during discharging. Electrochim. Acta 2015, 155, 279–287. [Google Scholar] [CrossRef]
- Yang, X.; Ye, Q.; Cheng, P.; Zhao, T.S. Effects of the electric field on ion crossover in vanadium redox flow batteries. Appl. Energy 2015, 145, 306–319. [Google Scholar] [CrossRef]
- Won, S.; Oh, K.; Ju, H. Numerical analysis of vanadium crossover effects in all-vanadium redox flow batteries. Electrochim. Acta 2015, 177, 310–320. [Google Scholar] [CrossRef]
- Oh, K.; Won, S.; Ju, H. A comparative study of species migration and diffusion mechanisms in all-vanadium redox flow batteries. Electrochim. Acta 2015, 181, 238–247. [Google Scholar] [CrossRef]
- Wang, Y.; Cho, S.C. Analysis and three-dimensional modeling of vanadium flow batteries. J. Electrochem. Soc. 2014, 161, A1200–A1212. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Ling, C.; Birgersson, E.; Vynnycky, M.; Han, M. Verified reduction of dimensionality for an all-vanadium redox flow battery model. J. Power Sources 2015, 279, 345–350. [Google Scholar] [CrossRef]
- Xiong, B.; Zhao, J.; Wei, Z.; Skyllas-Kazacos, M. Extended Kalman filter method for state of charge estimation of vanadium redox flow battery using thermal-dependent electrical model. J. Power Sources 2014, 262, 50–61. [Google Scholar] [CrossRef]
- Chen, C.L.; Yeoh, H.K.; Chakrabarti, M.H. An enhancement to Vynnycky’s model for the all-vanadium redox flow battery. Electrochim. Acta 2014, 120, 167–179. [Google Scholar] [CrossRef]
- Ma, X.; Zhang, H.; Xing, F. A three-dimensional model for negative half cell of the vanadium redox flow battery. Electrochim. Acta 2011, 58, 238–246. [Google Scholar] [CrossRef]
- Mohamed, M.; Ahmad, H.; Seman, M.A.; Razali, S.; Najib, M. Electrical circuit model of a vanadium redox flow battery using extended Kalman filter. J. Power Sources 2013, 239, 284–293. [Google Scholar]
- Yin, C.; Gao, Y.; Guo, S.; Tang, H. A coupled three dimensional model of vanadium redox flow battery for flow field designs. Energy 2014, 74, 886–895. [Google Scholar]
- Yan, Y.; Li, Y.; Skyllas-Kazacos, M.; Bao, J. Modelling and simulation of thermal behaviour of vanadium redox flow battery. J. Power Sources 2016, 322, 116–128. [Google Scholar] [CrossRef]
- Wandschneider, F.; Finke, D.; Grosjean, S.; Fischer, P.; Pinkwart, K.; Tübke, J.; Nirschl, H. Model of a vanadium redox flow battery with an anion exchange membrane and a Larminie-correction. J. Power Sources 2014, 272, 436–447. [Google Scholar] [CrossRef]
- Wandschneider, F.; Küttinger, M.; Noack, J.; Fischer, P.; Pinkwart, K.; Tübke, J.; Nirschl, H. A coupled-physics model for the vanadium oxygen fuel cell. J. Power Sources 2014, 259, 125–137. [Google Scholar] [CrossRef]
- Shinkle, A.A.; Sleightholme, A.E.; Thompson, L.T.; Monroe, C.W. Electrode kinetics in non-aqueous vanadium acetylacetonate redox flow batteries. J. Appl. Electrochem. 2011, 41, 1191–1199. [Google Scholar] [CrossRef]
- Bard, A.J.; Faulkner, L.R. Electrochemical Methods: Fundamentals and Applications, 2nd ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2000. [Google Scholar]
| Term | Positive Electrode | Negative Electrode |
|---|---|---|
| Mass conservation equation of V(II), SV(II) | — | j/F |
| Mass conservation equation of V(III), SV(III) | j/F | -j/F |
| Mass conservation equation of V(IV), SV(IV) | -j/F | — |
| Parameter | Value |
|---|---|
| Cell length, Hcell | 0.026 m |
| Cell width, wcell | 0.026 m |
| Electrode thickness, Le | 0.0036 m |
| Membrane thickness, Lm | 0.000125 m |
| Electrode porosity [39], ε | 0.883 |
| Electrode spicific surface area *, a | 1.75 × 104 m2/m3 |
| Electrode permeability [39] | 6.0 × 10−11 |
| Electrode electrical conductivity [39] | 220 S m−1 |
| Temperature, T | 300 K |
| Membrane conductivity [32] | 17 S m−1 |
| Areal contact resistance [42] | 74 mΩ cm−2 |
| Term | Value |
|---|---|
| Standard rate constant of negative reaction, k− [53] | 1.25 × 10−5 m s−1 |
| Standard rate constant of positive reaction, k+ [53] | 1.18 × 10−5 m s−1 |
| Cathodic transfer coefficient of negative electrode, α− | 0.5 |
| Cathodic transfer coefficient of positive electrode, α+ | 0.5 |
| [32] | −1.75 V |
| [32] | 0.45 V |
| Term | Value at 10% SOC |
|---|---|
| 10 mol m−3 | |
| 90 mol m−3 | |
| 90 mol m−3 | |
| 10 mol m−3 | |
| 490 mol m−3 | |
| 510 mol m−3 | |
| 500 mol m−3 | |
| 500 mol m−3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, S.; Lu, Y. Numerical Parametric Investigation of Nonaqueous Vanadium Redox Flow Batteries. Batteries 2022, 8, 75. https://doi.org/10.3390/batteries8080075
Huang S, Lu Y. Numerical Parametric Investigation of Nonaqueous Vanadium Redox Flow Batteries. Batteries. 2022; 8(8):75. https://doi.org/10.3390/batteries8080075
Chicago/Turabian StyleHuang, Shaopei, and Yujuan Lu. 2022. "Numerical Parametric Investigation of Nonaqueous Vanadium Redox Flow Batteries" Batteries 8, no. 8: 75. https://doi.org/10.3390/batteries8080075
APA StyleHuang, S., & Lu, Y. (2022). Numerical Parametric Investigation of Nonaqueous Vanadium Redox Flow Batteries. Batteries, 8(8), 75. https://doi.org/10.3390/batteries8080075