Abstract
Both the binder and solid–electrolyte interface play an important role in improving the cycling stability of electrodes for Na-ion batteries. In this study, a novel tetrabutylammonium (TBA) alginate binder is used to prepare a Na0.67MnO2 electrode for sodium-ion batteries with improved electrochemical performance. The ageing of the electrodes is characterized. TBA alginate-based electrodes are compared to polyvinylidene fluoride- (PVDF) and Na alginate-based electrodes and show favorable electrochemical performance, with gravimetric capacity values of up to 164 mAh/g, which is 6% higher than measured for the electrode prepared with PVDF binder. TBA alginate-based electrodes also display good rate capability and improved cyclability. The solid–electrolyte interface of TBA alginate-based electrodes is similar to that of PVDF-based electrodes. As the only salt of alginic acid soluble in non-aqueous solvents, TBA alginate emerges as a good alternative to PVDF binder in battery applications where the water-based processing of electrode slurries is not feasible, such as the demonstrated case with Na0.67MnO2.
1. Introduction
Energy storage systems that are made from abundant materials are essential for the transition to a more sustainable economy. Although lithium-ion batteries (LIBs) are by far the most popular battery technology, the low availability of lithium, as well as use of cobalt and other rare metals raise questions on the sustainability and long-term viability of LIBs as the only energy storage solution. With the high abundance of sodium and relative similarity to LIBs, sodium-ion batteries (SIBs) are emerging as a suitable battery technology for stationary energy storage [1]. The widespread adoption of the SIB technology, however, is hampered by many challenges, including the relatively low energy density or service life compared to LIBs. Lower energy density cathodes, such as Na2FeP2O7 or NaFePO4, are generally stable during cycling [2,3], while the cycle life of many of the higher energy density cathodes (e.g., transition metal oxides NaxMO2 where M—transition metal) has so far been subpar [4], so advances in material and electrode engineering are still required.
In most battery electrodes, a binder is used to assure a mechanically stable contact between the current collector, active material, and electron-conducting additives. The good electrochemical stability in the 0–5 V range, chemical resistance in electrolyte, durable adhesion to current collector, and good binding between PVDF and active electrode materials make polyvinylidene fluoride (PVDF) one of the most popular binder choices for LIBs, so PVDF is also often used in SIBs. However, PVDF-based electrodes cannot tolerate the large volume changes that some of the current electrode materials undergo upon charging and discharging [5]. Moreover, in SIBs, PVDF is shown to undergo defluorination if no additional source of fluorine is available [6], so alternative binders are starting to emerge. Organic compounds, such as carboxymethyl cellulose (CMC), styrene butadiene rubber (SBR) and alginate compounds have been reported [7]. Due to their improved mechanical properties and chemical stability, these new binders often offer better cycle life, higher capacity, and rate capability.
Among alginate compounds, sodium alginate [8], the sodium salt of alginic acid, a compound that is obtained from the cell walls of brown algae, is by far the most studied. It can extend the cycle life of many battery materials due to its higher mechanical strength (as shown in studies on Si anode for LIBs [9,10]), as well as by forming a more uniform protective coating on the particles of the active material [11,12,13]. An added benefit is Na alginate’s solubility in water, enabling preparation of aqueous electrode slurries and thus ensuring environmentally friendlier preparation of electrodes. Unfortunately, organic solvents are not able to dissolve Na alginate. This makes the Na alginate binder incompatible with many of the SIB electrodes that decompose rapidly if exposed to air or water, promoting the search for a compound that would combine the favourable properties of Na alginate while also enabling the preparation of anhydrous electrode slurries.
The instability in water is perhaps best exemplified by layered metal oxide cathodes, such as P2-type Na0.67MnO2 [14]. Capacity-wise, P2-type Na0.67MnO2 is a very competitive SIB cathode, with high practical gravimetric capacity in the range of 160–200 mAh/g [15]. At the average discharge potential of 2.75 V vs. Na/Na+, this results in material energy density of up to 550 Wh/kg. Actual measured capacities are a subject to the chosen voltage range, which is in turn often a compromise between specific charge capacity and cycle life. First investigated in 1999 by Paulsen et al. [16], it has received increased attention from the scientific community over the last five years. Luo et al. showed a capacity of roughly 160 mAh/g and cycle life decrease of roughly 50% over 500 full charge-discharge cycles over a narrow 2.0–3.8 V vs. Na/Na+ range as well as improvement when substituting some Mn with Fe and Ni [17]. Lyu et al. demonstrated that the cycle life of Na0.67MnO2 can be significantly improved when switching to solid electrolyte [18]. More recently, materials engineered on nanoscale, such as nanoplatelets [19] and other nanostructures [20,21], have displayed increased rate capability, albeit with only marginal improvements in cycle life and overall capacity. On average, Na0.67MnO2 lost around 15% of its capacity over first 100 charge-discharge cycles. Doping has yielded improvements regarding cycle life and stability in air. For example, doping with Co [22], Ni, and Mg [23] has been somewhat effective. Creating higher charge-discharge potentials by substituting Mn with Ni, Cu, or Fe [24] have yielded promising results. A more detailed review on recent progress in development of cathodes for SIBs can be found in references [25,26].
The underlying reason why Mn in Na0.67MnO2 is often partly replaced by Ni, Fe, Ti, Al, Mg, and other metals [21,23,27,28,29,30] is the attempt to improve its cycle life. The cycle life of pristine P2-type Na0.67MnO2 is known to be relatively bad due to the gliding and reordering of crystalographic planes at the highly charged states, that lead to P2-P2′, P2-O2, or P2-OP4 phase transitions [21,29]. Doping and Mn substitution lead to altered interlayer spacing and thus altered O-O repulsion and O-Na attraction forces that lead to changes in the structural stability of Na0.67MnO2 [31].
The same effect of changing interlayer spacing take place upon exposing Na0.67MnO2 to water. However, the structural changes happen to such an extreme degree that the P2-type Na0.67MnO2 structure eventually disintegrates. Upon exposure to water, Na+ in Na0.67MnO2 is displaced by H+, changing the interlayer spacing, causing crystallographic planes to glide and leading to the phase transformation of Na0.67MnO2 [24,32]. Ultimately, proton intercalation increases interlayer distance to the point where water molecules can be inserted in the interlayer space [24,33], leading to decomposition of the original P2-type crystal structure, and the cathode loses its capacity for reversible extraction and insertion of Na. There have been signs of a certain water exposure having a positive effect on the stability and rate capability of Na0.67MnO2 due to increased interlayer spacing [31]. However, without any modifications, treating pristine Na0.67MnO2 in water or even just storing it in air leads to the significant worsening of electrochemical properties. Similar effects are observed in polyanionic Na-ion cathodes.
Although aqueous electrode preparation shows promise in some areas, the described challenges promote a search for alternative binders that are stable in contact with the active materials, do not decompose in the voltage range of 2.0–4.3 V, and are soluble in non-aqueous solvents to assure the stability of the active material. Here, for the first time, we demonstrate tetrabutylammonium (TBA) alginate, a TBA salt of alginic acid [34], as a suitable alternative to PVDF and Na alginate. TBA alginate is soluble in water as well as dimethyl sulfoxide (DMSO) [34], N,N-dimethylformamide [35], and other polar aprotic solvents containing tetrabutylammonium fluoride [36], making it the only currently reported alginate that is soluble in non-aqueous solvents. As shown in this work, TBA alginate can open new opportunities for the non-aqueous processing of SIB cathode slurries and demonstrates promising capacity and cyclability improvements over the PVDF-based electrodes.
2. Results
2.1. Morphology and Structure of Na0.67MnO2
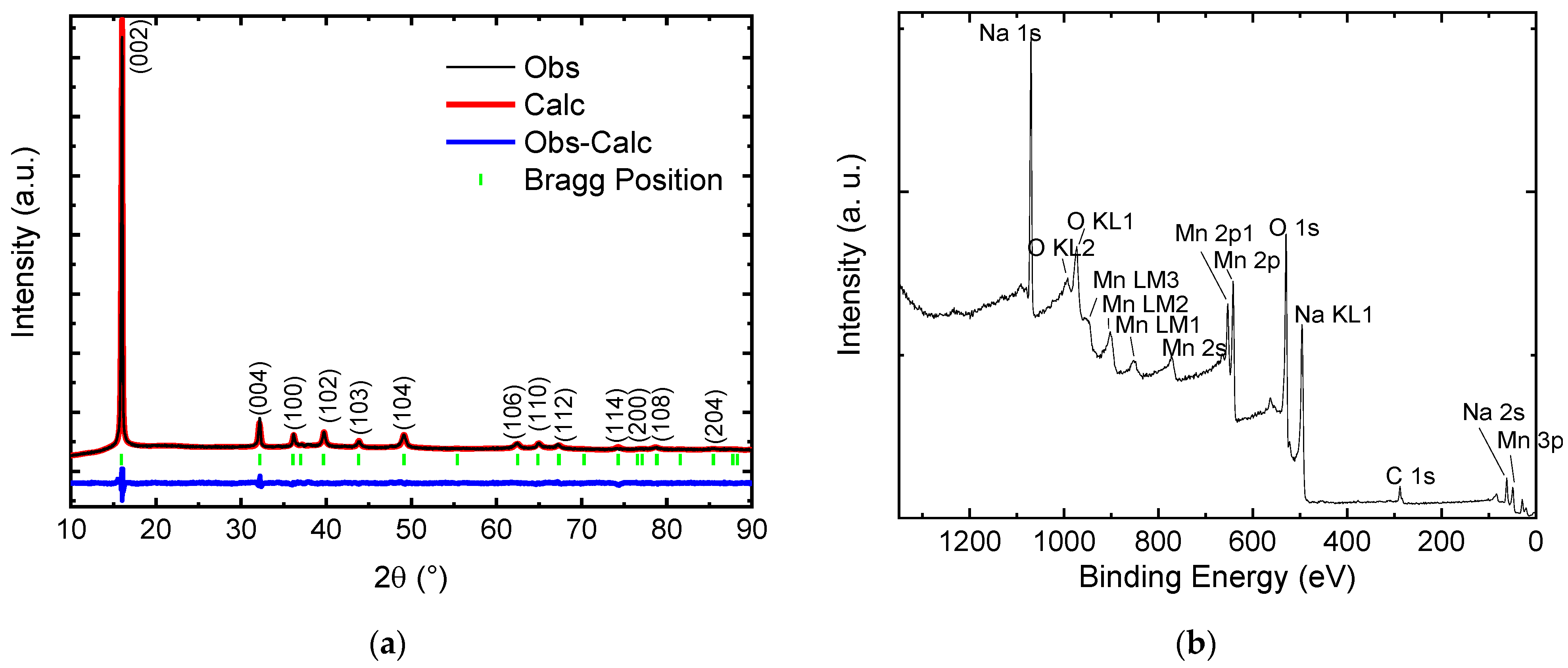
The Na-Mn molar ratio in the as-obtained material corresponds to Na0.66±0.02MnO2 according to ICP-MS results. The synthesized powder is black and has the P2-type layered structure of Na0.67MnO2, as verified by x-ray diffraction (XRD) and shown in Figure 1a. Rietveld analysis of the XRD pattern confirms the correspondence to the hexagonal crystal lattice with P63/mmc space group (space group no. 193) as first observed by Paulsen et al. [16]. The refined lattice parameters are a = 2.87217 Å and c = 11.11251 Å with no impurity peaks detected. Rwp = 4.6% and RB = 2.4% factors are reasonably small.
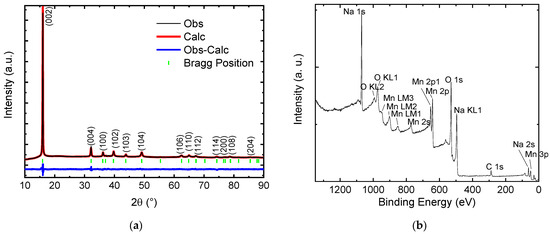

Figure 1.
Structural, compositional and morphological data of the synthesized Na0.67MnO2 cathode: (a) XRD data and Rietveld refinement—all peaks can be indexed with the layered P2-type structure of Na0.67MnO2 as reported by Paulsen et al. [16]; (b) XPS survey shows presence of all elements present in Na0.67MnO2; (c,d) SEM images indicate a grain size of 1–5 μm.
XPS results of the as-prepared Na0.67MnO2 confirm the presence of Na, Mn and O along with trace amounts of carbon (Figure 1b). Individual peaks of Na, Mn, O, and C are shown in Supplementary Information, Figure S1. Mn 2p signal is analysed, and the deconvoluted peaks are consistent with Mn signal in Na0.67MnO2 [23]. The energy separation between the Mn 2p3/2 and Mn 2p1/2 states is 11.3 eV, similar to that observed in LiMn2O4 spinels [37]. To determine the oxidation state of manganese species, one can use Mn 3s peaks [38,39,40,41]. It has been shown that the separation between the peaks in MnO is in the range of 5.7–5.8 eV, in Mn2O3—5.4–5.5 eV, while in MnO2 the separation can be observed in the range of 4.5–4.9 eV. The experimental values measured here are 83.7 eV and 88.8 eV, corresponding to E = 5.1 eV. The obtained results suggest that the valence state of the manganese species in the material corresponds to a combination of Mn3+ and Mn4+ (Supplementary Information, Figure S1e). Detailed XPS spectra along with additional discussion are provided in Supplementary Information, Section S1.
Figure 1c,d show the morphology of the as-synthesized powder. Grain size ranges from 1 to 5 μm, and crystallites often have sharp edges. The presence of NaHCO3, NaOH, Na2CO3, or any other impurity could not be detected by XRD.
2.2. Synthesis and Characterization of TBA Alginate
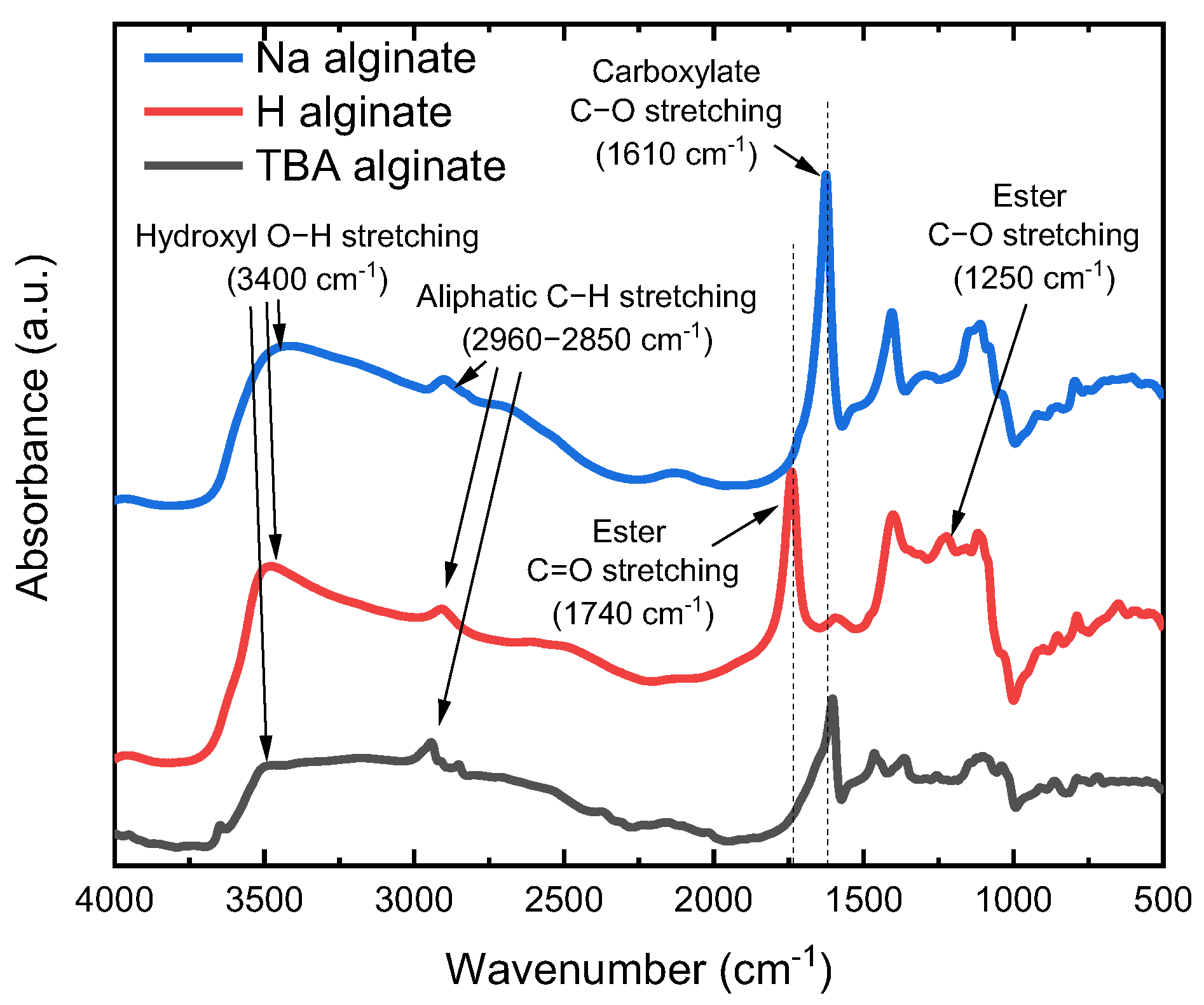
The obtained TBA alginate is a slightly yellowish powder, which can be dissolved in DMF (concentration 2 wt.%), ultimately obtaining a solution with no residue present. FTIR data (Figure 2) are in good agreement with the results of Schleeh et al. [42]. After Na alginate is converted to alginic acid, a peak at 1610 cm−1 characteristic to carboxylate C-O stretching in Na alginate disappears, while the peak at 1740 cm−1 corresponding to saturated COOH becomes visible. As expected, with the synthesis of TBA alginate, the peak of COO− at 1610 cm−1 reappears. While we do not determine the degree of substitution of TBA (DSTBA) in the scope of this paper, we note that a similar synthesis leads to DSTBA values in the range of 0.80–0.95 [42].
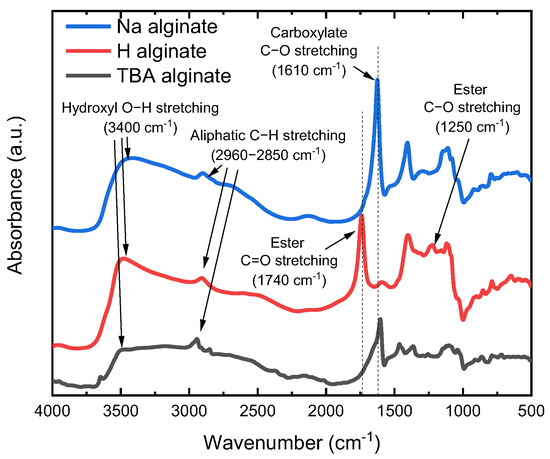
Figure 2.
FTIR spectra of powders of alginic acid (H alginate), Na alginate and TBA alginate.
2.3. Preparation of Electrodes
Na0.67MnO2 electrodes were prepared by using TBA alginate and two other binders for reference, PVDF and Na alginate. A homogenous slurry can be obtained within the first 30 min of mixing when using PVDF binder. The mixing is nevertheless continued for 4 h to match the mixing times of electrodes based on alginate chemistries, since slurries with alginate binders were found to require mixing for longer, in order to obtain homogenous electrodes. Although none are used in this study, employing surfactants might be advised in the future work in order to speed up the electrode preparation. As water is quicker to evaporate than NMP or DMF, Na alginate-based electrodes were dried in room conditions initially to avoid electrode cracking. In our experiments, appearance of cracks correlated with significantly worse long term cycling results, so extended drying times (and somewhat longer water exposure) are preferred over a cracked electrode.
Regarding the coating conditions, we found no difference in electrochemical properties (rate capability and long-term cycling) between electrodes coated and handled in Ar-filled glovebox and ambient conditions, likely due to the very limited air-exposure time. See details in Supplementary Information, figures and discussion in Chapter S2.
SEM images of obtained electrodes (Figure 3) indicate that the electrodes are homogeneous, and particles of active material are evenly dispersed within the matrix of the binder and carbon black electron-conducting additive.


Figure 3.
SEM of Na0.67MnO2 electrodes prepared with (a,d) PVDF; (b,e) Na alginate and (c,f) TBA alginate binders. The length of the scale bar is 10 μm for images in the top row and 3 μm for images in the bottom row.
2.4. Electrochemical Properties and Cycling Behaviour
2.4.1. Electrochemical Properties in Standard SIB Half-Cells
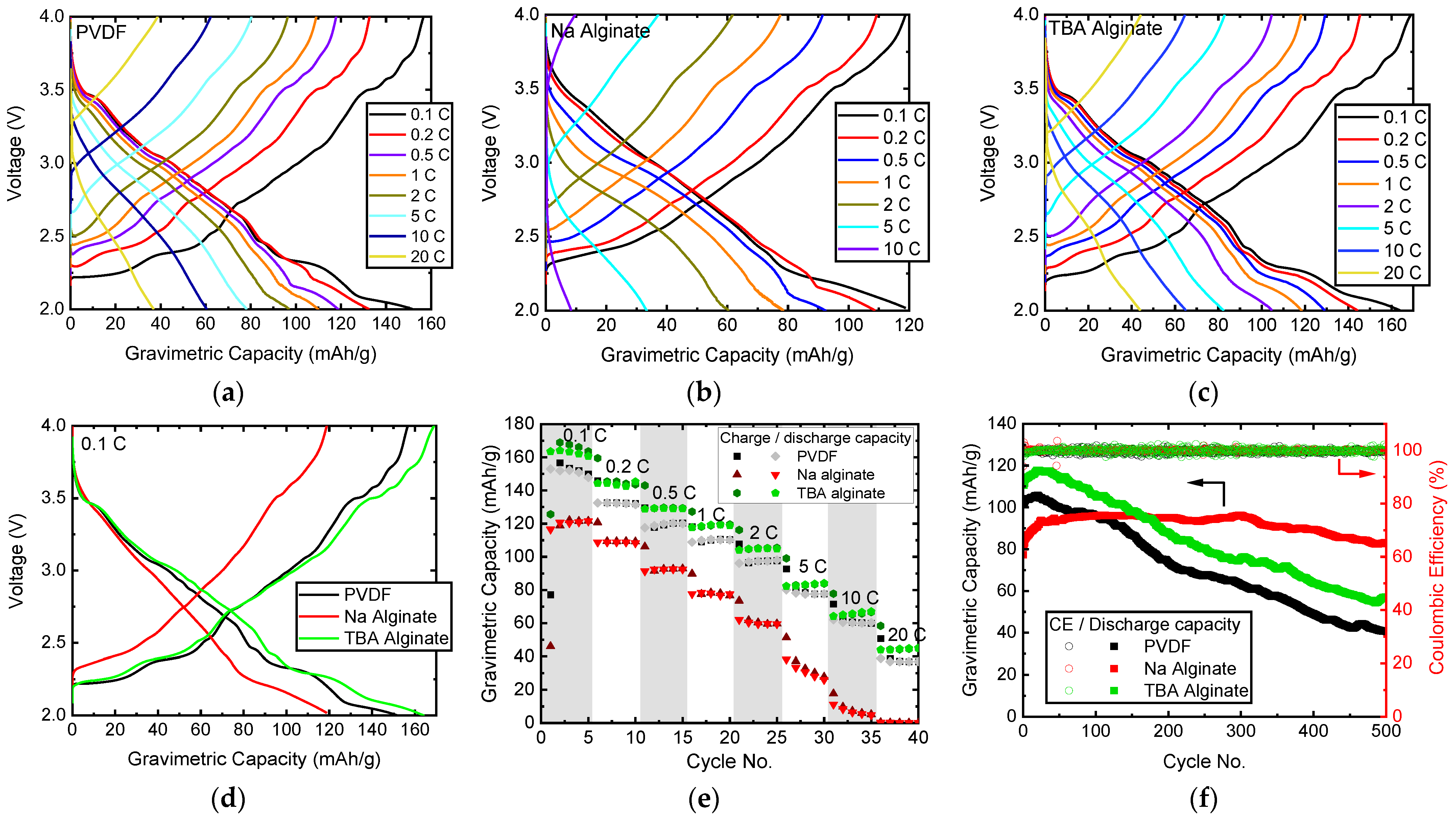
First, a baseline is established by assessing the electrodes prepared with PVDF and Na alginate binders. While PVDF-based Na0.67MnO2 displays charge capacity of roughly 155 mAh/g, as shown in Figure 4a, the capacity decreases to 120 mAh/g if Na alginate binder is used (Figure 4b). This is a result of the aqueous preparation route, as Na0.67MnO2 is known to be unstable in water and humidity [24]. We find that the water-unstable Na0.67MnO2 has been partially decomposed by exchanging Na+ with H+ and ultimately intercalating H2O between the Mn-O layers, as confirmed by XRD in Figure 5. While one can speculate that some of the intercalated hydrogen can later be displaced with Na again, it has been shown that air-exposed electrodes display decreased rate capability, gravimetric capacity, and worse cyclability. This is indeed what we see in this study (Figure 4), as the gravimetric capacity of Na alginate-based electrodes is significantly reduced.
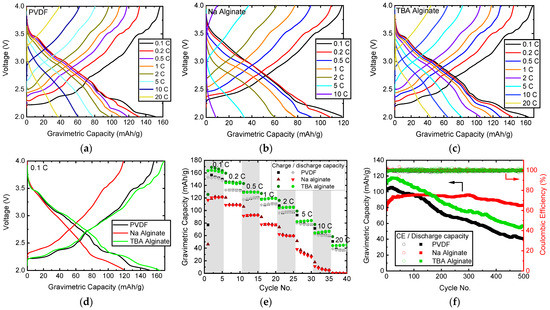
Figure 4.
Electrochemical properties of Na0.67MnO2 electrodes prepared with PVDF, Na alginate and TBA alginate binders: charge-discharge curves of Na0.67MnO2 electrodes with (a) PVDF binder, (b) Na alginate binder, (c) TBA alginate binder; (d) charge-discharge curves at 0.1 C (17.5 mA/g); (e) rate capability of Na0.67MnO2 electrodes; (f) cycle life of Na0.67MnO2 electrodes cycled at 1 C; 1 C corresponds to 175 mA/g.
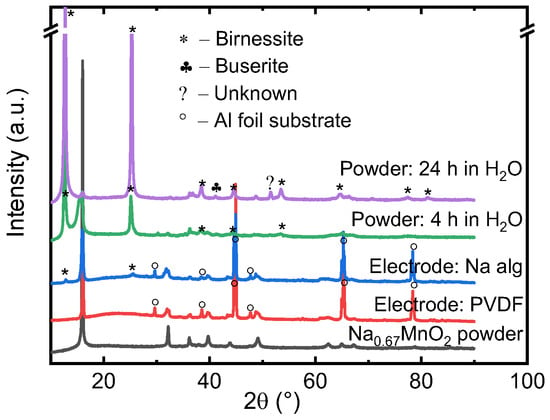
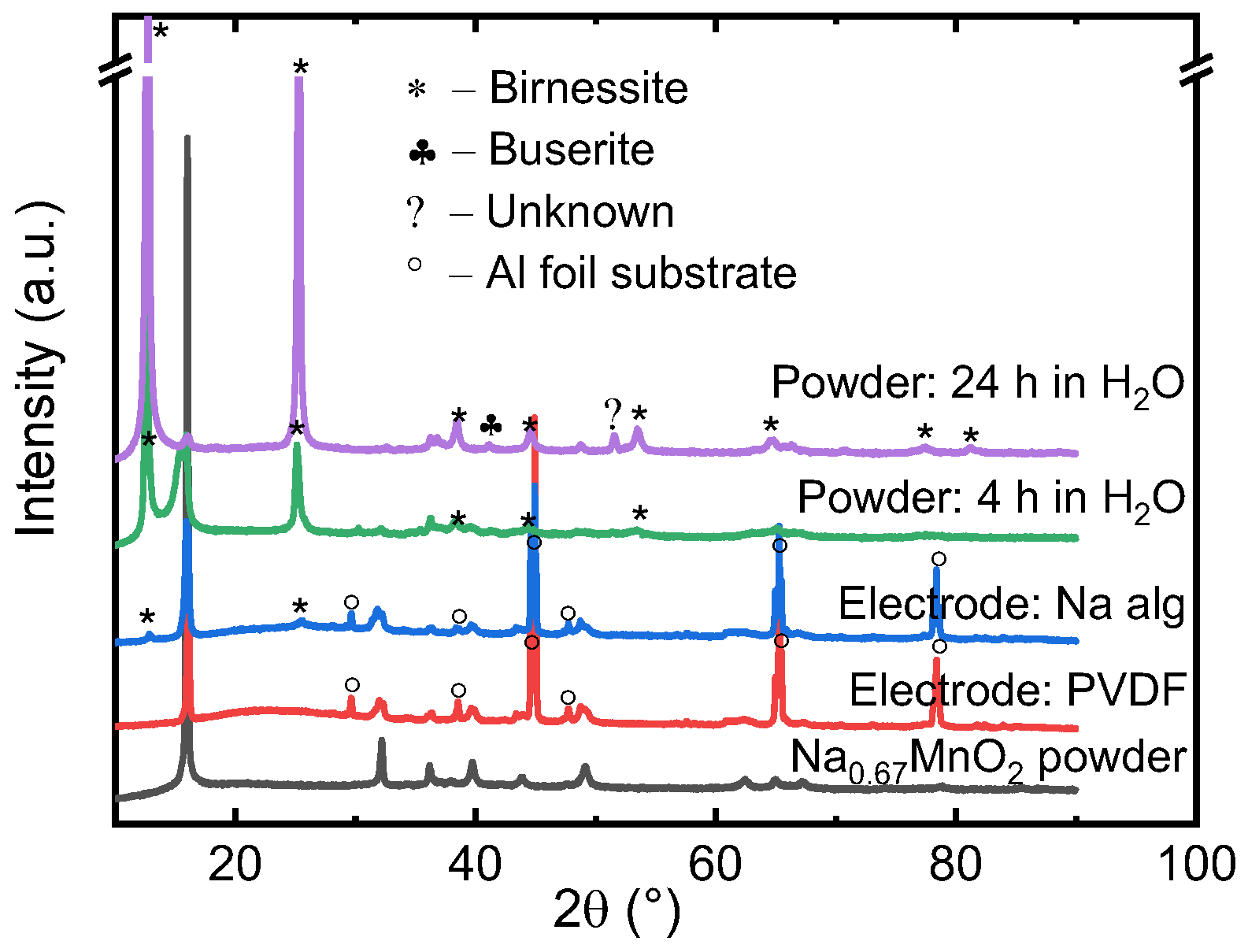
Figure 5.
XRD of Na0.67MnO2 electrodes prepared with PVDF and Na alginate binders, and Na0.67MnO2 powder—pristine and after 4 h and 24 h of water exposure.
The phase associated with water-exposure of NaxMO2 (M – transition metal) is birnessite, and it has an increased interlayer distance compared to P2-type Na0.67MnO2 [43]. Further intercalation of water is associated with formation of the buserite phase. Despite the hydrated (birnessite) phase being electrochemically active and capable of storing sodium with a gravimetric charge of at least 84 mAh/g [44], the value is significantly lower than the roughly 160 mAh/g observed for pristine P2-type Na0.67MnO2. As shown in Figure 5, we found that within 4 h of water exposure (4 h is the time needed to ensure proper mixing of electrode slurry), birnessite impurities have already become apparent. If water exposure is continued, after 24 h almost none of the original structure is retained, and most peaks can be attributed to birnessite or buserite phases. A third phase also appears. Although the lack of additional peaks means that we are not able to pinpoint the impurity phase with a reasonable accuracy, the peak is not associated with Na2CO3 or NaHCO3. The connection to NaOH or its hydrates, however, cannot be ruled out.
When using TBA alginate binder, we are able to eliminate the exposure to water while still using a salt of alginic acid as a binder. Results in Figure 4c show that electrodes prepared with TBA alginate binder display 6% higher gravimetric capacity, up to 164 mAh/g, with good rate capability when compared to PVDF and Na alginate binders (Figure 4e). In general, the results indicate a good application potential of TBA alginate as a binder for Na-ion batteries. Judged from the charge-discharge curves at 0.1 C (Figure 4d), the overvoltage and thus internal resistances of PVDF and TBA alginate-based Na0.67MnO2 electrodes are similar, while binders with Na alginate display noticeably higher overvoltage and less sharp voltage plateaus, likely a consequence of the partially transformed crystal structure. We are able to observe all voltage plateaus characteristic to Na0.67MnO2 for both TBA alginate and PVDF-based electrodes (Figure 4d).
The measured specific discharge capacity of TBA alginate-based Na0.67MnO2 electrode is 164 mAh/g at 17.6 mA/g current. This exceeds most of the reported capacities measured within similar voltage window, even doped P2-type Na0.67MO2 materials (M—transition metal oxide) [11,17,21,23,45,46]. Our literature survey also suggests that on average, P2-type Na0.67MO2 compounds see their discharge capacity decrease by 15% over the first 100 cycles. Depending on how this is calculated, we see the discharge capacity decrease by 8–10%, which is at least a 5% reduction in ageing rate. Of course when compared to state-of-art in LIBs, there is still much room for improvement with regard to cycle life. However, we expect that combining TBA alginate binder with Na0.67MO2 [22,31] or other types of materials for SIBs with higher stability due to doping or other structural modifications can further increase the cycling stability and gravimetric capacity.
The cycling performance of all electrodes, shown in Figure 4f, indicate similarities between electrodes prepared with PVDF and TBA-alginate binders, although the measured absolute gravimetric charge capacity values are higher for TBA alginate-based electrodes. The capacity decreased to 80% of the initial value at cycle 162 for the PVDF-based electrode and cycle 197 for the TBA alginate-based electrode. There is also an initial increase of capacity for both PVDF and TBA alginate-based electrodes, which is higher for TBA alginate. This type of behaviour during first cycles is not uncommon [17,47,48], and is likely related to the formation of a stable SEI on the surface of the electrode, as it has previously been shown to be electrolyte-dependent [48].
As shown in Supplementary Figure S3, the evolution of charge-discharge curves follows a similar pattern for both PVDF and TBA alginate-based electrodes, where the plateaus and other features clearly visible in the initial cycles become less distinct over the cycling and transform somewhat already within the first 20 cycles. This is likely due to irreversible structural changes, induced by discharging in the range of 2.2–2.0 V [15]. The charge-discharge curves of Na alginate-based electrodes are slightly more washed-out.
Somewhat surprisingly, we see the cycling behaviour of Na alginate-based electrode to be significantly improved over PVDF and TBA alginate-based electrodes. The capacity rises form initial 81 mAh/g to 96 mAh/g at 1 C. After 160 cycles the capacity of Na alginate-based electrode surpasses that of TBA alginate-based electrode. After 500 cycles, the capacity of Na alginate-based electrode is still maintained at 82.8 mAh/g or 102% of the initial value. It has been shown that Na alginate can effectively coat the particles of active material, thus preventing several processes: cracking of electrodes, increase in charge-transfer resistance (due to a more stable SEI) and the detachment of electrode from the current collector [11]. It is likely that these are also the reasons behind the improved cycle life of Na alginate-based electrodes in our study. What is somewhat surprising is that this behaviour is not mirrored in TBA alginate-based electrodes, indicating functional differences between TBA alginate and Na alginate.
In order to rule out the improved cyclability of Na alginate-based electrodes coming from the water exposure, we prepared a PVDF-based electrode where Na0.67MnO2 had first been exposed to water in a similar way as while preparing the water-based slurry with Na alginate binder. The electrochemical measurements do not indicate improved cycling performance (see Chapter S4 and Figure S4 in Supplementary Information). Hence, it can be concluded that improvements in long term cycling observed for Na alginate-based electrodes are indeed a consequence of the binder and not the water-exposure of the active material.
The initial Coulombic efficiency is an important parameter for evaluating the performance of electrodes. Measured Coulombic efficiencies are summarised in Table 1. The large values of the initial Coulombic efficiency are not surprising, as Na0.67MnO2 is initially sodium deficient. Assuming that 0.67 parts of Na can be extracted from the electrode, while one full part of Na is inserted in the discharge part of the cycle, the initial Coulombic efficiency should indeed be 149%, in good agreement with the values measured for PVDF and TBA alginate-based electrodes. The initial Coulombic efficiency of Na alginate is somewhat higher, which is consistent with the expected Na loss during aqueous processing of Na0.67MnO2 [24]. Estimated from the observed Coulombic efficiency, the initial stoichiometry of active material in alginate-based electrode is around Na0.4MnO2.

Table 1.
Coulombic efficiency as a function of cycle and binder material.
For both alginate-based electrodes, the stabilised Coulombic efficiency is very close to 100. Coulombic efficiency of PVDF-based electrodes also stabilises above 99%, although the determined values are slightly lower than those for alginate electrodes.
2.4.2. TBA Alginate Binder in Other Cell Chemistries
In order to understand the extent to which the improvement of electrochemical properties with TBA alginate binder can be beneficial to other cell chemistries, PVDF, Na alginate, and TBA alginate electrodes were measured in two other cells𠅃SIB half-cell without SEI-stabilising additive added to the electrolyte and LIB half-cell with LiFePO4 cathode.
As the electrolyte chemistries used in Na-ion batteries are more diverse than in LIBs, it is important to note that the measurements reported so far have been performed in NaClO4 electrolyte (1 M NaClO4 salt in propylene carbonate (PC)) containing 5 wt.% fluoroethylene carbonate (FEC) additive. FEC is known to stabilise the electrode–electrolyte interface and prevent the dissolution of PVDF, functioning as an additional source of fluorine that forms NaF interfacial layer [49]. Na alginate can extend the cycle life of electrodes due to uniformly coating the electrode surfaces [11] and preventing parasitic reactions between the electrolyte and electrode. Na alginate is in general not as vulnerable to decomposition as PVDF [50]. To clarify where TBA alginate binder lies in this spectrum, long-term cycling tests were performed in electrochemical cells with 1 M NaClO4 electrolyte in PC without FEC additive. In such conditions, all electrodes display significantly worse cycle life than measured with the FEC-containing electrolyte (Supplementary Information, Chapter S5, Figure S5). The capacities of PVDF, TBA alginate, and Na alginate-based electrodes decrease below 80% of the initial value after 27, 16, and 288 cycles respectively. Since NaF is known to be a crucial part of the SEI [49] and TBA alginate does not contain fluorine, the results are perhaps not too surprising. The obtained data again outline the importance of a stable SEI layer and indicate that the TBA alginate does not provide as strong of an effect on stabilising the SEI layer as Na alginate, where a protective particle coating effect has been suggested [51]. As at no point does the capacity of TBA alginate-based electrode exceed that of PVDF-based, it is likely that the rapid decomposition of the electrode leaves a notable mark already during the first full charge-discharge cycle.
Both initial Coulombic efficiencies (CEs) as well as stabilised CEs are significantly lower than those measured in 1 M NaClO4 electrolyte with 5 wt.% FEC additive (Supplementary Information, Table S1 in Chapter S5). TBA alginate sees the lowest CE, pointing to the most intense parasitic reactions. Na alginate, on the other hand, has the highest CE, consistent with the relatively good long-term cycling results. The results put TBA and sodium salts of alginic acid in stark contrast and again indicate that there are notable functional differences between both alginate binders.
We also carried out tests with the LiFePO4 cathode for Li-ion batteries and found the results to be very similar (Supplementary Information, Chapter S6). PVDF outperforms TBA alginate binder in the LIB cell chemistry with 1M LiPF6 electrolyte salt in the ethylene carbonate (EC)–dimethyl carbonate (DMC) mixture. Although the reasons for this require further investigation, we hypothesize that this behaviour could be due to instability of TBA alginate in the electrolyte used in LIB cell (1 M LiPF6 in EC/DMC), resulting in the reduced mechanical integrity of the electrode and subsequent loss of electric contact between the current collector and active particles.
2.5. Analysis of Electrode-Electrolyte Interphase
To better understand the mechanisms for the apparently different behaviour between the three binders, we carried out electrochemical impedance (EIS) studies as well as SEM and XPS characterization of the cycled electrodes.
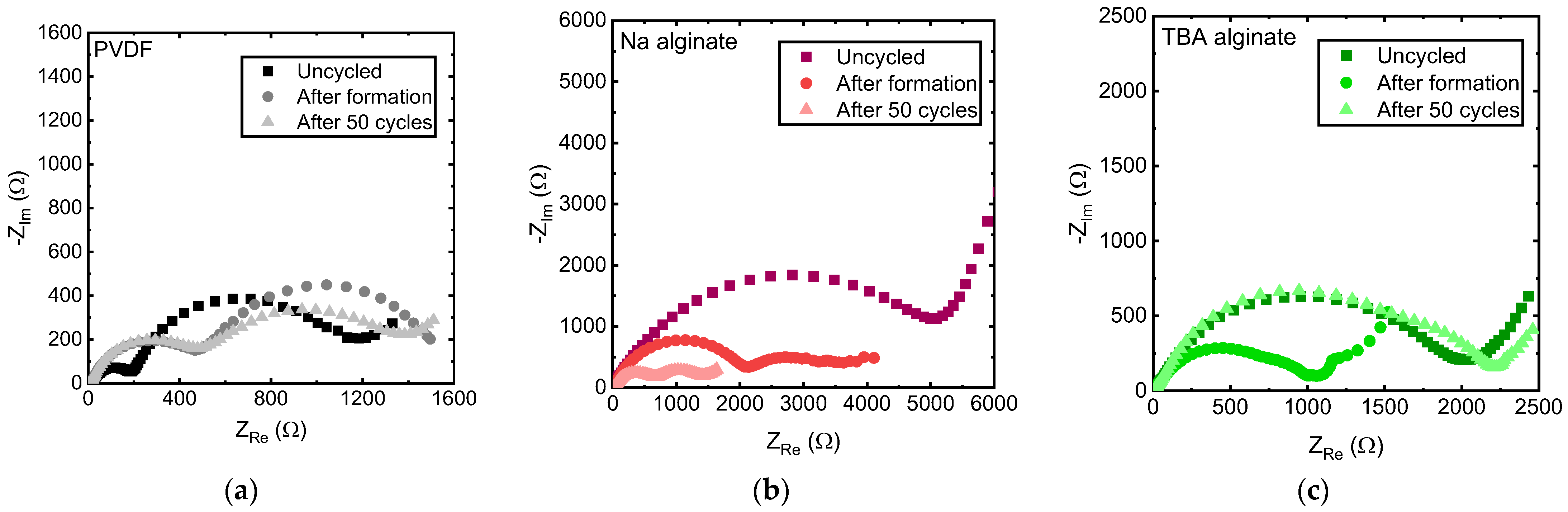
EIS results, shown in Figure 6, show the evolution of internal resistances. EIS data of uncycled cells, cells after formation (3 full charge-discharge cycles at a slow C-rate), and after 50 cycles at 1 C (175 mA/g) are shown. Two arcs are observed for all electrodes. The first corresponds to SEI resistance while the second to charge transfer processes [52,53], although the precise interpretation of these types of EIS results is still being disputed and might be more ambiguous [54,55]. As Gaberscek et al. have shown [54], in some cases, the high frequency arc can also correspond to electrode-current collector resistance rather than the SEI.
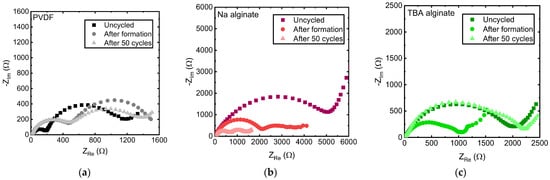
Figure 6.
EIS spectra of Na0.67MnO2 electrodes prepared with (a) PVDF, (b) Na alginate and (c) TBA alginate binders, measured 3 h after assembly of the cell, after 3 formation cycles at a slow rate and after 50 cycles at 1 C (175 mA/g).
Looking at the EIS spectra individually (Figure 6a–c), we find that the internal resistances of PVDF-based electrode change relatively little, with SEI resistance increasing after formation, and charge transfer resistance slightly reducing after 50 cycles. The second semicircle of the EIS spectrum of the PVDF-based electrode flattens with cycling, pointing to the arising inhomogeneities, likely on the electrode’s surface. Na alginate-based Na0.67MnO2 electrode undergoes significant changes, with both resistances notably shrinking during cycling. Both charge transfer and SEI resistances of TBA alginate-based electrode initially shrink similarly to the Na alginate-based electrode. However, after 50 cycles, an increase in the SEI resistance is observed.
When the EIS spectra are compared for similarly aged PVDF, Na alginate, and TBA alginate based Na0.67MnO2 electrodes, consistent with the lower gravimetric capacity and higher overvoltage values, Na alginate has the largest electrode resistance. However, it shrinks upon cycling, ultimately nearing that of the PVDF-based electrode. TBA alginate-based electrode displays significantly larger resistance.
SEM measurements of cycled electrodes, shown in Figure 7, indicate a considerably changed electrode morphology of PVDF-based electrodes, likely arising from a growth of a porous SEI. The surfaces of Na alginate and TBA alginate-based electrodes also display visible signs of SEI build-up (surface residue and smoother edges of grains can be observed). Cracks can be seen in Na alginate-based electrodes at some sites. However, this does not seem to be a widespread phenomenon. All in all, however, the all electrodes maintain their integrity, as the shape, size, or distribution of the active particles has not undergone any notable changes.

Figure 7.
SEM of Na0.67MnO2 electrodes cycled at 1 C for 500 cycles, prepared with (a,d) PVDF; (b,e) Na alginate and (c,f) TBA alginate binders. The length of the scale bar is 10 μm for images in the top row and 3 μm for images in the bottom row.
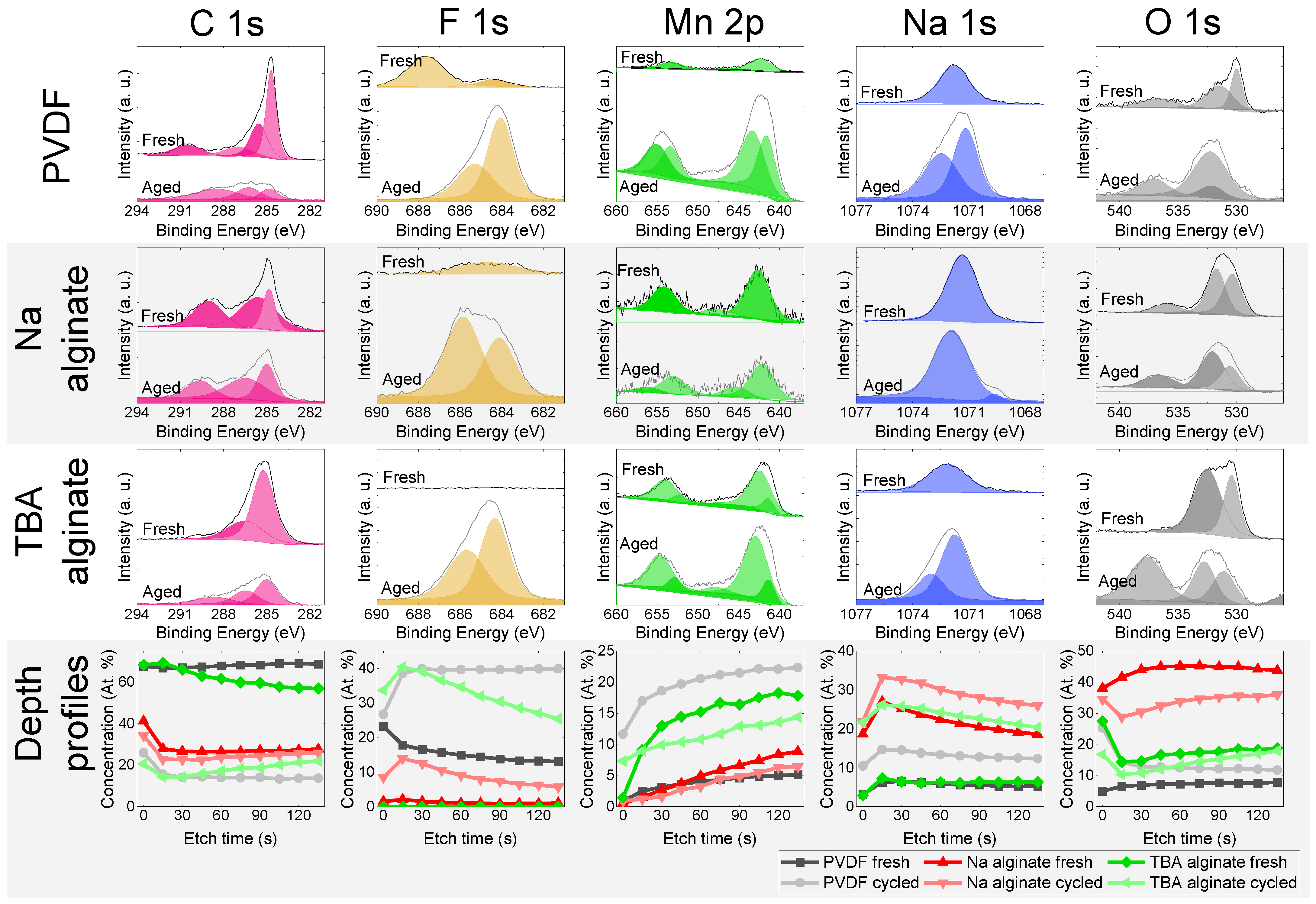
Surfaces of cycled (500 cycles at 1 C) and uncycled electrodes were analysed with XPS (Figure 8). Typically, upon cycling, the solid–electrolyte interface (SEI) starts to grow, consuming some of the sodium and often increasing the internal resistance. The SEI is typically composed of NaF, Na2O, Na2CO3, and other products of electrolyte and electrode decomposition.
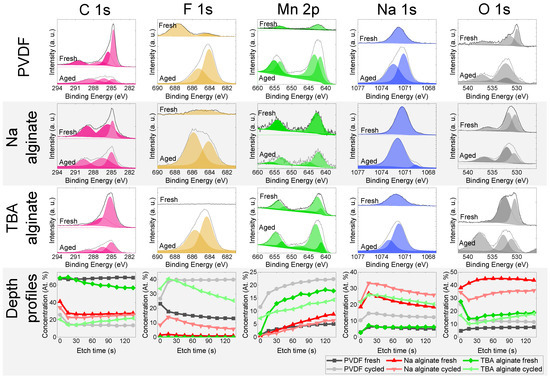
Figure 8.
XPS results of Na0.67MnO2 electrodes for fresh and aged (cycled) electrodes that have undergone 500 charge-discharge cycles at 1 C (175 mA/g) rate—individual spectra and compositional analysis.
NaF is a prominent part of the SEI. The XPS peak corresponding to Na-F bonding at 684.3 eV is observed for all the cycled electrodes. This is the result of the desirable decomposition of FEC electrolyte additive. The peak for C-F bonding (CF2 in PVDF binder) at 688.2 eV is observed in fresh PVDF-based electrodes. Depth profiles of F indicate a considerable amount of F trapped in the SEI. The disappearance of C-F peak at 688.2 eV in the aged PVDF-based electrode again confirms the considerable thickness of the SEI, while the depth distribution of fluorine indicates a relatively higher fluorine content in TBA alginate and PVDF-based electrodes compared to Na alginate-based electrodes.
C1s peak can be deconvoluted into several peaks. Located at 284.8 eV is the peak associated with the C–C bonds of adventitious carbon and acetylene black. The peak at approximately 285.8 eV corresponds to hydrocarbon bonding (C-H), while peaks at 287.2, 289.2 and 290.7 eV are associated with C–O/C=O, Na2CO3 and R–CO3/CFx (x ≥ 2), respectively [49,56]. The shape and intensity of PVDF electrode changes significantly upon cycling, signalling a significant SEI growth, while that of Na alginate-based electrode remains virtually unchanged. The intensity of the TBA alginate electrode’s peaks is lower for the aged electrode, but the shape remains unchanged and signals presence of mostly C–C and C–O/C=O bonds.
Mn signal measured for most electrodes is weaker in aged electrodes. This points to the formation of a SEI where Mn is not present. An exception to this is the PVDF-based electrodes, where the Mn signal in cycled electrode intensifies. The dissolution of Mn was previously observed In Na0.67MnO2 [57] and is likely more prevalent due to a more inhomogeneous SEI. This is in agreement with Zou et al. who have also reported formation of MnF2 on the surface of a PVDF-based electrodes [21].
The signal of Na changes significantly when comparing fresh and cycled TBA alginate and PVDF alginate-based electrodes, again pointing towards a notable SEI growth, while the Na 1s spectrum of Na alginate-based electrode stays relatively similar for fresh and aged electrodes. Peaks of ROCO2Na (1070.3 eV), Na2CO3 (1071.5 eV), and NaF (1072.8 eV) are observed for Na. After the cycling, the peaks of Na shift to the right for PVDF and TBA alginate-based electrodes, pointing to an increasing intensity of ROCO2Na functional groups (1070.3 eV), although an appearance of NaF (1072.8 eV) is also observed. For Na alginate-based electrode, the shift is in the other direction, signifying a considerable prevalence of Na2CO3 (also evident from the C 1s spectrum). The Na concentration has increased considerably after cycling for PVDF and TBA alginate-based electrodes due to the SEI growth, while it remains similar for Na alginate-based electrode due to the binder containing significant amount of Na.
Oxygen signal contains several bands peaking at around 530 eV and 536 eV. The latter is the KLL line of Na. Oxygen spectra consist of peaks at 530.1, 531.8 eV, and 532.6 eV, corresponding to Na-O-Mn, Na2CO3, and RCH2ONa respectively. While for the PVDF and Na alginate-based electrodes a large proportion comes from Na0.67MnO2 and Na2CO3, XPS data from TBA alginate-based electrode contain a considerably larger number of signals coming from RCH2ONa.
To sum up, the results of the XPS point to the formation of a SEI that is Na- and F-rich and contains several organic compounds not seen in the fresh electrodes. Although TBA alginate does not contain fluorine, judging from the XPS results, it forms Na- and F-containing compounds on its surface that are similar to those observed on PVDF-based electrode. In general, the difference between SEI formed on PVDF-based electrode and SEI formed on TBA alginate-based electrode is not large. On the other hand, Na alginate electrodes have less NaF in the SEI and generally undergo smaller compositional changes upon cycling.
3. Discussion
The set of results from structural, morphological, and interfacial characterisations combined with an array of electrochemical tests in different systems provide the first insight into the working mechanisms of TBA alginate binder.
Qualitatively, there are more functional similarities between TBA alginate and PVDF than between TBA and Na alginate binders. Electrodes based on PVDF and TBA alginate binders both follow a similar capacity decay pattern during the long-term charge-discharge cycling. Internal electrode resistances measured with EIS increase slightly during cycling, and electrodes based on both binders have a considerable amount of Na-F bonding in the SEI after cycling in 1 M NaClO4 electrolyte with 5 wt.% FEC additive. XPS results indicate that the SEI grown on both TBA alginate- and PVDF-based electrodes contain considerable amounts of Na-F and RCH2ONa compounds.
Perhaps the most notable difference between PVDF and TBA alginate-based electrodes is the porosity of the SEI, as evident in the SEM images. This is likely the reason why the observed SEI resistance measured by EIS is comparably largest for PVDF-based electrodes. Given the similar chemical composition of SEIs and good integrity of electrodes evidenced before and after long-term cycling experiments, it is likely that the improvement of the electrochemical properties of TBA alginate-based Na0.67MnO2 electrodes comes from an improved homogeneity of the SEI.
Interestingly, considerable functional differences exist between TBA and Na alginate electrodes. As evident from the XPS spectra (especially C, O and Na), Na alginate undergoes considerably less surface change upon ageing, pointing to an already-stable protective layer before cycling. The SEI layer seems to get richer in fluorine, with the SEI composition not limited only to NaF, and with it the resistance of the electrode reduces, as evidenced by EIS. Furthermore, when the Na alginate-based electrode is cycled in the electrolyte without FEC additive, the lack of NaF interfacial layer does not seem to affect the cycle life as much as that of the PVDF and TBA alginate-based electrodes. It can be hypothesized that Na alginate forms a more homogenous protective coating on the surface of the electrodes and thus prevents some of the undesirable decomposition reactions. The reason behind the radically different interfacial behaviours between TBA and Na alginate-based electrodes, however, remains a potentially interesting topic for future studies.
Naturally, challenges remain in optimising the process of mixing and coating the electrode slurries. In the present study, it took considerably longer to obtain a homogenous TBA alginate-based slurry than a comparable PVDF-based one. An interesting subject of practical future studies could be the performance of TBA alginate binder in conjunction with active materials that have an inherently better cycle life and do not suffer from Mn2+ dissolution and oxygen evolution as much as Na0.67MnO2 does. If work on Na0.67MnO2 is to be continued, several pathways could be considered for increasing cycle life of the cathode, as currently it falls below 50% of initial after 500 cycles. These pathways would most likely have to deal with mitigating the P2-P2′ and P2-OP4 phase transitions in the active material that have been shown to have a significant impact on the cycle life [21,29]. Approaches to stabilize the structure by modifying the crystal lattice [31] or via the doping or substitution of Mn or other elements have provided good results [46,58]. Further alterations of electrode formulations, i.e., electron-conducting additives [59], protective coatings [60], or the formation of various nanostructured materials, e.g., nanocomposites [61] or nanostructures with high specific surface area [62], could also be considered. Our study also opens up potential to further investigation and optimisation of the electrode slurries.
From a practical point of view, the use of TBA alginate binder instead of PVDF is appealing, as this research demonstrates the superior electrochemical performance of the TBA alginate-based Na0.67MnO2 electrode when compared to PVDF binder-based electrodes, while measurements of electrode surface and SEI prior to and after cycling suggest these electrodes also possess good interfacial stability.
4. Materials and Methods
Na0.67MnO2 was prepared by solid-state synthesis. Stoichiometric amounts of Mn2O3 (≥99%, Sigma-Aldrich, Darmstadt, Germany) and Na2CO3 (≥99%, Sigma-Aldrich, Germany) were ball-milled for 2 h. The mixture was pressed into pellets and heated at 900 °C for 24 h in air. The samples were then cooled down to 500 °C, taken out of the furnace, and transferred to an Ar-filled glove box for further cooling and processing to avoid unnecessary contact with moisture, oxygen, and CO2.
A procedure similar to that used by Babak et al. [34] and Pawar et al. [36] was followed. Thus, 120 mL 0.6 M HCl was mixed with 120 mL 96% ethanol. Na-alginate was added to the mixture and stirred for 12 h at 4 °C to obtain alginic acid. The mixture was filtered under vacuum and washed with alcohol and acetone. The solid product was dried in vacuum drying oven at 60 °C for 12 h.
1 g of fully dried alginic acid was dispersed in 150 mL water, then 1.5 M tetrabutylammonium hydroxide (TBA-OH) solution was added dropwise with continuous stirring at room temperature until the alginic acid was dissolved and the pH was adjusted to 8 ± 1. The mixture was dried in vacuum oven at 60 °C for 24 h until fully dry solid product was obtained.
The crystal structure of Na0.67MnO2 was characterized by X-ray diffraction (XRD) using Rigaku MiniFlex600 X-ray diffractometer with Cu Kα radiation at room temperature. The patterns were recorded in the 2θ range of 10–90°. Rietveld refinement was conducted with Bruker TOPAS software. An Agilent 8900 ICP-QQQ Inductively Coupled Plasma Mass Spectrometer (ICP-MS) equipped with a MicroMist nebulizer was used to determine Na and Mn ratio in NaMnO2. Before measurements, NaMnO2 was dissolved in nitric acid. Quantification of Na and Mn was done by 5-point calibration graph method in He mode. Scanning electron microscopy (SEM) images of Na0.67MnO2 powder and electrodes were taken by Thermo Scientific (Waltham, MA, USA) Helios 5 UX scanning electron microscope. Powders of the Na alginate, alginic acid and TBA alginate were analyzed with Fourier transformation infrared spectroscopy (FTIR) using Bruker Vertex 80v vacuum FTIR spectrometer.
X-ray photoelectron spectroscopy (XPS) was carried out for Na0.67MnO2 powder as well as battery electrodes before and after cycling (post-mortem) using ThermoFisher ESCALAB Xi+ instrument with monochromatic Al Kα X-ray source. The samples were transferred from the argon-filled glovebox to the XPS instrument in an inert gas transfer vessel. The instrument’s binding energy scale was calibrated to give a binding energy at 932.6 eV for Cu 2p3/2 line of freshly etched metallic copper. A charge compensation system was used, with the surface of the sample irradiated with a flood of electrons to produce nearly neutral surface charge. The experimental data has been referenced to C 1s adventitious carbon. The spectra were recorded by using an X-ray beam size 650 × 10 microns, a pass energy of 20 eV and step size 0.1 eV.
The electrodes were prepared with wet slurry-coating method. The active material (Na0.67MnO2) was mixed with carbon black and binder (PVDF, Na alginate or TBA alginate) in a weight ratio of 75:15:10. Solvents N-methyl-2-pyrrolidone (NMP), water or N,N-dimethylformamide (DMF) were used respectively, the substances were mixed using mill mixer Retsch MM200. The obtained slurry was coated on an Al foil using a doctor blade and dried in a vacuum drying oven at 80 °C for 24 h.
The electrochemical performance was tested in Swagelok type cells assembled in Ar-filled glove box. Metallic sodium was used as counter electrode. Electrochemical cells were assembled, using glass fiber filter (Whatman GF/B) as separator and 1 M NaClO4 solution in propylene carbonate (PC) with 5 wt.% fluoroethylene carbonate (FEC) as electrolyte. For selected measurements, described further in the text, the use of FEC additive was avoided. Electrochemical measurements were carried out by using Biologic VMP3 potentiostat/galvanostat. Galvanostatic charge-discharge curves were obtained in constant current mode between 2 and 4 V. We assume 1 C charge/discharge current to correspond to 175 mA/g, based on the stoichiometry Na0.67MnO2.
Selected measurements with the PVDF and alginate binders were carried out in LIB cells following a procedure that is similar to the one described above. LIB cells consisted of metallic Li counter electrode, LiFePO4 (LFP, battery grade, MTI Corp., Richmond, CA, USA) electrode (LiFePO4:carbon black:binder ratio 75:15:10), Whatman GF/B separator and electrolyte, 1 M LiPF6 in a mixture of ethylene carbonate (EC) and diethyl carbonate (DEC). The volume ratio of solvents was 1:1. Tests were carried out in the voltage range of 2.7–4.0 V.
For a post-mortem study, Na0.67MnO2 electrode was cycled at 1 C rate (175 mA/g) for 500 full charge-discharge cycles. To avoid contamination of the electrode surface, the experiments were carried out in an electrochemical cell without a separator. Cells were disassembled in an Ar-filled glovebox. Electrodes were rinsed in PC twice, 2 min each time, and dried before transferring to XPS in an inert-gas transfer tool to avoid air exposure. SEM images of the cycled electrodes were taken by using a Thermo Scientific Helios 5 UX scanning electron microscope.
5. Conclusions
Na0.67MnO2 electrode with TBA alginate binder for SIBs has been successfully prepared and characterised. The prepared Na0.67MnO2 electrode has a gravimetric capacity of up to 164 mAh/g (6% higher than electrode prepared with the current state-of-art binder—PVDF) and good rate capability and cyclability. To the best of our knowledge, this is the first study reporting TBA alginate as a binder in a battery application and is consequentially also the first alginate binder-based electrode prepared from a non-aqueous slurry. We conclude that TBA alginate-based electrodes, while somewhat tedious to prepare, display improved electrochemical performance when compared to PVDF-based electrodes. Therefore, TBA alginate is a good potential alternative to PVDF in battery applications where water-based processing of slurries is not feasible, such as the demonstrated case with Na0.67MnO2 cathode.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/batteries8010006/s1, Figure S1: XPS spectra of Na0.67MnO2, Figure S2: Electrochemical measurements of Na0.67MnO2 electrodes prepared in air and in argon-filled glovebox, Figure S3: Charge-discharge curves at 175 mA/g (1 C) over 500 cycles in 1 M NaClO4 electrolyte in PC with 5 wt.% FEC additive for electrodes prepared with PVDF, Na alginate and TBA alginate binder. Charge-discharge curves in same electrolyte without FEC additive for electrodes prepared with PVDF, Na alginate and TBA alginate, Figure S4: Electrochemical properties of Na0.67MnO2 active material treated for 2 h in deionized water, prepared with PVDF binder, Figure S5: Long term cycling results at 1 C (175 mA/g) rate for Na0.67MnO2 electrodes prepared with PVDF, Na alginate and TBA alginate binders in 1 M NaClO4 electrolyte without FEC additive, Figure S6: Electrochemical performance of PVDF-, Na alginate- and TBA alginate-based LiFePO4 electrodes, Table S1: Coulombic efficiencies of Na0.67MnO2 electrodes with different binders in electrolyte without FEC additive; measurements during the first five cycles and averaged value from cycles 50–10.
Author Contributions
Conceptualization, G.K.; methodology, G.K. and B.K.; software for evaluation of data, J.H.; validation, B.K. and G.K.; formal analysis, G.K., B.K., A.S., A.V. and P.K.; investigation, B.K., P.K., A.S. and A.V.; resources, G.K. and J.H.; writing—original draft preparation, G.K., B.K. and A.S.; writing—review and editing, all authors; visualization, G.K.; supervision, G.K.; project administration, G.K.; funding acquisition, G.K. and G.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by State Education Development Agency, Republic of Latvia, grant number 1.1.1.2/VIAA/1/16/166, “Advanced Materials for Sodium Ion Batteries”. Institute of Solid-State Physics, University of Latvia as the Centre of Excellence has received funding from the European Union’s Horizon 2020 Framework Program H2020-WIDESPREAD-01-2016-2017-TeamingPhase2 under grant agreement No. 739508, project CAMART2.
Institutional Review Board Statement
Not applicable.
Data Availability Statement
Data supporting reporting results available by request: gints.kucinskis@cfi.lu.lv.
Conflicts of Interest
The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Vaalma, C.; Buchholz, D.; Weil, M.; Passerini, S. A cost and resource analysis of sodium-ion batteries. Nat. Rev. Mater. 2018, 3, 18013. [Google Scholar] [CrossRef]
- Jin, T.; Li, H.; Zhu, K.; Wang, P.-F.; Liu, P.; Jiao, L. Polyanion-type cathode materials for sodium-ion batteries. Chem. Soc. Rev. 2020, 49, 2342–2377. [Google Scholar] [CrossRef]
- Kucinskis, G.; Nesterova, I.; Sarakovskis, A.; Bikse, L.; Hodakovska, J.; Bajars, G. Electrochemical performance of Na2FeP2O7/C cathode for sodium-ion batteries in electrolyte with fluoroethylene carbonate additive. J. Alloys Compd. 2022, 895, 162656. [Google Scholar] [CrossRef]
- Lyu, Y.; Liu, Y.; Yu, Z.-E.; Su, N.; Liu, Y.; Li, W.; Li, Q.; Guo, B.; Liu, B. Recent advances in high energy-density cathode materials for sodium-ion batteries. Sustain. Mater. Technol. 2019, 21, e00098. [Google Scholar] [CrossRef]
- Zhao, X.; Niketic, S.; Yim, C.-H.; Zhou, J.; Wang, J.; Abu-Lebdeh, Y. Revealing the Role of Poly(vinylidene fluoride) Binder in Si/Graphite Composite Anode for Li-Ion Batteries. ACS Omega 2018, 3, 11684–11690. [Google Scholar] [CrossRef]
- Kubota, K.; Komaba, S. Review—Practical Issues and Future Perspective for Na-Ion Batteries. J. Electrochem. Soc. 2015, 162, A2538–A2550. [Google Scholar] [CrossRef]
- Bresser, D.; Buchholz, D.; Moretti, A.; Varzi, A.; Passerini, S. Alternative binders for sustainable electrochemical energy storage—The transition to aqueous electrode processing and bio-derived polymers. Energy Environ. Sci. 2018, 11, 3096–3127. [Google Scholar] [CrossRef] [Green Version]
- Kovalenko, I.; Zdyrko, B.; Magasinski, A.; Hertzberg, B.; Milicev, Z.; Burtovyy, R.; Luzinov, I.; Yushin, G. A Major Constituent of Brown Algae for Use in High-Capacity Li-Ion Batteries. Science 2011, 334, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Ge, M.; Rong, J.; Fang, X.; Zhou, C. Porous Doped Silicon Nanowires for Lithium Ion Battery Anode with Long Cycle Life. Nano Lett. 2012, 12, 2318–2323. [Google Scholar] [CrossRef] [PubMed]
- Sethuraman, V.A.; Nguyen, A.; Chon, M.J.; Nadimpalli, S.P.V.; Wang, H.; Abraham, D.P.; Bower, A.F.; Shenoy, V.B.; Guduru, P.R. Stress Evolution in Composite Silicon Electrodes during Lithiation/Delithiation. J. Electrochem. Soc. 2013, 160, A739–A746. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Jiang, K.; Zhang, X.; Zhang, X.; Guo, S.; Zhou, H. Sodium Alginate Enabled Advanced Layered Manganese-Based Cathode for Sodium-Ion Batteries. ACS Appl. Mater. Interfaces 2019, 11, 26817–26823. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; Lu, X.; Yu, X.; Hu, Y.-S.; Li, H.; Yang, X.-Q.; Chen, L. Sodium Storage and Transport Properties in Layered Na2Ti3O7 for Room-Temperature Sodium-Ion Batteries. Adv. Energy Mater. 2013, 3, 1186–1194. [Google Scholar] [CrossRef]
- Darjazi, H.; Staffolani, A.; Sbrascini, L.; Bottoni, L.; Tossici, R.; Nobili, F. Sustainable Anodes for Lithium- and Sodium-Ion Batteries Based on Coffee Ground-Derived Hard Carbon and Green Binders. Energies 2020, 13, 6216. [Google Scholar] [CrossRef]
- Wang, P.-F.; You, Y.; Yin, Y.-X.; Guo, Y.-G. Layered Oxide Cathodes for Sodium-Ion Batteries: Phase Transition, Air Stability, and Performance. Adv. Energy Mater. 2018, 8, 1701912. [Google Scholar] [CrossRef]
- Kumakura, S.; Tahara, Y.; Kubota, K.; Chihara, K.; Komaba, S. Sodium and Manganese Stoichiometry of P2-Type Na2/3MnO2. Angew. Chemie Int. Ed. 2016, 55, 12760–12763. [Google Scholar] [CrossRef]
- Paulsen, J.M.; Dahn, J.R. Studies of the layered manganese bronzes, Na2/3[Mn1−xMx]O2 with M=Co, Ni, Li, and Li2/3[Mn1−xMx]O2 prepared by ion-exchange. Solid State Ion. 1999, 126, 3–24. [Google Scholar] [CrossRef]
- Luo, C.; Langrock, A.; Fan, X.; Liang, Y.; Wang, C. P2-type transition metal oxides for high performance Na-ion battery cathodes. J. Mater. Chem. A 2017, 5, 18214–18220. [Google Scholar] [CrossRef]
- Lyu, Y.-Q.; Yu, J.; Wu, J.; Effat, M.B.; Ciucci, F. Stabilizing Na-metal batteries with a manganese oxide cathode using a solid-state composite electrolyte. J. Power Sources 2019, 416, 21–28. [Google Scholar] [CrossRef]
- Zhou, Z.; Li, J.; Luo, Z.; He, Z.; Zheng, J.; Li, Y.; Mao, J.; Dai, K.; Yan, C.; Sun, Z. Na2/3MnO2 nanoplates with exposed active planes as superior electrochemical performance sodium-ion batteries. Ionics 2021, 27, 5187–5196. [Google Scholar] [CrossRef]
- Jung, E.; Park, Y.; Park, K.; Kwon, M.-S.; Park, M.; Sinha, A.K.; Lee, B.-H.; Kim, J.; Lee, H.S.; Chae, S.I.; et al. Synthesis of nanostructured P2-Na2/3MnO2 for high performance sodium-ion batteries. Chem. Commun. 2019, 55, 4757–4760. [Google Scholar] [CrossRef]
- Zuo, W.; Qiu, J.; Liu, X.; Zheng, B.; Zhao, Y.; Li, J.; He, H.; Zhou, K.; Xiao, Z.; Li, Q.; et al. Highly-stable P2–Na0.67MnO2 electrode enabled by lattice tailoring and surface engineering. Energy Storage Mater. 2020, 26, 503–512. [Google Scholar] [CrossRef]
- Konarov, A.; Kim, H.J.; Voronina, N.; Bakenov, Z.; Myung, S.-T. P2-Na2/3MnO2 by Co Incorporation: As a Cathode Material of High Capacity and Long Cycle Life for Sodium-Ion Batteries. ACS Appl. Mater. Interfaces 2019, 11, 28928–28933. [Google Scholar] [CrossRef] [PubMed]
- Hemalatha, K.; Jayakumar, M.; Bera, P.; Prakash, A.S. Improved electrochemical performance of Na0.67MnO2 through Ni and Mg substitution. J. Mater. Chem. A 2015, 3, 20908–20912. [Google Scholar] [CrossRef]
- Zuo, W.; Qiu, J.; Liu, X.; Ren, F.; Liu, H.; He, H.; Luo, C.; Li, J.; Ortiz, G.F.; Duan, H.; et al. The stability of P2-layered sodium transition metal oxides in ambient atmospheres. Nat. Commun. 2020, 11, 3544. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Li, X.; Tang, K.; Wang, D.; Long, M.; Gao, H.; Chen, W.; Liu, C.; Liu, H.; Wang, G. Recent progress of emerging cathode materials for sodium ion batteries. Mater. Chem. Front. 2021, 5, 3735–3764. [Google Scholar] [CrossRef]
- Abraham, K.M. How Comparable Are Sodium-Ion Batteries to Lithium-Ion Counterparts? ACS Energy Lett. 2020, 5, 3544–3547. [Google Scholar] [CrossRef]
- Yabuuchi, N.; Kajiyama, M.; Iwatate, J.; Nishikawa, H.; Hitomi, S.; Okuyama, R.; Usui, R.; Yamada, Y.; Komaba, S. P2-type Nax[Fe1/2Mn1/2]O2 made from earth-abundant elements for rechargeable Na batteries. Nat. Mater. 2012, 11, 512–517. [Google Scholar] [CrossRef] [PubMed]
- Billaud, J.; Singh, G.; Armstrong, A.R.; Gonzalo, E.; Roddatis, V.; Armand, M.; Rojo, T.; Bruce, P.G. Na0.67Mn1−xMgxO2 (0 ≤ x ≤ 0.2): A high capacity cathode for sodium-ion batteries. Energy Environ. Sci. 2014, 7, 1387–1391. [Google Scholar] [CrossRef]
- Liu, X.; Zuo, W.; Zheng, B.; Xiang, Y.; Zhou, K.; Xiao, Z.; Shan, P.; Shi, J.; Li, Q.; Zhong, G.; et al. P2-Na0.67AlxMn1−xO2: Cost-Effective, Stable and High-Rate Sodium Electrodes by Suppressing Phase Transitions and Enhancing Sodium Cation Mobility. Angew. Chemie Int. Ed. 2019, 58, 18086–18095. [Google Scholar] [CrossRef]
- Clément, R.J.; Bruce, P.G.; Grey, C.P. Review—Manganese-Based P2-Type Transition Metal Oxides as Sodium-Ion Battery Cathode Materials. J. Electrochem. Soc. 2015, 162, A2589–A2604. [Google Scholar] [CrossRef]
- Zuo, W.; Liu, X.; Qiu, J.; Zhang, D.; Xiao, Z.; Xie, J.; Ren, F.; Wang, J.; Li, Y.; Ortiz, G.F.; et al. Engineering Na+-layer spacings to stabilize Mn-based layered cathodes for sodium-ion batteries. Nat. Commun. 2021, 12, 4903. [Google Scholar] [CrossRef] [PubMed]
- Monyoncho, E.; Bissessur, R. Unique properties of α-NaFeO2: De-intercalation of sodium via hydrolysis and the intercalation of guest molecules into the extract solution. Mater. Res. Bull. 2013, 48, 2678–2686. [Google Scholar] [CrossRef]
- Duffort, V.; Talaie, E.; Black, R.; Nazar, L.F. Uptake of CO2 in Layered P2-Na0.67Mn0.5Fe0.5O2: Insertion of Carbonate Anions. Chem. Mater. 2015, 27, 2515–2524. [Google Scholar] [CrossRef]
- Babak, V.G.; Skotnikova, E.A.; Lukina, I.G.; Pelletier, S.; Hubert, P.; Dellacherie, E. Hydrophobically Associating Alginate Derivatives: Surface Tension Properties of Their Mixed Aqueous Solutions with Oppositely Charged Surfactants. J. Colloid Interface Sci. 2000, 225, 505–510. [Google Scholar] [CrossRef]
- Leone, G.; Torricelli, P.; Chiumiento, A.; Facchini, A.; Barbucci, R. Amidic alginate hydrogel for nucleus pulposus replacement. J. Biomed. Mater. Res. Part A 2008, 84A, 391–401. [Google Scholar] [CrossRef]
- Pawar, S.N.; Edgar, K.J. Chemical Modification of Alginates in Organic Solvent Systems. Biomacromolecules 2011, 12, 4095–4103. [Google Scholar] [CrossRef]
- Ramana, C.V.; Massot, M.; Julien, C.M. XPS and Raman spectroscopic characterization of LiMn2O4 spinels. Surf. Interface Anal. 2005, 37, 412–416. [Google Scholar] [CrossRef]
- Murray, J.W.; Dillard, J.G.; Giovanoli, R.; Moers, H.; Stumm, W. Oxidation of Mn(II): Initial mineralogy, oxidation state and ageing. Geochim. Cosmochim. Acta 1985, 49, 463–470. [Google Scholar] [CrossRef]
- Junta, J.L.; Hochella, M.F. Manganese (II) oxidation at mineral surfaces: A microscopic and spectroscopic study. Geochim. Cosmochim. Acta 1994, 58, 4985–4999. [Google Scholar] [CrossRef]
- Lee, G.; Song, K.; Bae, J. Permanganate oxidation of arsenic(III): Reaction stoichiometry and the characterization of solid product. Geochim. Cosmochim. Acta 2011, 75, 4713–4727. [Google Scholar] [CrossRef]
- Namgung, S.; Chon, C.-M.; Lee, G. Formation of diverse Mn oxides: A review of bio/geochemical processes of Mn oxidation. Geosci. J. 2018, 22, 373–381. [Google Scholar] [CrossRef]
- Schleeh, T.; Madau, M.; Roessner, D. Synthesis enhancements for generating highly soluble tetrabutylammonium alginates in organic solvents. Carbohydr. Polym. 2014, 114, 493–499. [Google Scholar] [CrossRef]
- Han, M.H.; Sharma, N.; Gonzalo, E.; Pramudita, J.C.; Brand, H.E.A.; López del Amo, J.M.; Rojo, T. Moisture exposed layered oxide electrodes as Na-ion battery cathodes. J. Mater. Chem. A 2016, 4, 18963–18975. [Google Scholar] [CrossRef]
- Shan, X.; Guo, F.; Charles, D.S.; Lebens-Higgins, Z.; Abdel Razek, S.; Wu, J.; Xu, W.; Yang, W.; Page, K.L.; Neuefeind, J.C.; et al. Structural water and disordered structure promote aqueous sodium-ion energy storage in sodium-birnessite. Nat. Commun. 2019, 10, 4975. [Google Scholar] [CrossRef] [PubMed]
- Hemalatha, K.; Jayakumar, M.; Prakash, A.S. Influence of the manganese and cobalt content on the electrochemical performance of P2-Na0.67MnxCo1−xO2 cathodes for sodium-ion batteries. Dalt. Trans. 2018, 47, 1223–1232. [Google Scholar] [CrossRef] [PubMed]
- Peng, B.; Sun, Z.; Jiao, S.; Wang, G.; Zhang, G. Electrochemical Performance Optimization of Layered P2-Type Na0.67MnO2 through Simultaneous Mn-Site Doping and Nanostructure Engineering. Batter. Supercaps 2020, 3, 147–154. [Google Scholar] [CrossRef]
- Liu, L.; An, M.; Yang, P.; Zhang, J. Superior cycle performance and high reversible capacity of SnO2/graphene composite as an anode material for lithium-ion batteries. Sci. Rep. 2015, 5, 9055. [Google Scholar] [CrossRef]
- Pandit, B.; Rondiya, S.R.; Dzade, N.Y.; Shaikh, S.F.; Kumar, N.; Goda, E.S.; Al-Kahtani, A.A.; Mane, R.S.; Mathur, S.; Salunkhe, R.R. High Stability and Long Cycle Life of Rechargeable Sodium-Ion Battery Using Manganese Oxide Cathode: A Combined Density Functional Theory (DFT) and Experimental Study. ACS Appl. Mater. Interfaces 2021, 13, 11433–11441. [Google Scholar] [CrossRef]
- Vogt, L.O.; El Kazzi, M.; Jämstorp Berg, E.; Pérez Villar, S.; Novák, P.; Villevieille, C. Understanding the Interaction of the Carbonates and Binder in Na-Ion Batteries: A Combined Bulk and Surface Study. Chem. Mater. 2015, 27, 1210–1216. [Google Scholar] [CrossRef]
- Luque, G.L.; Li, Y.; Zeng, X.; Luo, X.; Leiva, E.P.M.; Chen, Z.; Amine, K. Impact of alginate and fluoroethylene carbonate on the electrochemical performance of SiO–SnCoC anode for lithium-ion batteries. J. Solid State Electrochem. 2019, 23, 397–405. [Google Scholar] [CrossRef]
- Ling, L.; Bai, Y.; Wang, Z.; Ni, Q.; Chen, G.; Zhou, Z.; Wu, C. Remarkable Effect of Sodium Alginate Aqueous Binder on Anatase TiO2 as High-Performance Anode in Sodium Ion Batteries. ACS Appl. Mater. Interfaces 2018, 10, 5560–5568. [Google Scholar] [CrossRef]
- Barsoukov, E.; Macdonald, J.R. Impedance Spectroscopy: Theory, Experiment and Applications, 2nd ed.; Barsoukov, E., Macdonald, J.R., Eds.; Wiley: Hoboken, NJ, USA, 2005; ISBN 9780471647492. [Google Scholar]
- Dees, D.; Gunen, E.; Abraham, D.; Jansen, A.; Prakash, J. Alternating Current Impedance Electrochemical Modeling of Lithium-Ion Positive Electrodes. J. Electrochem. Soc. 2005, 152, A1409. [Google Scholar] [CrossRef]
- Atebamba, J.-M.; Moskon, J.; Pejovnik, S.; Gaberscek, M. On the Interpretation of Measured Impedance Spectra of Insertion Cathodes for Lithium-Ion Batteries. J. Electrochem. Soc. 2010, 157, A1218. [Google Scholar] [CrossRef]
- Gaberšček, M. Understanding Li-based battery materials via electrochemical impedance spectroscopy. Nat. Commun. 2021, 12, 6513. [Google Scholar] [CrossRef]
- Shkrob, I.A.; Zhu, Y.; Marin, T.W.; Abraham, D. Reduction of Carbonate Electrolytes and the Formation of Solid-Electrolyte Interface (SEI) in Lithium-Ion Batteries. 1. Spectroscopic Observations of Radical Intermediates Generated in One-Electron Reduction of Carbonates. J. Phys. Chem. C 2013, 117, 19255–19269. [Google Scholar] [CrossRef]
- Caballero, A.; Hernán, L.; Morales, J.; Sánchez, L.; Santos Peña, J.; Aranda, M.A.G. Synthesis and characterization of high-temperature hexagonal P2-Na0.6MnO2 and its electrochemical behaviour as cathode in sodium cells. J. Mater. Chem. 2002, 12, 1142–1147. [Google Scholar] [CrossRef]
- Hou, H.; Gan, B.; Gong, Y.; Chen, N.; Sun, C. P2-Type Na0.67Ni0.23Mg0.1Mn0.67O2 as a High-Performance Cathode for a Sodium-Ion Battery. Inorg. Chem. 2016, 55, 9033–9037. [Google Scholar] [CrossRef]
- Zhang, Q.; Yu, Z.; Du, P.; Su, C. Carbon Nanomaterials Used as Conductive Additives in Lithium Ion Batteries. Recent Pat. Nanotechnol. 2010, 4, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Somo, T.R.; Mabokela, T.E.; Teffu, D.M.; Sekgobela, T.K.; Ramogayana, B.; Hato, M.J.; Modibane, K.D. A Comparative Review of Metal Oxide Surface Coatings on Three Families of Cathode Materials for Lithium Ion Batteries. Coatings 2021, 11, 744. [Google Scholar] [CrossRef]
- Kucinskis, G.; Bajars, G.; Bikova, K.; Kaprans, K.; Kleperis, J. Microstructural Influence on Electrochemical Properties of LiFePO4/C/Reduced Graphene Oxide Composite Cathode. Russ. J. Electrochem. 2019, 55, 517–523. [Google Scholar] [CrossRef]
- Liu, J.; Tang, K.; Song, K.; van Aken, P.A.; Yu, Y.; Maier, J. Electrospun Na3V2(PO4)3/C nanofibers as stable cathode materials for sodium-ion batteries. Nanoscale 2014, 6, 5081. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).