Titanium Activation in Prussian Blue Based Electrodes for Na-ion Batteries: A Synthesis and Electrochemical Study




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
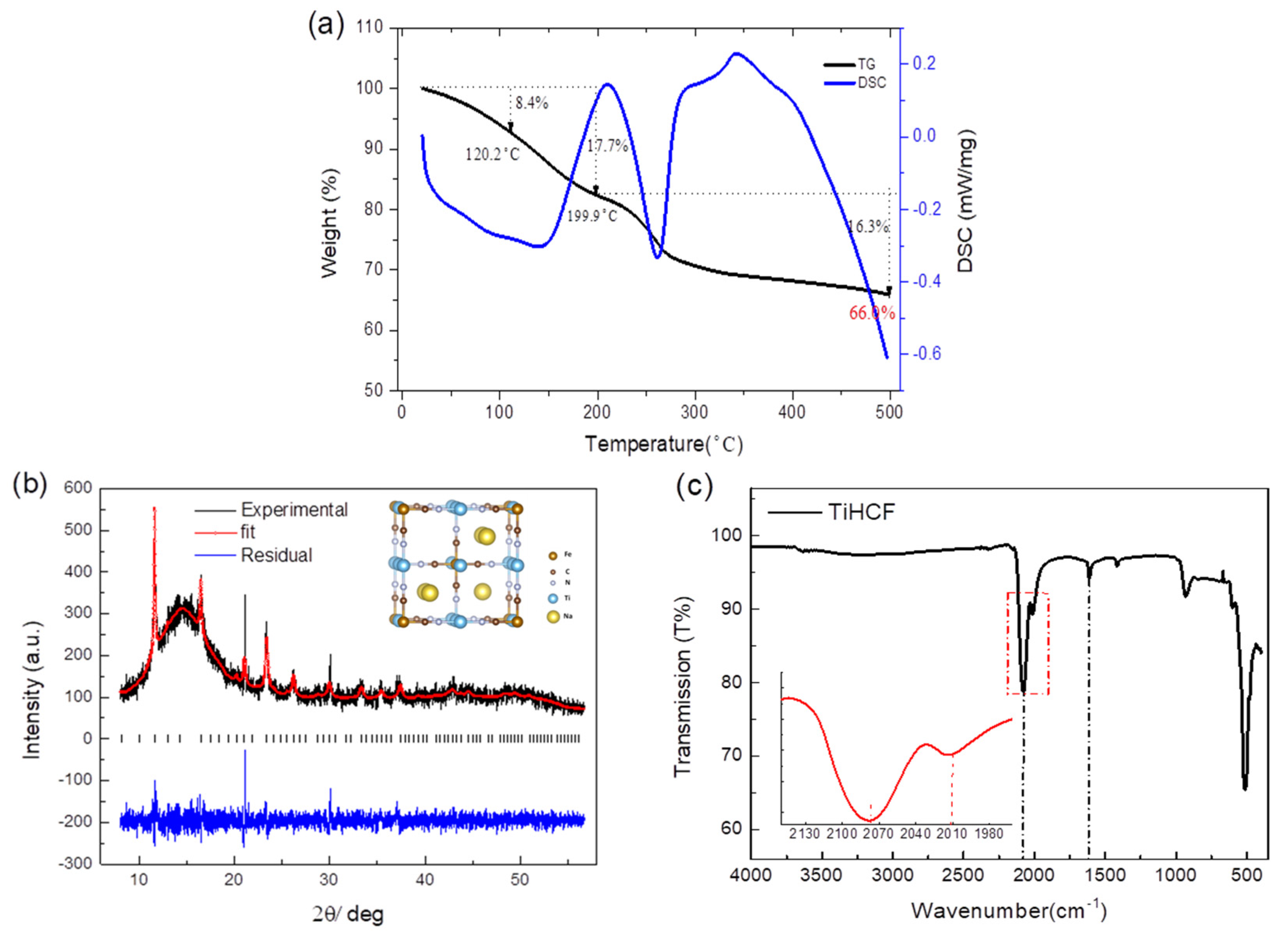
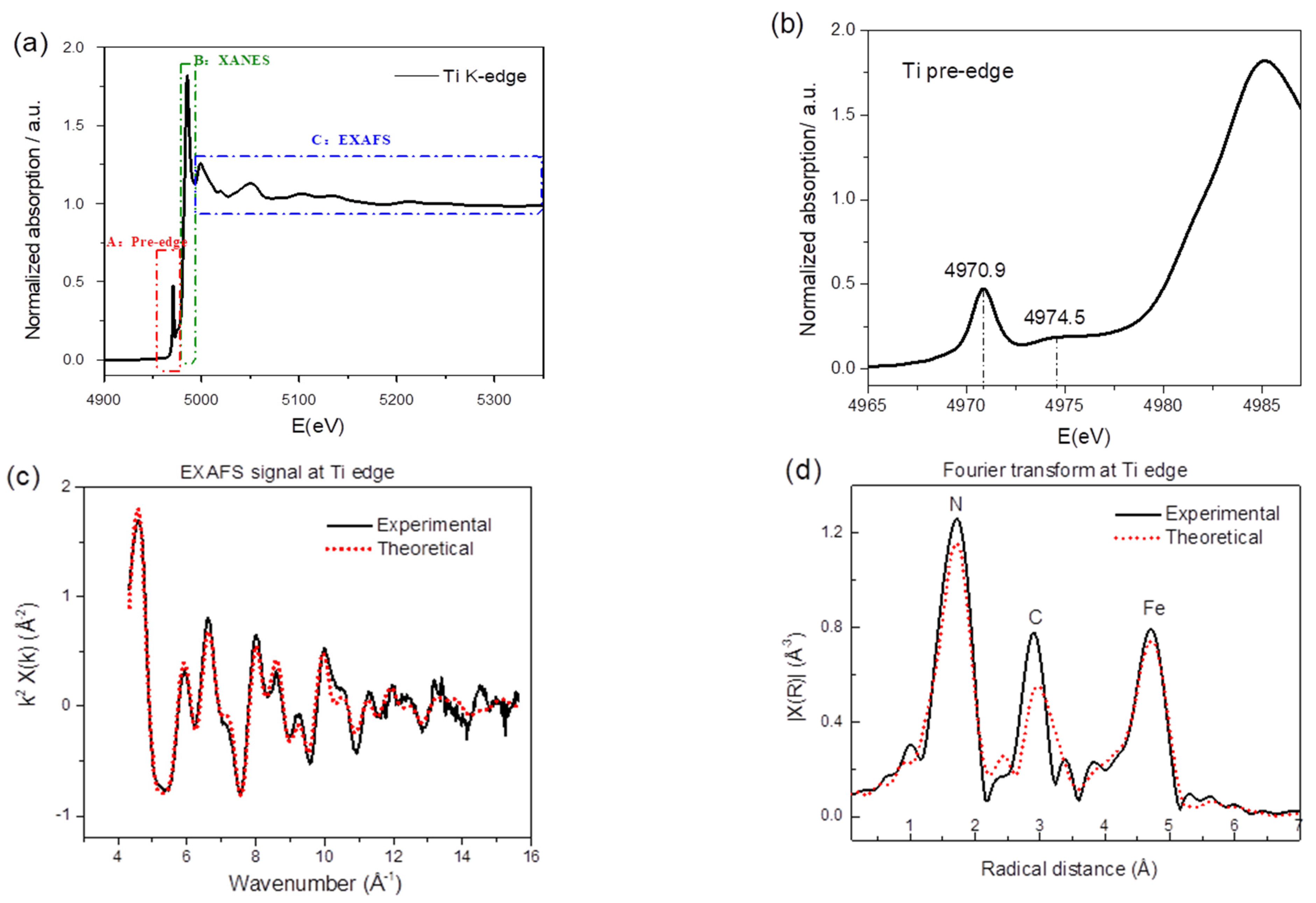
2.1. Preparation and Characterization of TiHCF
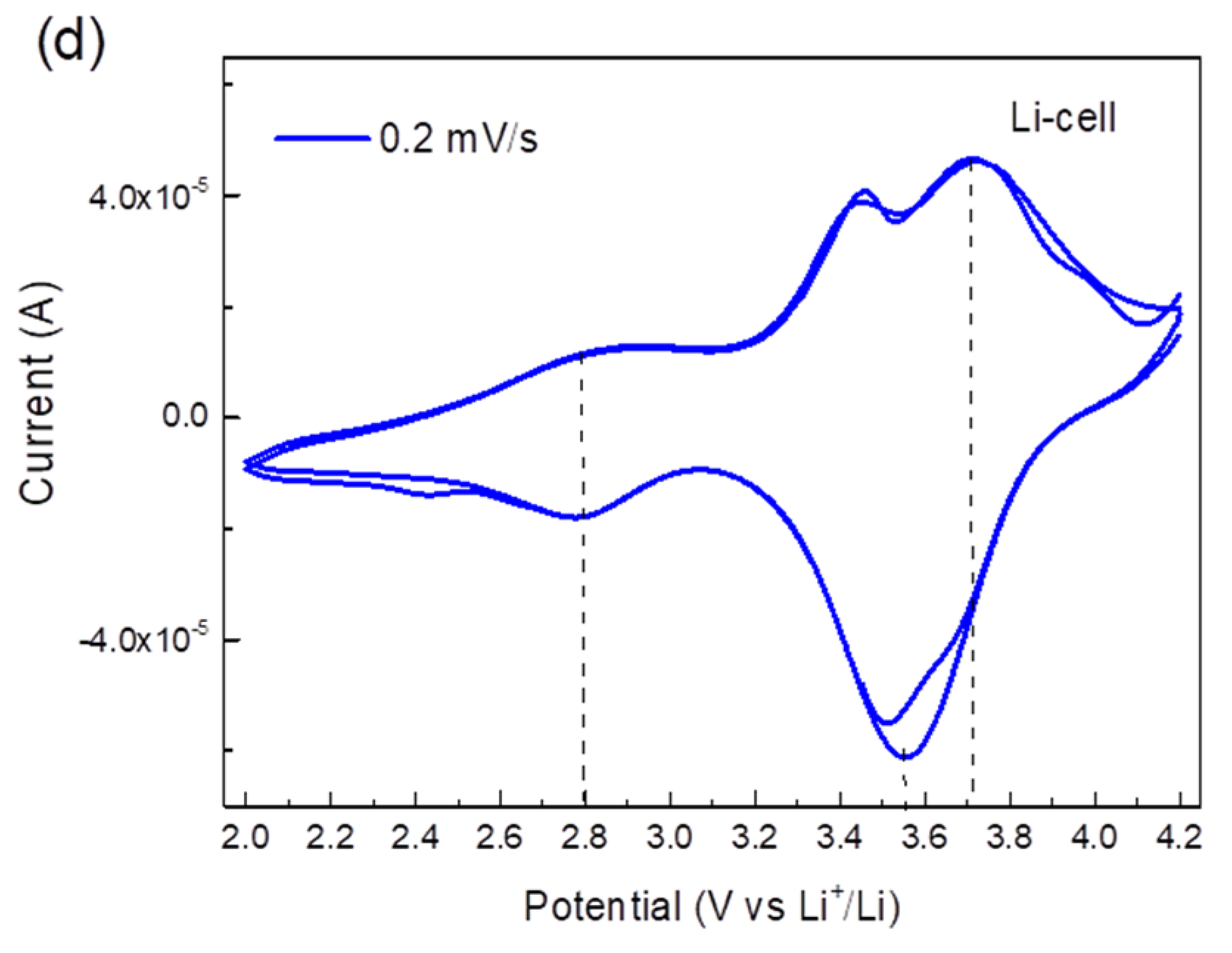
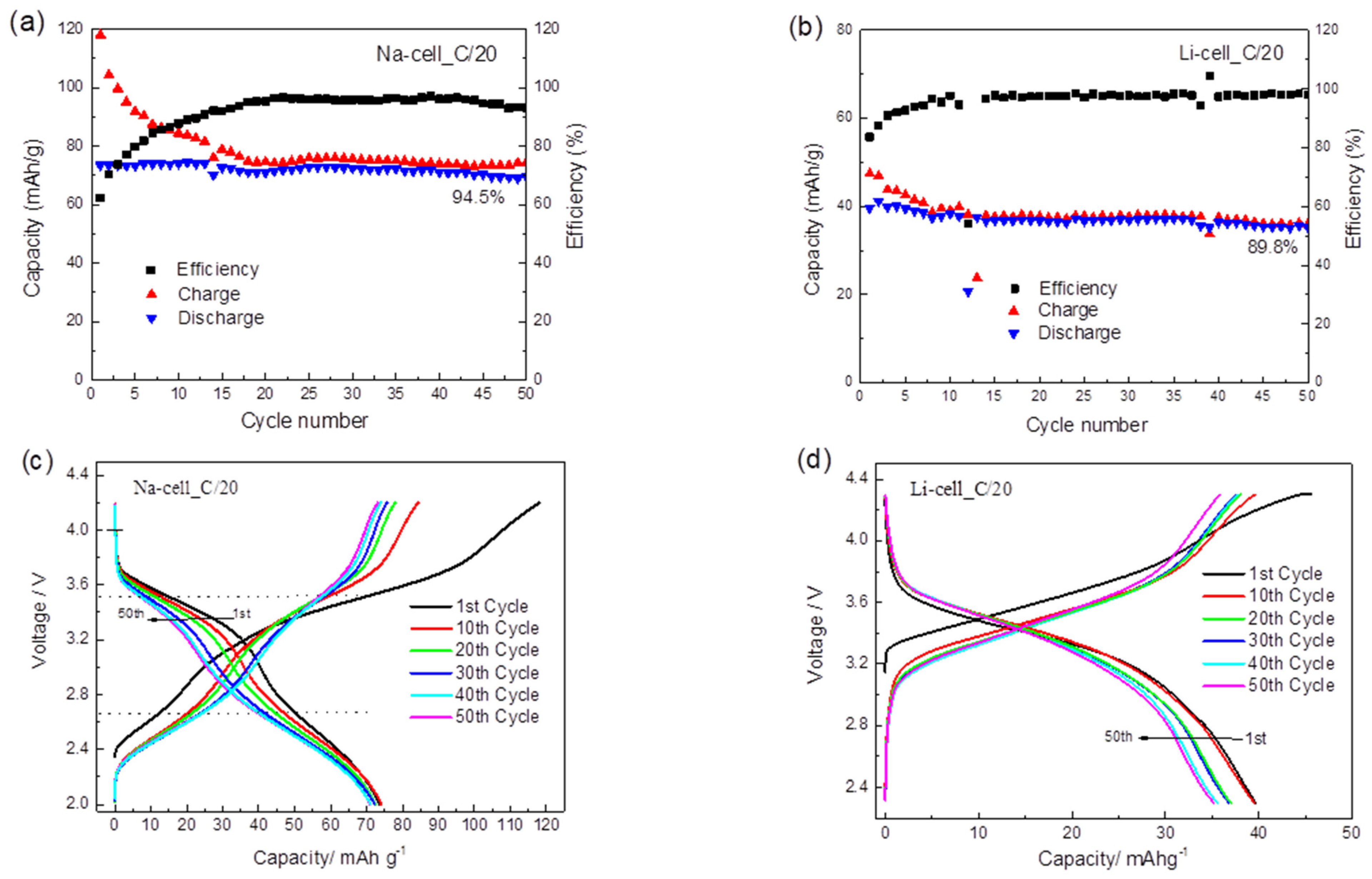
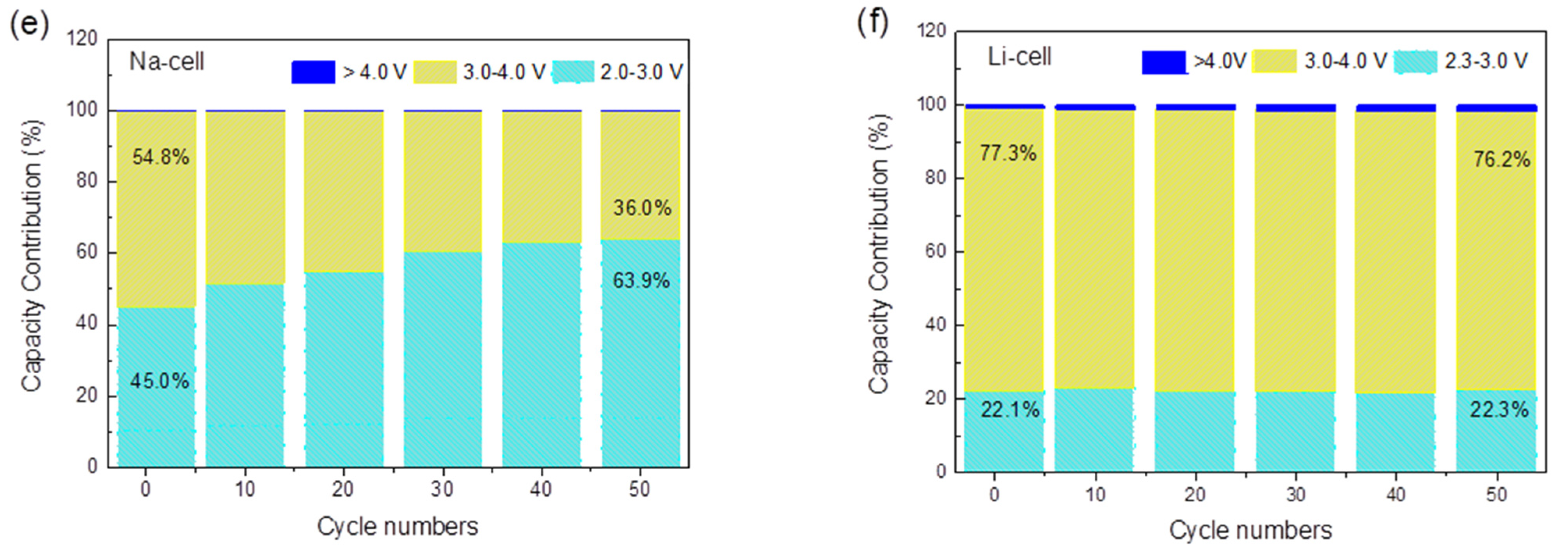
2.2. Electrochemical Application of TiHCF
3. Materials and Methods
3.1. Material Synthesis
3.2. Electrode Preparation and Electrochemical Tests
3.3. Characterization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Neff, V.D. Electrochemical oxidation and reduction of thin films of Prussian blue. J. Electrochem. Soc. 1978, 125, 886. [Google Scholar] [CrossRef]
- Carpenter, M.K.; Conell, R.S.; Simko, S.J. Electrochemistry and electrochromism of vanadium hexacyanoferrate. Inorg. Chem. 1990, 29, 845–850. [Google Scholar] [CrossRef]
- Kulesza, P.J.; Malik, M.A.; Zamponi, S.; Berrettoni, M.; Marassi, R. Electrolyte-cation-dependent coloring, electrochromism and thermochromism of cobalt (II) hexacyanoferrate (III, II) films. J. Electroanal. Chem. 1995, 397, 287–292. [Google Scholar] [CrossRef]
- Zhou, D.M.; Ju, H.X.; Chen, H.Y. Catalytic oxidation of dopamine at a microdisk platinum electrode modified by electrodeposition of nickel hexacyanoferrate and Nafion. J. Electroanal. Chem. 1996, 408, 219–223. [Google Scholar] [CrossRef]
- Chen, S.M. Characterization and electrocatalytic properties of cobalt hexacyanoferrate. Electrochim. Acta 1998, 43, 3359–3369. [Google Scholar] [CrossRef]
- Eftekhari, A. Aluminum electrode modified with manganese hexacyanoferrate as a chemical sensor for hydrogen peroxide. Talanta 2001, 55, 395–402. [Google Scholar] [CrossRef]
- Garcıa, T.; Casero, E.; Lorenzo, E.; Pariente, F. Electrochemical sensor for sulfite determination based on iron hexacyanoferrate film modified electrodes. Sens. Actuators B 2005, 106, 803–809. [Google Scholar] [CrossRef]
- Matsuda, T.; Moritomo, Y. Thin Film Electrode of Prussian Blue Analogue for Li-ion Battery. Appl. Phys. Express 2011, 4, 047101. [Google Scholar] [CrossRef]
- Wang, L.; Lu, Y.; Liu, J.; Xu, M.; Cheng, J.; Zhang, D.; Goodenough, J.B. A Superior Low-Cost Cathode for a Na-Ion Battery. Angew. Chem. Int. Ed. 2013, 52, 1964. [Google Scholar] [CrossRef]
- Eftekhari, A. Potassium secondary cell based on Prussian blue cathode. J. Power Sources 2004, 126, 221–228. [Google Scholar] [CrossRef]
- Mizuno, Y.; Okubo, M.; Hosono, E.; Kudo, T.; Ohishi, K.; Okazawa, A.; Kojima, N.; Kurono, R.; Nishimura, S.; Yamada, A. Electrochemical Mg2+ intercalation into a bimetallic CuFe Prussian blue analog in aqueous electrolytes. J. Mater. Chem. A 2013, 1, 13055–13059. [Google Scholar] [CrossRef]
- Trcoli, R.; La Mantia, F. An Aqueous Zinc-Ion Battery Based on Copper Hexacyanoferrate. ChemSusChem 2015, 8, 481–485. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Pan, G.L.; Li, G.R.; Gao, X.P. Copper hexacyanoferrate nanoparticles as cathode material for aqueous Al-ion batteries. J. Mater. Chem. A 2015, 3, 959. [Google Scholar] [CrossRef]
- Kuperman, N.; Padigi, P.; Goncher, G.; Evans, D.; Thiebes, J.; Solanki, R. High performance Prussian blue cathode fornonaqueous Ca-ion intercalation battery. J. Power Sources 2017, 342, 414–418. [Google Scholar] [CrossRef]
- Hu, Y.X.; Ye, D.L.; Hu, B.H.; Zhu, X.B.; Wang, S.C.; Li, L.L.; Peng, S.J.; Wang, L.Z. A binder-free and free-standing cobalt Sulfide@Carbon nanotube cathode material for aluminum-ion batteries. Adv. Mater. 2018, 30, 1703824. [Google Scholar] [CrossRef] [PubMed]
- Keggins, J.F.; Miles, F.D. Structures and Formulæ of the Prussian Blues and Related Compounds. Nature 1936, 137, 577–578. [Google Scholar] [CrossRef]
- Giorgetti, M.; Guadagnini, L.; Tonelli, D.; Minicucci, M.; Aquilanti, G. Structural characterization of electrodeposited copper hexacyanoferrate films by using a spectroscopic multi-technique approach. Phys. Chem. Chem. Phys. 2012, 14, 5527–5537. [Google Scholar] [CrossRef]
- Wessells, C.D.; Peddada, S.V.; Huggins, R.A.; Cui, Y. Nickel Hexacyanoferrate Nanoparticle Electrodes for Aqueous Sodium and Potassium Ion Batteries. Nano Lett. 2011, 11, 5421–5425. [Google Scholar] [CrossRef]
- Wessells, C.D.; Huggins, R.A.; Cui, Y. Copper hexacyanoferrate battery electrodes with long cycle life and high power. Nat. Commun. 2011, 2, 550. [Google Scholar] [CrossRef]
- Matsuda, T.; Takachi, M.; Moritomo, Y. A sodium manganese ferrocyanide thin film for Na-ion batteries. Chem. Commun. 2013, 49, 2750. [Google Scholar] [CrossRef]
- Wang, L.; Song, J.; Qiao, R.M.; Wray, L.A.; Hossain, M.A.; Chuang, Y.D.; Yang, W.L.; Lu, Y.H.; Evans, D.; Lee, J.J.; et al. Rhombohedral Prussian White as Cathode for Rechargeable Sodium-Ion Batteries. J. Am. Chem. Soc. 2015, 137, 2548–2554. [Google Scholar] [CrossRef] [PubMed]
- You, Y.; Yu, X.Q.; Yin, Y.X.; Nam, K.W.; Guo, Y.G. Sodium iron hexacyanoferrate with high Na content as a Na-rich cathode material for Na-ion batteries. Nano Res. 2015, 8, 117. [Google Scholar] [CrossRef]
- Takachi, M.; Matsuda, T.; Moritomo, Y. Cobalt Hexacyanoferrate as Cathode Material for Na+ Secondary Battery. Appl. Phys. Express 2013, 6, 025802. [Google Scholar] [CrossRef]
- Mullaliu, A.; Asenbauer, J.; Aquilanti, G.; Passerini, S.; Giorgetti, M. Highlighting the Reversible Manganese Electroactivity in Na-Rich Manganese Hexacyanoferrate Material for Li- and Na-Ion Storage. Small Methods 2020, 4, 1900529. [Google Scholar] [CrossRef]
- Qian, J.; Wu, C.; Cao, Y.; Ma, Z.; Huang, Y.; Ai, X.; Yang, H. Prussian blue cathode materials for sodium-ion batteries and other ion batteries. Adv. Energy Mater. 2018, 8, 1702619. [Google Scholar] [CrossRef]
- Lee, H.W.; Wang, R.Y.; Pasta, M.; Lee, S.W.; Liu, N.; Cui, Y. Manganese hexacyanomanganate open framework as a high-capacity positive electrode material for sodium-ion batteries. Nat. Commun. 2014, 5, 5280. [Google Scholar] [CrossRef]
- Pasta, M.; Wessells, C.D.; Liu, N.; Nelson, J.; McDowell, M.T.; Huggins, R.A.; Toney, M.F.; Cui, Y. Full open-framework batteries for stationary energy storage. Nat. Commun. 2014, 5, 3007. [Google Scholar] [CrossRef]
- Nie, P.; Shen, L.F.; Luo, H.F.; Ding, B.; Xu, G.Y.; Wang, J.; Zhang, X.G. Prussian blue analogues: A new class of anode materials for lithium ion batteries. J. Mater. Chem. A 2014, 2, 5852–5857. [Google Scholar] [CrossRef]
- Wheeler, S.; Capone, I.; Day, S.; Tang, C.; Pasta, M. Low-Potential Prussian blue Analogues for Sodium-Ion Batteries: Manganese Hexacyanochromate. Chem. Mater. 2019, 31, 2619–2626. [Google Scholar] [CrossRef]
- Guo, S.H.; Yu, H.J.; Liu, D.Q.; Tian, W.; Liu, X.Z.; Hanada, N.; Ishida, M.; Zhou, H.S. A novel tunnel Na0.61Ti0.48Mn0.52O2 cathode material for sodium-ion batteries. Chem. Commun. 2014, 50, 7998–8001. [Google Scholar] [CrossRef]
- Wang, Y.S.; Xiao, R.J.; Hu, Y.S.; Avdeev, M.; Chen, L.Q. P2-Na0.6 [Cr0.6Ti0.4] O2 cation-disordered electrode for high-rate symmetric rechargeable sodium-ion batteries. Nat. Commun. 2015, 6, 6954. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, R.; Lai, W. Na2/3Ni1/3Ti2/3O2: “Bi-Functional” Electrode Materials for Na-Ion Batteries. ECS Electrochem. Lett. 2014, 3, A23–A25. [Google Scholar] [CrossRef]
- Wang, Y.S.; Zhu, W.; Guerfi, A.; Kim, C.; Zaghib, K. Roles of Ti in Electrode Materials for Sodium-Ion Batteries. Front. Energy Res. 2019, 7, 28. [Google Scholar] [CrossRef]
- Li, J.; Liu, J.; Sun, Q.; Banis, M.N.; Sun, X.; Sham, T.K. Tracking the effect of sodium insertion/extraction in amorphous and anatase TiO2 nanotubes. J. Phys. Chem. C 2017, 121, 11773–11782. [Google Scholar] [CrossRef]
- Yang, G.Z.; Song, H.W.; Wu, M.M.; Wang, C.X. Porous NaTi2 (PO4)3 nanocubes: A high-rate nonaqueous sodium anode material with more than 10,000 cycle life. J. Mater. Chem. A 2015, 3, 18718–18726. [Google Scholar] [CrossRef]
- Guo, S.H.; Yi, J.; Sun, Y.; Zhou, H.S. Recent advances in titanium-based electrode materials for stationary sodium-ion batteries. Energy Environ. Sci. 2016, 9, 2978–3006. [Google Scholar] [CrossRef]
- Sun, X.; Ji, X.Y.; Zhou, Y.T.; Shao, Y.; Zang, Y.; Wen, Z.Y.; Chen, C.H. A new gridding cyanoferrate anode material for lithium and sodium ion batteries: Ti0.75Fe0.25[Fe(CN)6]0.96·1.9H2O with excellent electrochemical properties. J. Power Sources 2016, 314, 35–38. [Google Scholar] [CrossRef]
- Xie, M.; Huang, Y.; Xu, M.; Chen, R.; Zhang, X.; Li, L.; Wu, F. Sodium titanium hexacyanoferrate as an environmentally friendly and low-cost cathode material for sodium-ion batteries. J. Power Sources 2016, 302, 7–12. [Google Scholar] [CrossRef]
- Luo, Y.; Shen, B.; Guo, B.; Hu, L.; Xu, Q.; Zhan, R.; Zhang, Y.; Bao, S.; Xu, M. Potassium titanium hexacyanoferrate as a cathode material for potassiumion Batteries. J. Phys. Chem. Solids 2018, 122, 31–35. [Google Scholar] [CrossRef]
- Song, J.; Wang, L.; Lu, Y.; Liu, J.; Guo, B.; Xiao, P.; Lee, J.J.; Yang, X.Q.; Henkelman, G.; Goodenough, J.B. Removal of Interstitial H2O in Hexacyanometallates for a Superior Cathode of a Sodium-Ion Battery. J. Am. Chem. Soc. 2015, 137, 2658. [Google Scholar] [CrossRef]
- Kettle, S.F.A.; Diana, E.; Boccaleri, E.; Stanghellini, P.L. The Vibrational Spectra of the Cyanide Ligand Revisited. Bridging Cyanides. Inorg. Chem. 2007, 46, 7. [Google Scholar] [CrossRef] [PubMed]
- Giorgetti, M.; Berrettoni, M.; Filipponi, A.; Kulesza, P.J.; Marassi, R. Evidence of four-body contributions in the EXAFS spectrum of Na2Co[Fe(CN)6]. Chem. Phys. Lett. 1997, 275, 108–112. [Google Scholar] [CrossRef]
- Luca, V.; Djajanti, S.; Howe, R.F. Structural and Electronic Properties of Sol-Gel Titanium Oxides Studied by X-ray Absorption Spectroscopy. J. Phys. Chem. B 1998, 102, 10650–10657. [Google Scholar] [CrossRef]
- Brydsoni, R.; Sauer, H.; Engel, W.; Thomas, J.M.; Zeitler, E.; Kosugi, N.; Kuroda, H. Electron energy loss and x-ray absorption spectroscopy of rutile and anatase: A test of structural sensitivity. J. Phys. Condens. Matter 1989, 1, 797–812. [Google Scholar] [CrossRef]
- Beaurepair, E.; Lewonczu, S.; Ringeisse, J.; Parlebas, J.C.; Uozumi, T.; Okada, K.; Kotani, A. Comparison between BIS and Ti K-XAS for TiO2: Experimental and Theoretical Study. Europhys. Lett. 1993, 22, 463–467. [Google Scholar] [CrossRef]
- Mullaliu, A.; Conti, P.; Aquilanti, G.; Plaisier, J.R.; Stievano, L.; Giorgetti, M. Operando XAFS and XRD Study of a Prussian Blue Analogue Cathode Material: Iron Hexacyanocobaltate. Condens. Matter 2018, 3, 36. [Google Scholar] [CrossRef]
- Krysina, O.V.; Timchenko, N.A.; Koval, N.N.; Zubavichus, Y.V. Structure of the local environment of titanium atoms in multicomponent nitride coatings produced by plasma-ion techniques. J. Phys. Conf. Ser. 2016, 699, 012060. [Google Scholar] [CrossRef]
- Lu, Y.H.; Wang, L.; Cheng, J.G.; Goodenough, J.B. Prussian blue: A new framework of electrode materials for sodium batteries. Chem. Commun. 2012, 48, 6544. [Google Scholar] [CrossRef]
- Rodríguez-Carvajal, J. Recent advances in magnetic structure determination by neutron powder diffraction. Phys. B Phys. Condens. Matter 1993, 192, 55–69. [Google Scholar] [CrossRef]
- Ravel, B.; Newville, M. ATHENA, ARTEMIS, HEPHAESTUS: Data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron Radiat. 2005, 12, 537–541. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, M.; Mullaliu, A.; Passerini, S.; Giorgetti, M. Titanium Activation in Prussian Blue Based Electrodes for Na-ion Batteries: A Synthesis and Electrochemical Study. Batteries 2021, 7, 5. https://doi.org/10.3390/batteries7010005
Li M, Mullaliu A, Passerini S, Giorgetti M. Titanium Activation in Prussian Blue Based Electrodes for Na-ion Batteries: A Synthesis and Electrochemical Study. Batteries. 2021; 7(1):5. https://doi.org/10.3390/batteries7010005
Chicago/Turabian StyleLi, Min, Angelo Mullaliu, Stefano Passerini, and Marco Giorgetti. 2021. "Titanium Activation in Prussian Blue Based Electrodes for Na-ion Batteries: A Synthesis and Electrochemical Study" Batteries 7, no. 1: 5. https://doi.org/10.3390/batteries7010005
APA StyleLi, M., Mullaliu, A., Passerini, S., & Giorgetti, M. (2021). Titanium Activation in Prussian Blue Based Electrodes for Na-ion Batteries: A Synthesis and Electrochemical Study. Batteries, 7(1), 5. https://doi.org/10.3390/batteries7010005