Pre-Lithiation Strategies for Rechargeable Energy Storage Technologies: Concepts, Promises and Challenges


Abstract
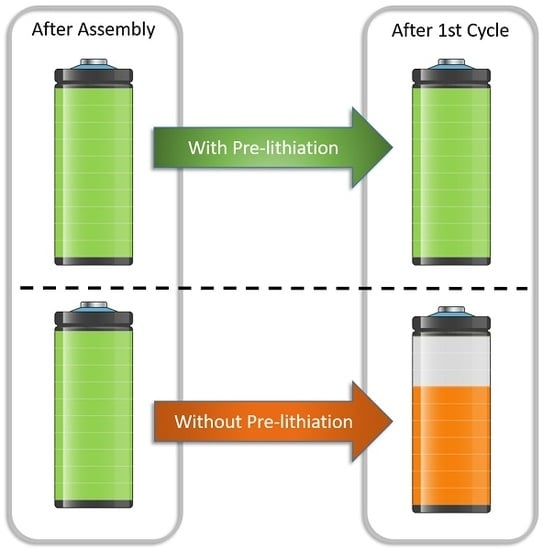
1. Introduction
2. Pre-Lithiation Concepts for the Negative Electrode in Lithium Ion Batteries
2.1. Concepts for Chemical Lithiation Using Active Reactants
2.2. Concepts for Electrochemical Pre-Lithiation
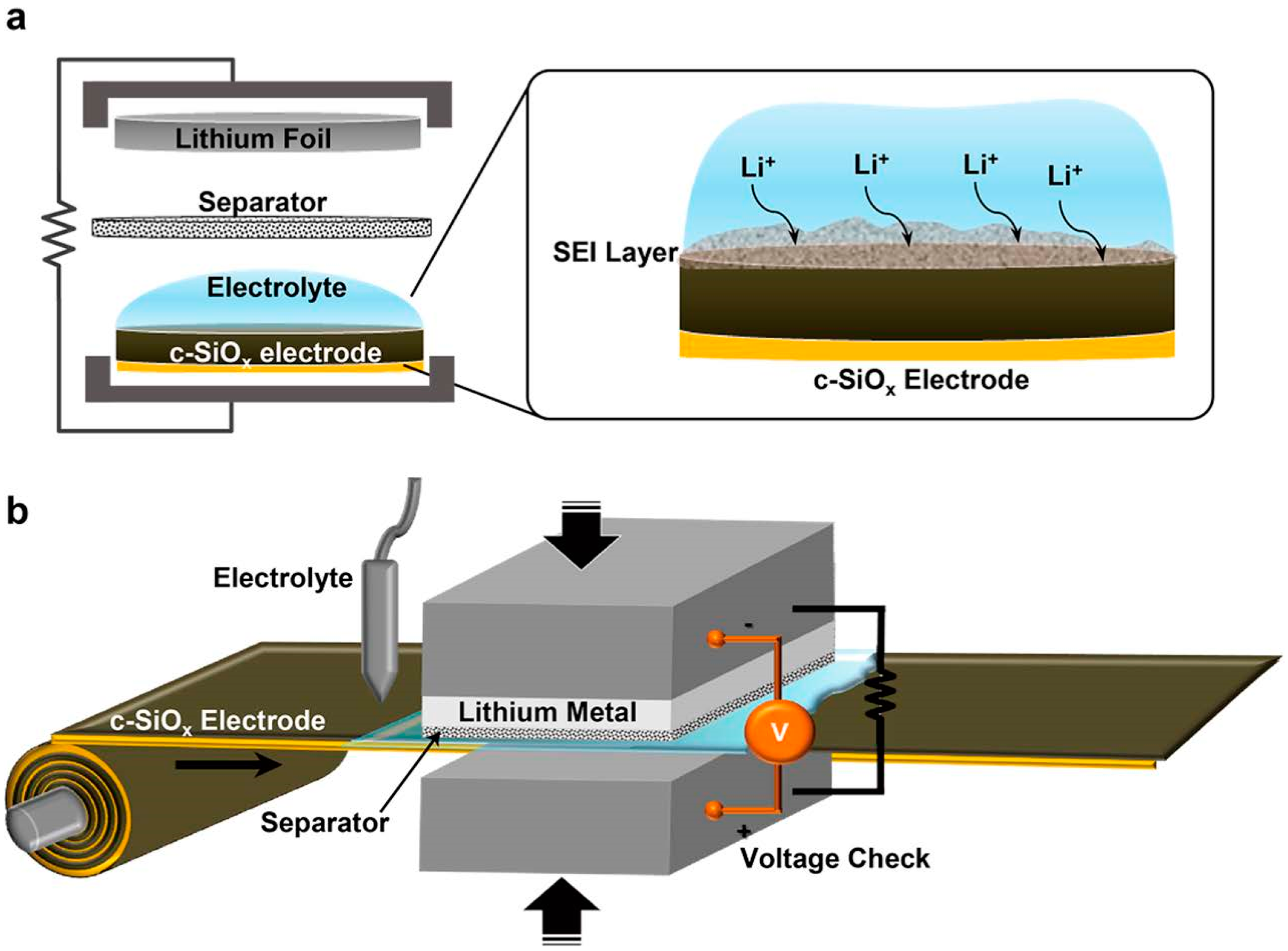
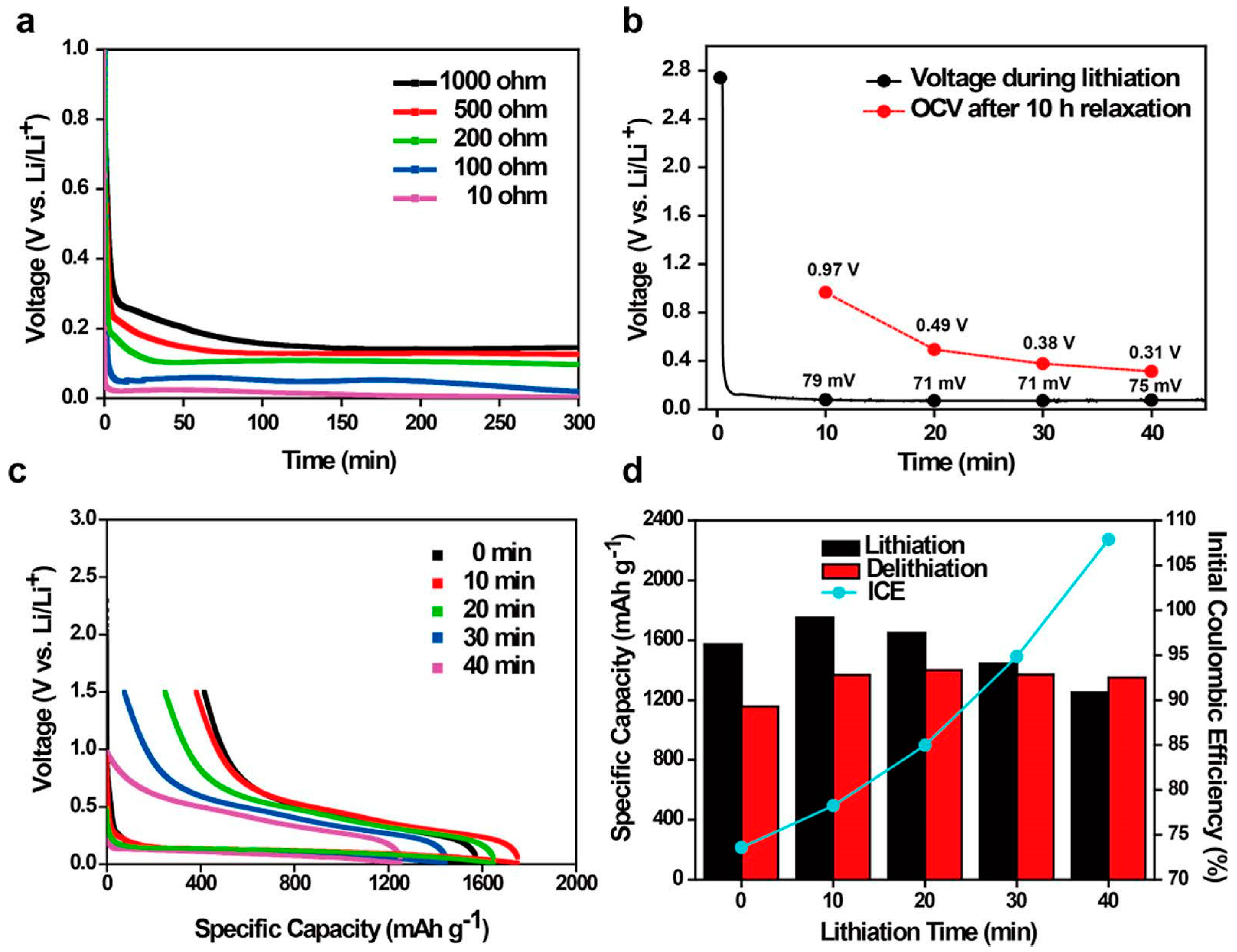
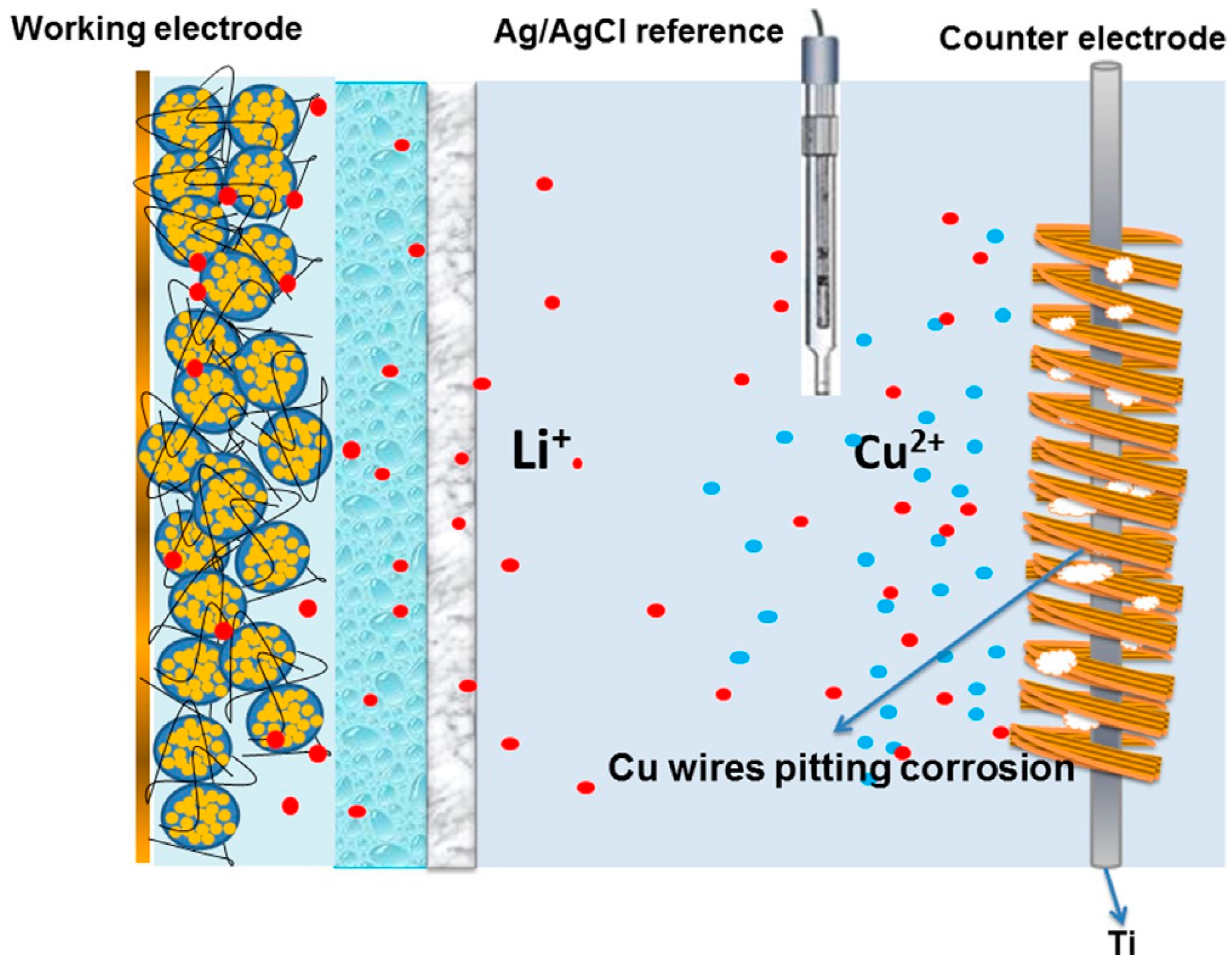
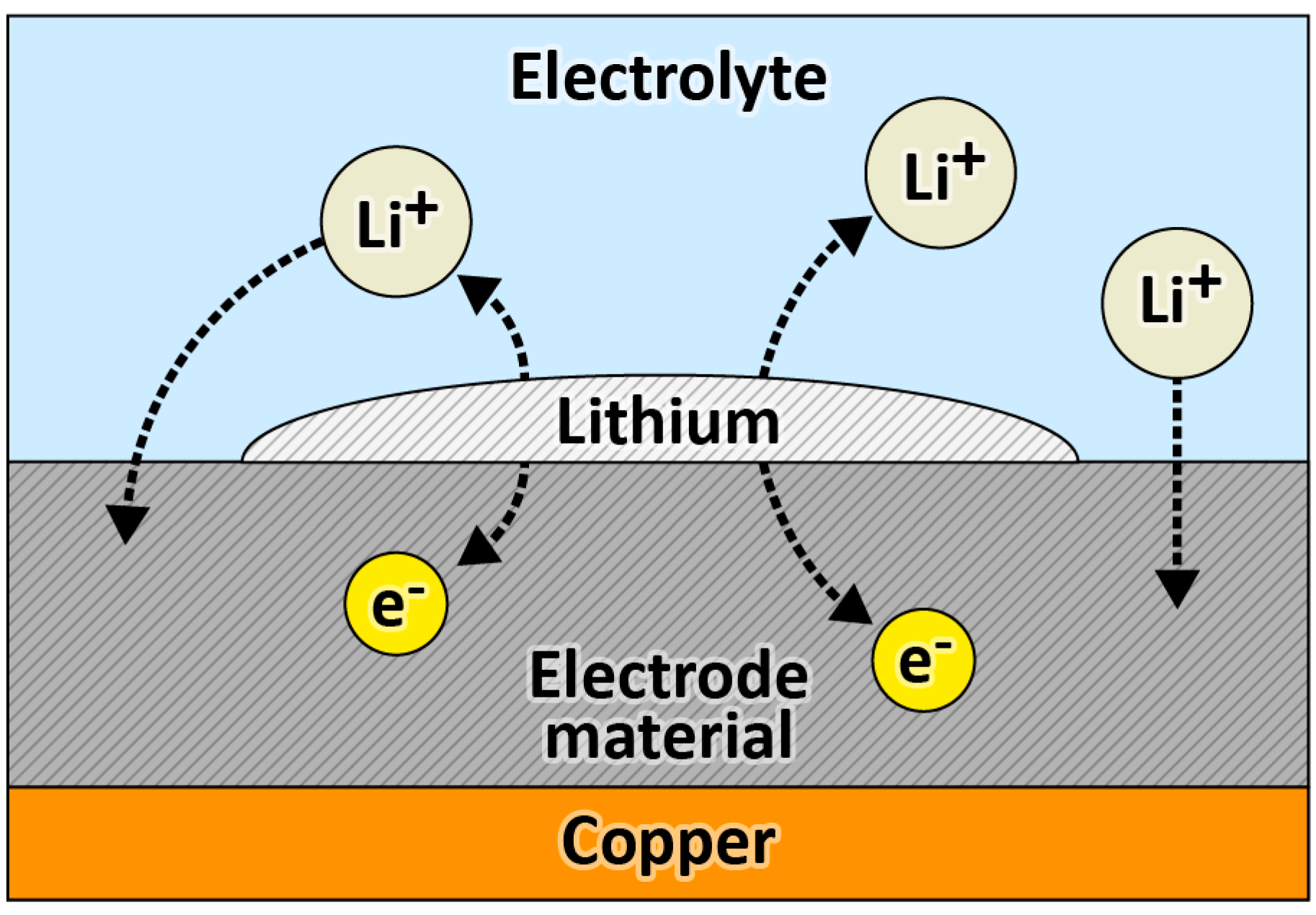
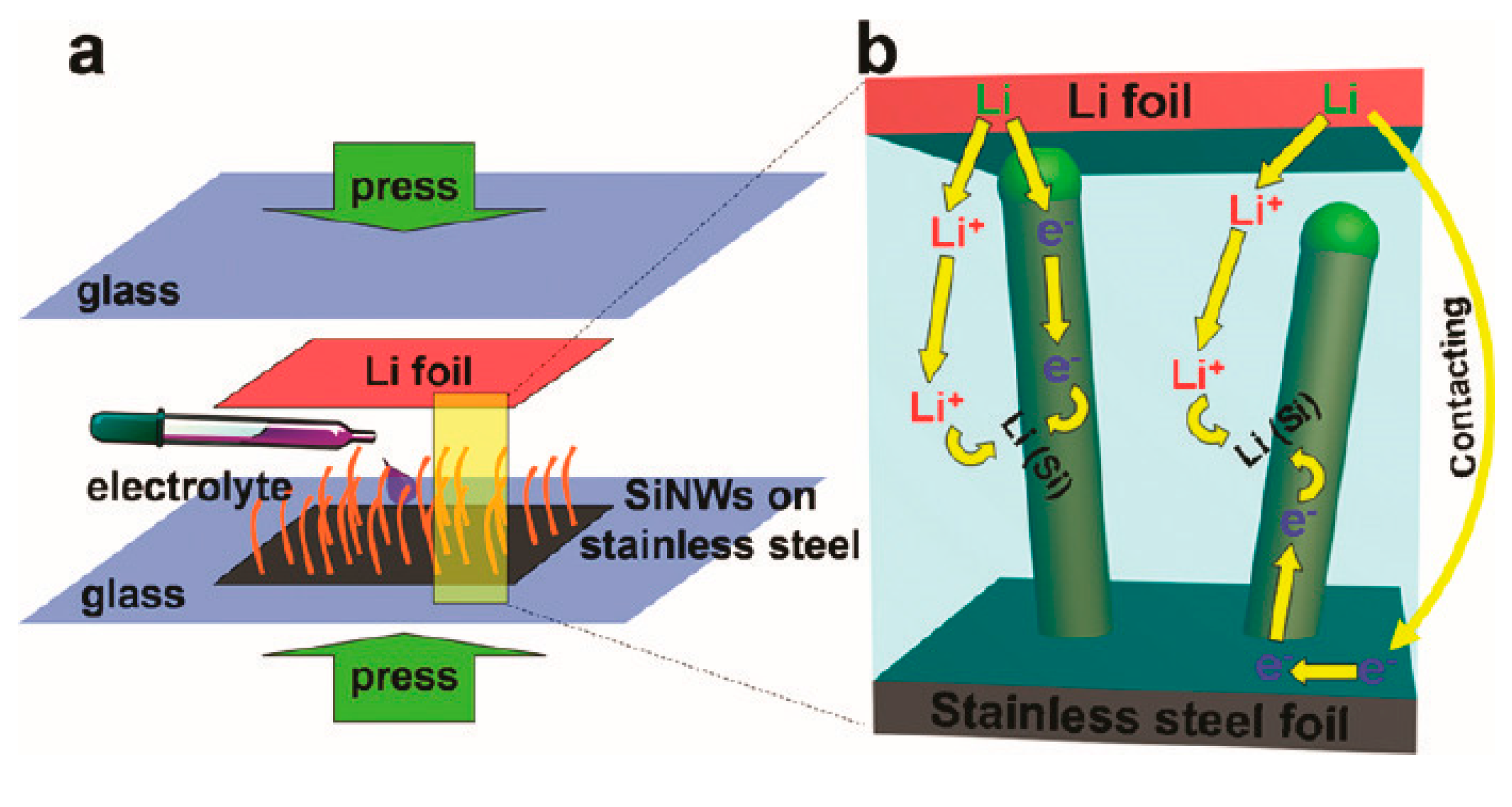
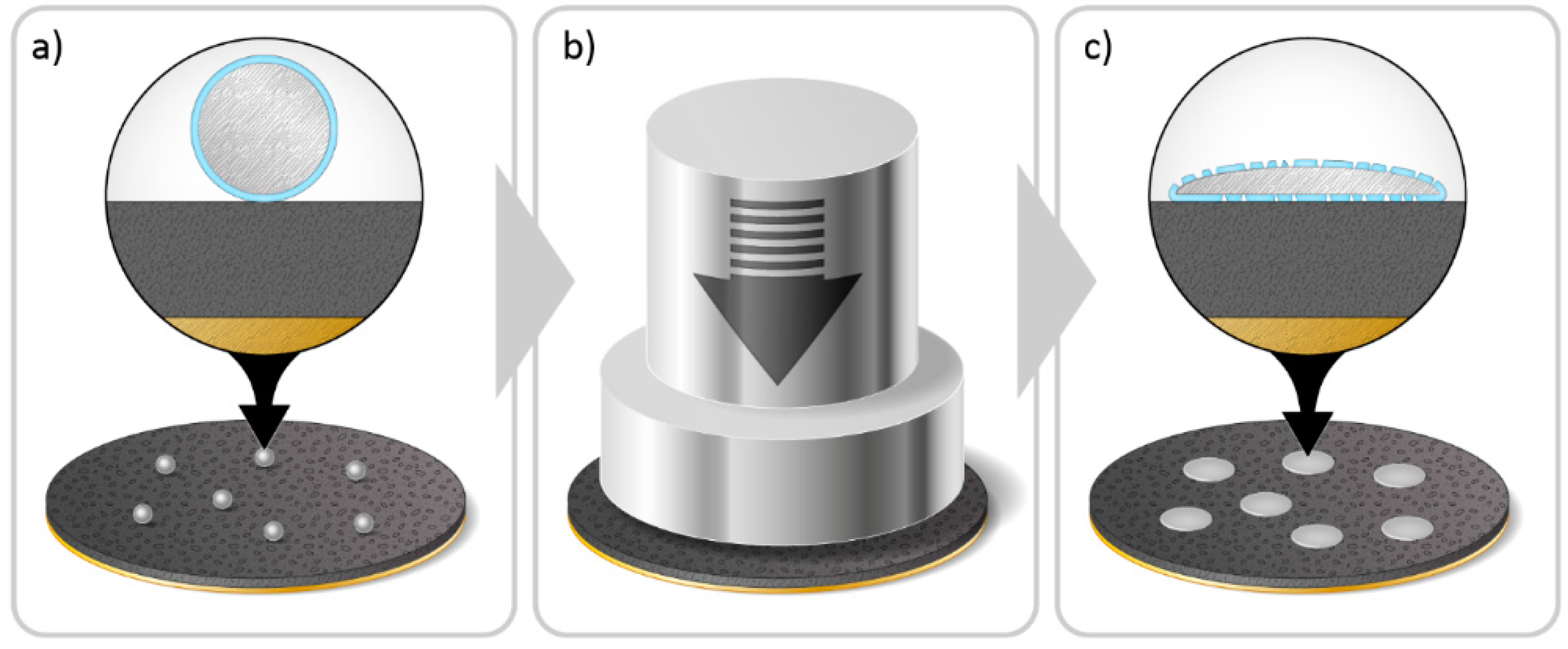
2.3. Pre-Lithiation by Direct Contact to Lithium Metal
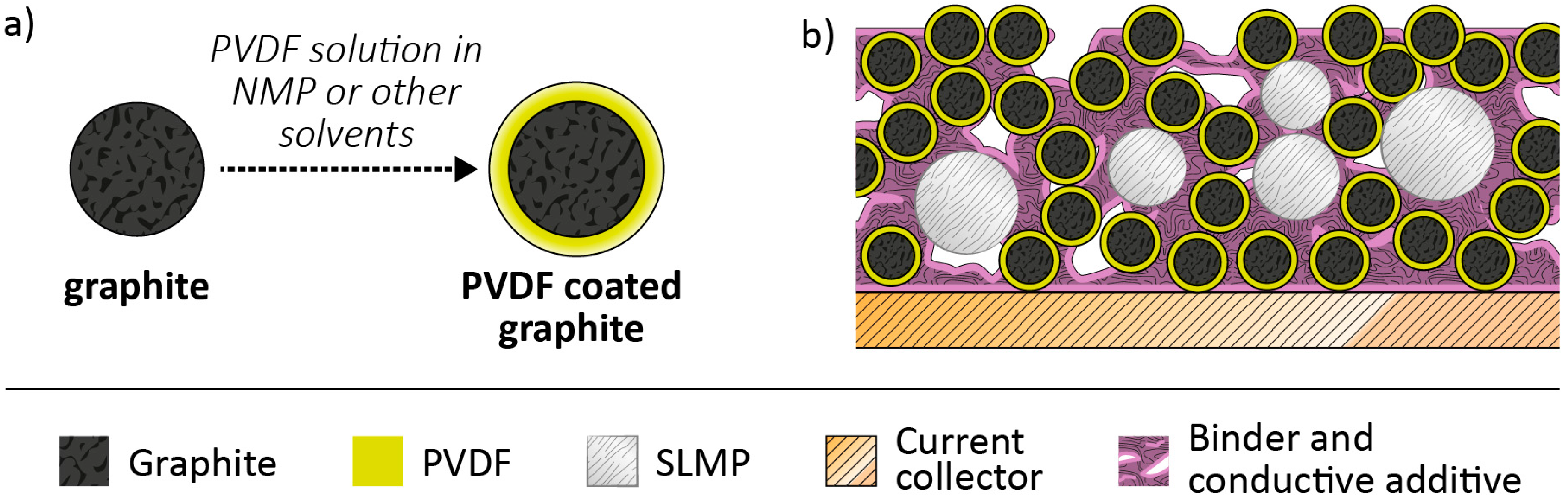
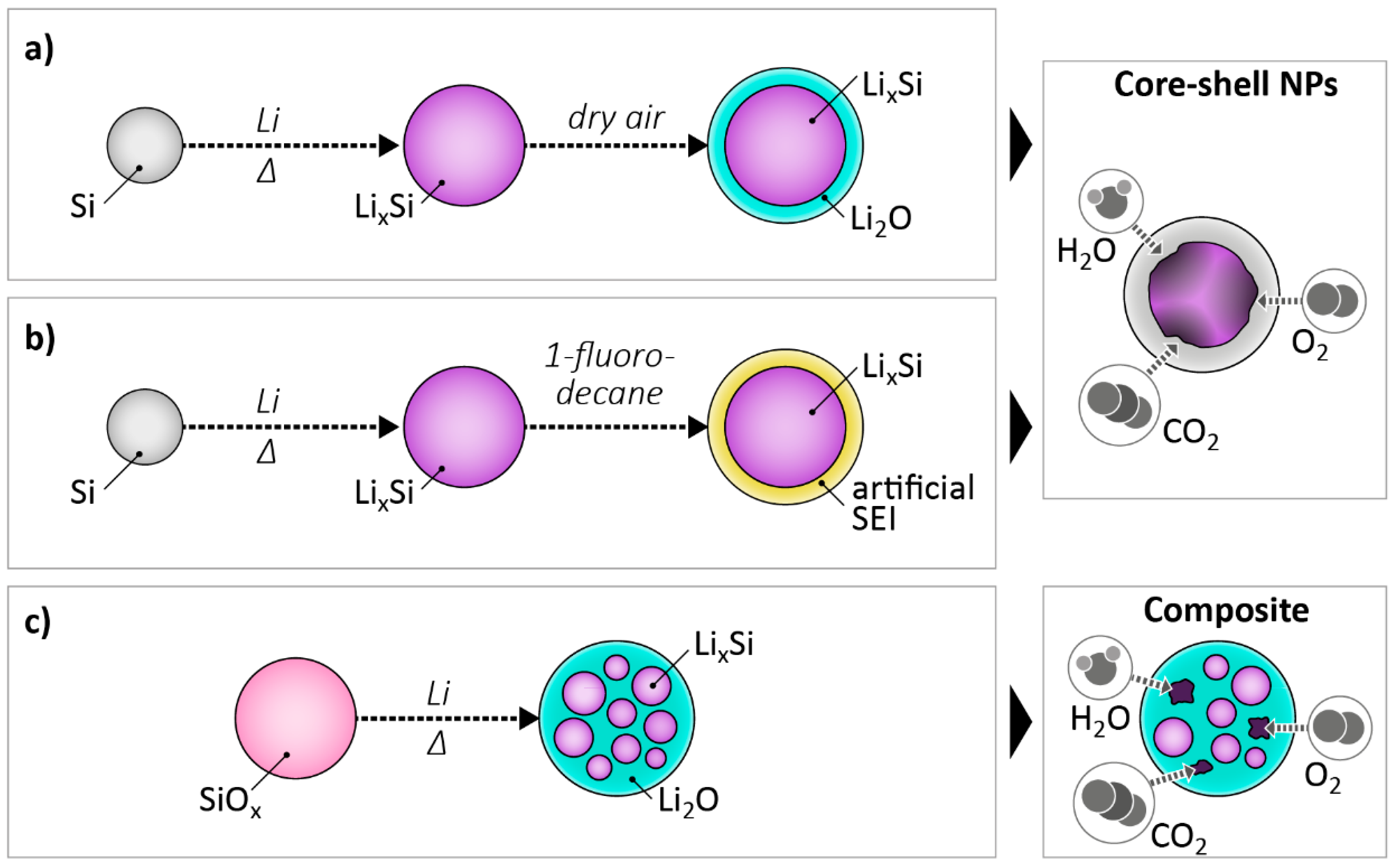
2.4. Pre-Lithiation by Use of Lithiated Active Materials as Negative Electrode Additives
2.5. Comparison of Different Pre-Lithiation Concepts for the Negative Electrode
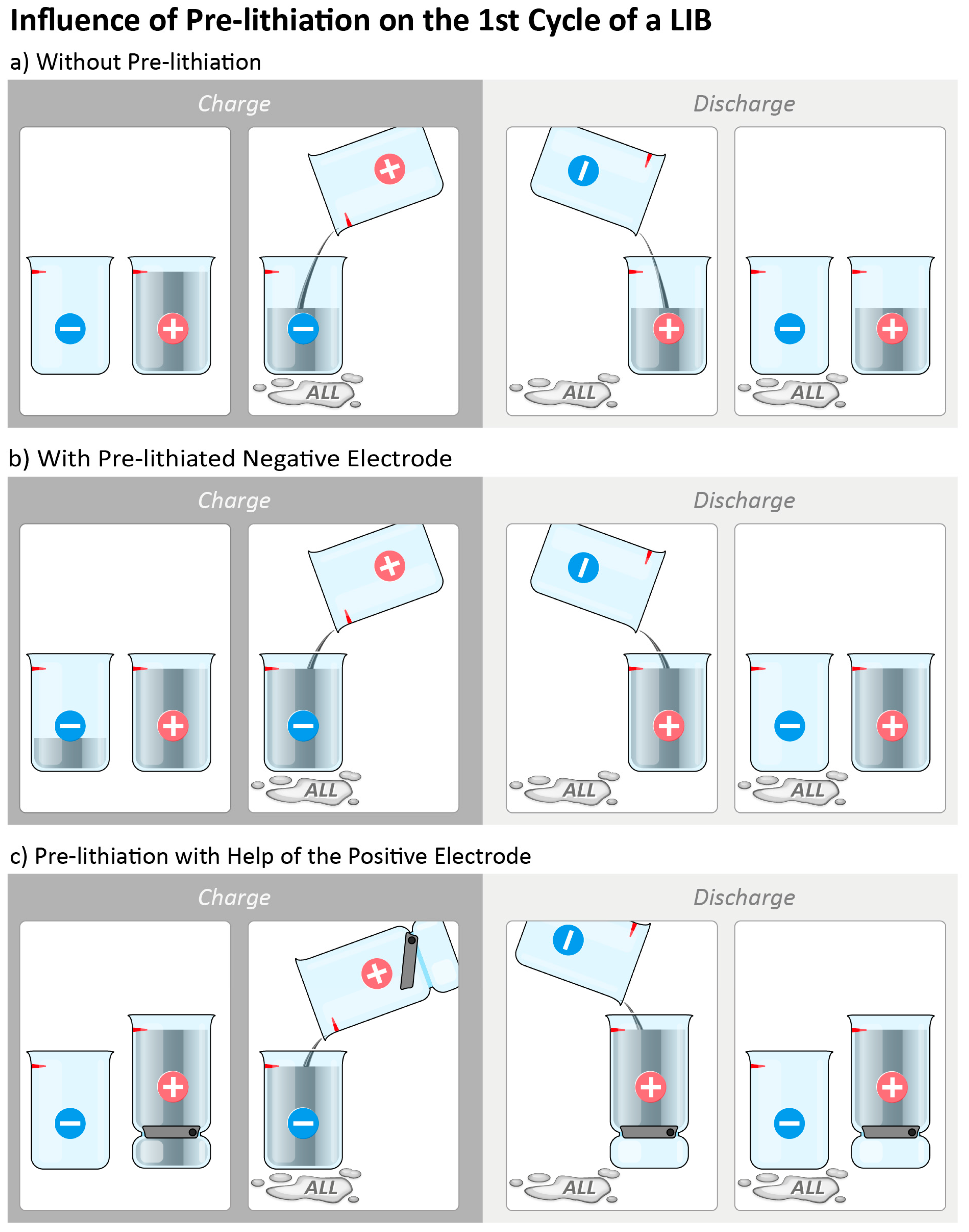
3. Pre-Lithiation with Help of the Positive Electrode
3.1. Pre-Lithiation by Using Positive Electrode Additives
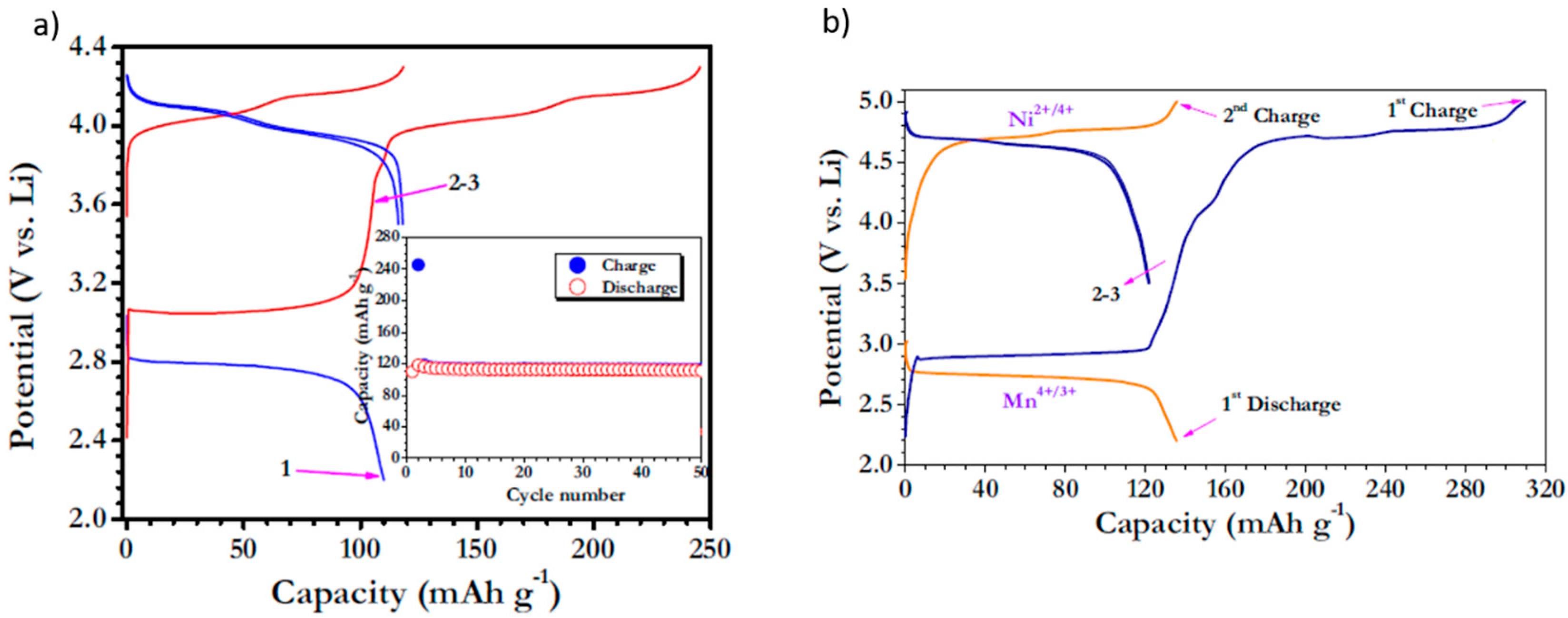
3.2. Over-Lithiated Positive Electrode Materials
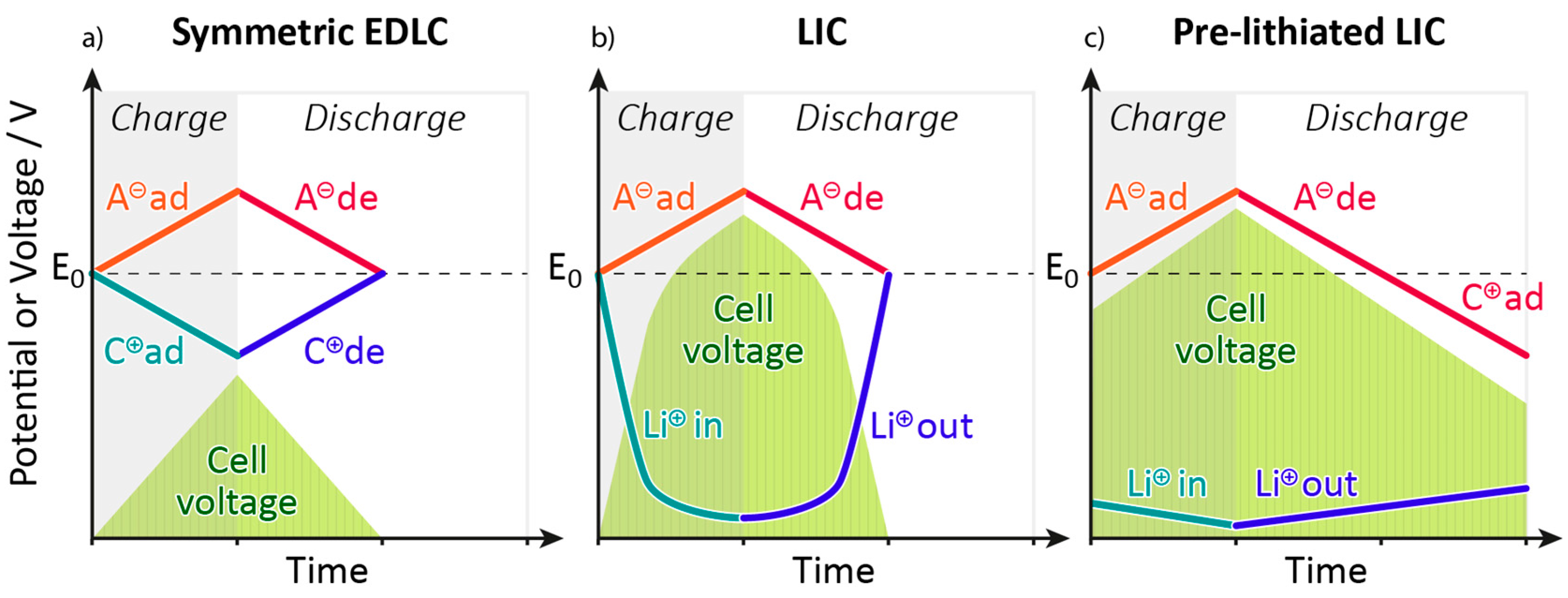
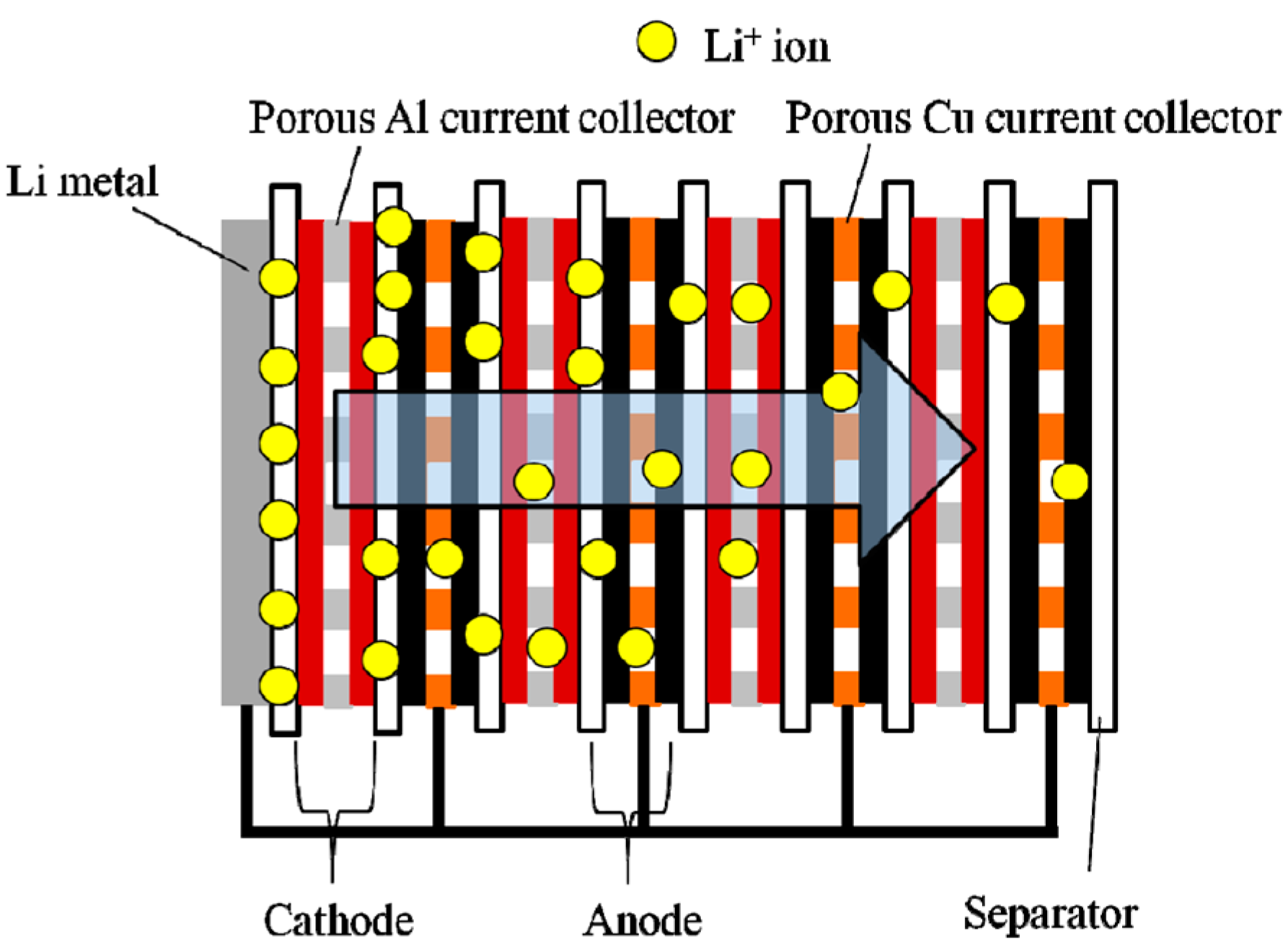
4. Pre-Lithiation in Lithium-Ion Capacitors
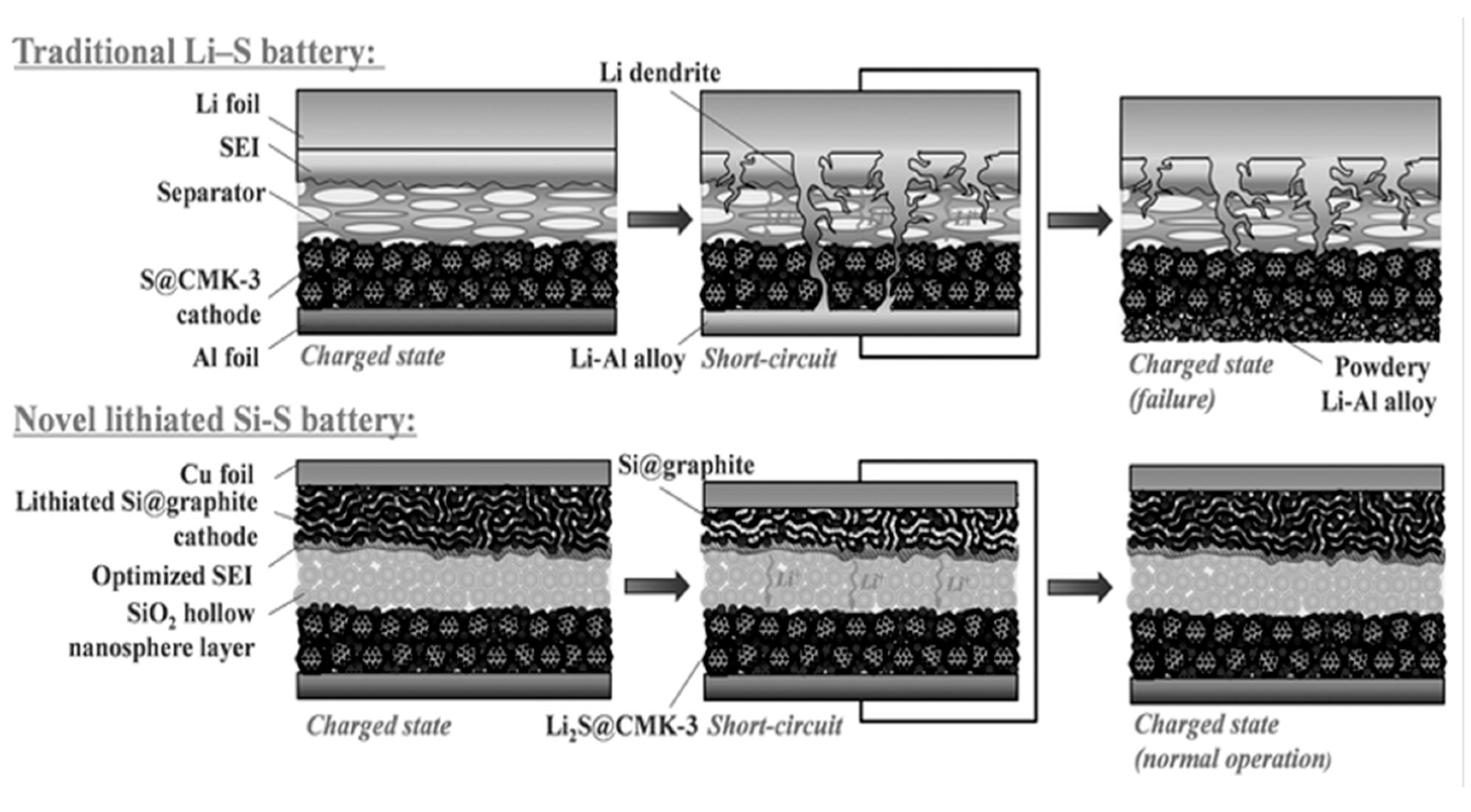
5. Pre-Lithiation for Rechargeable Li-Ion/Sulphur Batteries
5.1. Pre-Lithiation of the Negative Electrode–Charged State
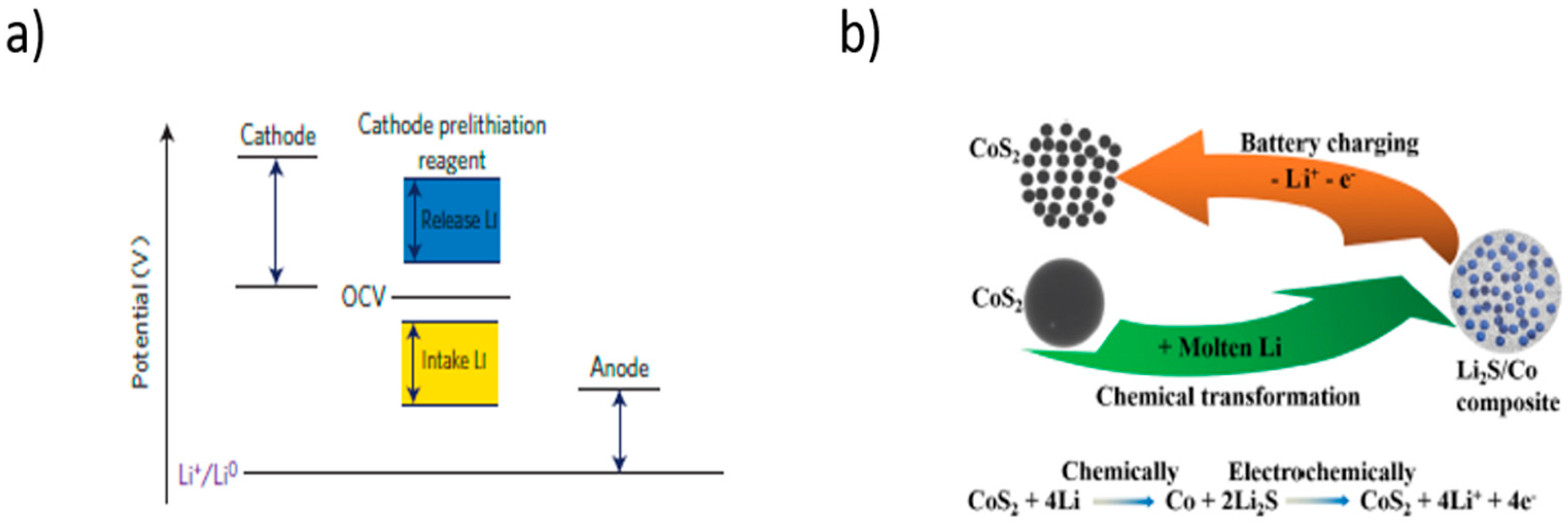
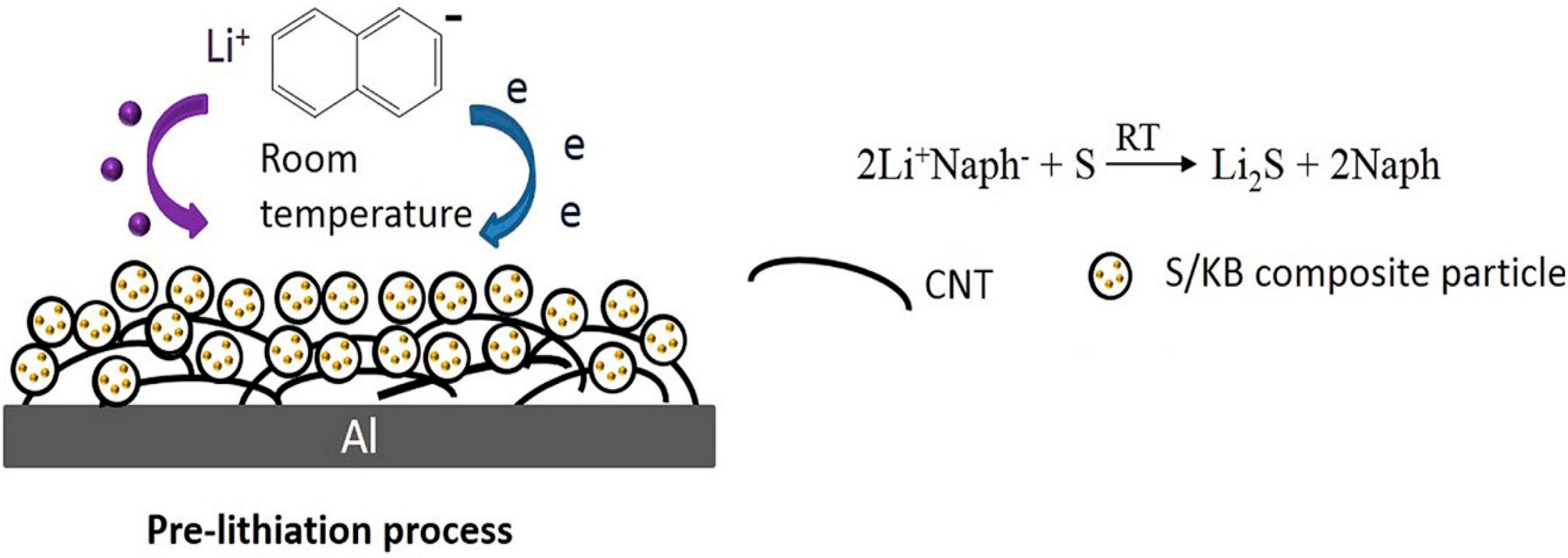
5.2. Pre-Lithiation of the Positive Electrode–Discharged State
6. Pre-Lithiation for Rechargeable Oxygen Batteries
7. Concluding Remarks and Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Andre, D.; Kim, S.J.; Lamp, P.; Lux, S.F.; Maglia, F.; Paschos, O.; Stiaszny, B. Future Generations of Cathode Materials: An Automotive Industry Perspective. J. Mater. Chem. A 2015, 3, 6709–6732. [Google Scholar] [CrossRef]
- Placke, T.; Kloepsch, R.; Duhnen, S.; Winter, M. Lithium Ion, Lithium Metal, and Alternative Rechargeable Battery Technologies: The Odyssey for High Energy Density. J. Solid State Electrochem. 2017, 21, 1939–1964. [Google Scholar] [CrossRef]
- Blomgren, G.E. The Development and Future of Lithium Ion Batteries. J. Electrochem. Soc. 2017, 164, A5019–A5025. [Google Scholar] [CrossRef]
- Crabtree, G.; Kocs, E.; Trahey, L. The Energy-Storage Frontier: Lithium-Ion Batteries and Beyond. MRS Bull. 2015, 40, 1067–1078. [Google Scholar] [CrossRef]
- Wagner, R.; Preschitschek, N.; Passerini, S.; Leker, J.; Winter, M. Current Research Trends and Prospects among the Various Materials and Designs Used in Lithium-Based Batteries. J. Appl. Electrochem. 2013, 43, 481–496. [Google Scholar] [CrossRef]
- Meister, P.; Jia, H.P.; Li, J.; Kloepsch, R.; Winter, M.; Placke, T. Best Practice: Performance and Cost Evaluation of Lithium Ion Battery Active Materials with Special Emphasis on Energy Efficiency. Chem. Mater. 2016, 28, 7203–7217. [Google Scholar] [CrossRef]
- Canepa, P.; Gautam, G.S.; Hannah, D.C.; Malik, R.; Liu, M.; Gallagher, K.G.; Persson, K.A.; Ceder, G. Odyssey of Multivalent Cathode Materials: Open Questions and Future Challenges. Chem. Rev. 2017, 117, 4287–4341. [Google Scholar] [CrossRef] [PubMed]
- Beltrop, K.; Beuker, S.; Heckmann, A.; Winter, M.; Placke, T. Alternative Electrochemical Energy Storage: Potassium-Based Dual-Graphite Batteries. Energy Environ. Sci. 2017, 10, 2090–2094. [Google Scholar] [CrossRef]
- Hwang, J.Y.; Myung, S.T.; Sun, Y.K. Sodium-Ion Batteries: Present and Future. Chem. Soc. Rev. 2017, 46, 3529–3614. [Google Scholar] [CrossRef] [PubMed]
- Ponrouch, A.; Frontera, C.; Barde, F.; Palacin, M.R. Towards a Calcium-Based Rechargeable Battery. Nat. Mater. 2016, 15, 169–172. [Google Scholar] [CrossRef] [PubMed]
- Besenhard, J.O.; Winter, M. Advances in Battery Technology: Rechargeable Magnesium Batteries and Novel Negative-Electrode Materials for Lithium Ion Batteries. ChemPhysChem 2002, 3, 155–159. [Google Scholar] [CrossRef]
- Vetter, J.; Novak, P.; Wagner, M.R.; Veit, C.; Moller, K.C.; Besenhard, J.O.; Winter, M.; Wohlfahrt-Mehrens, M.; Vogler, C.; Hammouche, A. Ageing Mechanisms in Lithium-Ion Batteries. J. Power Sources 2005, 147, 269–281. [Google Scholar] [CrossRef]
- Wohlfahrt-Mehrens, M.; Vogler, C.; Garche, J. Aging Mechanisms of Lithium Cathode Materials. J. Power Sources 2004, 127, 58–64. [Google Scholar] [CrossRef]
- Kleiner, K.; Ehrenberg, H. Challenges Considering the Degradation of Cell Components in Commercial Lithium-Ion Cells: A Review and Evaluation of Present Systems. Top. Curr. Chem. 2017, 375, 54–98. [Google Scholar] [CrossRef] [PubMed]
- Winter, M. The Solid Electrolyte Interphase—The Most Important and the Least Understood Solid Electrolyte in Rechargeable Li Batteries. Z. Phys. Chem. 2009, 223, 1395–1406. [Google Scholar] [CrossRef]
- Verma, P.; Maire, P.; Novak, P. A Review of the Features and Analyses of the Solid Electrolyte Interphase in Li-Ion Batteries. Electrochim. Acta 2010, 55, 6332–6341. [Google Scholar] [CrossRef]
- An, S.J.; Li, J.L.; Daniel, C.; Mohanty, D.; Nagpure, S.; Wood, D.L. The State of Understanding of the Lithium-Ion-Battery Graphite Solid Electrolyte Interphase (SEI) and its Relationship to Formation Cycling. Carbon 2016, 105, 52–76. [Google Scholar] [CrossRef]
- Michan, A.L.; Divitini, G.; Pell, A.J.; Leskes, M.; Ducati, C.; Grey, C.P. Solid Electrolyte Interphase Growth and Capacity Loss in Silicon Electrodes. J. Am. Chem. Soc. 2016, 138, 7918–7931. [Google Scholar] [CrossRef] [PubMed]
- Vogl, U.S.; Lux, S.F.; Das, P.; Weber, A.; Placke, T.; Kostecki, R.; Winter, M. The Mechanism of SEI Formation on Single Crystal Si(100), Si(110) and Si(111) Electrodes. J. Electrochem. Soc. 2015, 162, A2281–A2288. [Google Scholar] [CrossRef]
- Holtstiege, F.; Wilken, A.; Winter, M.; Placke, T. Running Out of Lithium? A Route to Differentiate between Capacity Losses and Active Lithium Losses in Lithium-Ion Batteries. Phys. Chem. Chem. Phys. 2017, 19, 25905–25918. [Google Scholar] [CrossRef] [PubMed]
- Winter, M.; Novak, P.; Monnier, A. Graphites for Lithium-Ion Cells: The Correlation of the First-Cycle Charge Loss with the Brunauer-Emmett-Teller Surface Area. J. Electrochem. Soc. 1998, 145, 428–436. [Google Scholar] [CrossRef]
- Placke, T.; Siozios, V.; Schmitz, R.; Lux, S.F.; Bieker, P.; Colle, C.; Meyer, H.W.; Passerini, S.; Winter, M. Influence of Graphite Surface Modifications on the Ratio of Basal Plane to “Non-Basal Plane” Surface Area and on the Anode Performance in Lithium Ion Batteries. J. Power Sources 2012, 200, 83–91. [Google Scholar] [CrossRef]
- Olivier, J.P.; Winter, M. Determination of the Absolute and Relative Extents of Basal Plane Surface Area and “Non-Basal Plane Surface” Area of Graphites and their Impact on Anode Performance in Lithium Ion Batteries. J. Power Sources 2001, 97–98, 151–155. [Google Scholar] [CrossRef]
- Kohs, W.; Santner, H.J.; Hofer, F.; Schrottner, H.; Doninger, J.; Barsukov, I.; Buqa, H.; Albering, J.H.; Moller, K.C.; Besenhard, J.O.; et al. A Study on Electrolyte Interactions with Graphite Anodes Exhibiting Structures with Various Amounts of Rhombohedral Phase. J. Power Sources 2003, 119, 528–537. [Google Scholar] [CrossRef]
- Zuo, X.X.; Zhu, J.; Muller-Buschbaum, P.; Cheng, Y.J. Silicon Based Lithium-Ion Battery Anodes: A Chronicle Perspective Review. Nano Energy 2017, 31, 113–143. [Google Scholar] [CrossRef]
- Obrovac, M.N.; Chevrier, V.L. Alloy Negative Electrodes for Li-Ion Batteries. Chem. Rev. 2014, 114, 11444–11502. [Google Scholar] [CrossRef] [PubMed]
- Winter, M.; Appel, W.K.; Evers, B.; Hodal, T.; Möller, K.C.; Schneider, I.; Wachtler, M.; Wagner, M.R.; Wrodnigg, G.H.; Besenhard, J.O. Studies on the Anode/Electrolyte Interface in Lithium Ion Batteries. Chem. Mon. 2001, 132, 473–486. [Google Scholar] [CrossRef]
- Krueger, S.; Kloepsch, R.; Li, J.; Nowak, S.; Passerini, S.; Winter, M. How Do Reactions at the Anode/Electrolyte Interface Determine the Cathode Performance in Lithium-Ion Batteries? J. Electrochem. Soc. 2013, 160, A542–A548. [Google Scholar] [CrossRef]
- Aravindan, V.; Lee, Y.S.; Madhavi, S. Best Practices for Mitigating Irreversible Capacity Loss of Negative Electrodes in Li-Ion Batteries. Adv. Energy Mater. 2017, 7, 1602607–1602624. [Google Scholar] [CrossRef]
- Kim, H.J.; Choi, S.; Lee, S.J.; Seo, M.W.; Lee, J.G.; Deniz, E.; Lee, Y.J.; Kim, E.K.; Choi, J.W. Controlled Prelithiation of Silicon Monoxide for High Performance Lithium-Ion Rechargeable Full Cells. Nano Lett. 2016, 16, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Forney, M.W.; Ganter, M.J.; Staub, J.W.; Ridgley, R.D.; Landi, B.J. Prelithiation of Silicon-Carbon Nanotube Anodes for Lithium Ion Batteries by Stabilized Lithium Metal Powder (SLMP). Nano Lett. 2013, 13, 4158–4163. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Lu, Z.D.; Liu, N.A.; Lee, H.W.; McDowell, M.T.; Cui, Y. Dry-Air-Stable Lithium Silicide-Lithium Oxide Core-Shell Nanoparticles as High-Capacity Prelithiation Reagents. Nat. Commun. 2014, 5, 5088–5095. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Wang, Z.H.; Lu, P.; Jiang, M.; Shi, F.F.; Song, X.Y.; Zheng, Z.Y.; Zhou, X.; Fu, Y.B.; Abdelbast, G.; et al. Toward Practical Application of Functional Conductive Polymer Binder for a High-Energy Lithium-Ion Battery Design. Nano Lett. 2014, 14, 6704–6710. [Google Scholar] [CrossRef] [PubMed]
- De la Llave, E.; Borgel, V.; Park, K.J.; Hwang, J.Y.; Sun, Y.K.; Hartmann, P.; Chesneau, F.F.; Aurbach, D. Comparison between Na-Ion and Li-Ion Cells: Understanding the Critical Role of the Cathodes Stability and the Anodes Pretreatment on the Cells Behavior. ACS Appl. Mater. Interfaces 2016, 8, 1867–1875. [Google Scholar] [CrossRef] [PubMed]
- Friesen, A.; Hildebrand, S.; Horsthemke, F.; Borner, M.; Klopsch, R.; Niehoff, P.; Schappacher, F.M.; Winter, M. Al2O3 Coating on Anode Surface in Lithium Ion Batteries: Impact on Low Temperature Cycling and Safety Behavior. J. Power Sources 2017, 363, 70–77. [Google Scholar] [CrossRef]
- Uhlmann, C.; Illig, J.; Ender, M.; Schuster, R.; Ivers-Tiffee, E. In Situ Detection of Lithium Metal Plating on Graphite in Experimental Cells. J. Power Sources 2015, 279, 428–438. [Google Scholar] [CrossRef]
- Liu, Q.; Du, C.; Shen, B.; Zuo, P.; Cheng, X.; Ma, Y.; Yin, G.; Gao, Y. Understanding Undesirable Anode Lithium Plating Issues in Lithium-Ion Batteries. RSC Adv. 2016, 6, 88683–88700. [Google Scholar] [CrossRef]
- Christensen, J.; Newman, J. Cyclable Lithium and Capacity Loss in Li-Ion Cells. J. Electrochem. Soc. 2005, 152, A818–A829. [Google Scholar] [CrossRef]
- Zheng, H.H.; Li, J.; Song, X.Y.; Liu, G.; Battaglia, V.S. A Comprehensive Understanding of Electrode Thickness Effects on the Electrochemical Performances of Li-Ion Battery Cathodes. Electrochim. Acta 2012, 71, 258–265. [Google Scholar] [CrossRef]
- Marinaro, M.; Weinberger, M.; Wohlfahrt-Mehrens, M. Toward Pre-Lithiated High Areal Capacity Silicon Anodes for Lithium-Ion Batteries. Electrochim. Acta 2016, 206, 99–107. [Google Scholar] [CrossRef]
- Cai, M.Y.; Sun, X.G.; Nie, Y.Y.; Chen, W.; Qiu, Z.W.; Chen, L.; Liu, Z.H.; Tang, H. Electrochemical Performance of Lithium-Ion Capacitors Using Pre-Lithiated Multiwalled Carbon Nanotubes as Anode. Nano 2017, 12, 1750051–1750060. [Google Scholar] [CrossRef]
- Zhang, J.; Shi, Z.Q.; Wang, C.Y. Effect of Pre-Lithiation Degrees of Mesocarbon Microbeads Anode on the Electrochemical Performance of Lithium-Ion Capacitors. Electrochim. Acta 2014, 125, 22–28. [Google Scholar] [CrossRef]
- Domi, Y.; Usui, H.; Iwanari, D.; Sakaguchi, H. Effect of Mechanical Pre-Lithiation on Electrochemical Performance of Silicon Negative Electrode for Lithium-Ion Batteries. J. Electrochem. Soc. 2017, 164, A1651–A1654. [Google Scholar] [CrossRef]
- Chang, S.; Moon, J.; Cho, K.; Cho, M. Multiscale Analysis of Prelithiated Silicon Nanowire for Li-Ion Battery. Comput. Mater. Sci. 2015, 98, 99–104. [Google Scholar] [CrossRef]
- Wang, Z.H.; Fu, Y.B.; Zhang, Z.C.; Yuan, S.W.; Amine, K.; Battaglia, V.; Liu, G. Application of Stabilized Lithium Metal Powder (SLMP®) in Graphite Anode—A High Efficient Prelithiation Method for Lithium-Ion Batteries. J. Power Sources 2014, 260, 57–61. [Google Scholar] [CrossRef]
- Schmitz, R.W.; Murmann, P.; Schmitz, R.; Muller, R.; Kramer, L.; Kasnatscheew, J.; Isken, P.; Niehoff, P.; Nowak, S.; Roschenthaler, G.V.; et al. Investigations on Novel Electrolytes, Solvents and SEI Additives for use in Lithium-Ion Batteries: Systematic Electrochemical Characterization and Detailed Analysis by Spectroscopic Methods. Prog. Solid State Chem. 2014, 42, 65–84. [Google Scholar] [CrossRef]
- Cekic-Laskovic, I.; von Aspern, N.; Imholt, L.; Kaymaksiz, S.; Oldiges, K.; Rad, B.R.; Winter, M. Synergistic Effect of Blended Components in Nonaqueous Electrolytes for Lithium Ion Batteries. Top. Curr. Chem. 2017, 375, 37. [Google Scholar] [CrossRef] [PubMed]
- Scott, M.G.; Whitehead, A.H.; Owen, J.R. Chemical Formation of a Solid Electrolyte Interface on the Carbon Electrode of a Li-Ion Cell. J. Electrochem. Soc. 1998, 145, 1506–1510. [Google Scholar] [CrossRef]
- Tabuchi, T.; Yasuda, H.; Yamachi, M. Li-Doping Process for LixSiO-Negative Active Material Synthesized by Chemical Method for Lithium-Ion Cells. J. Power Sources 2005, 146, 507–509. [Google Scholar] [CrossRef]
- Tabuchi, T.; Yasuda, H.; Yamachi, M. Mechanism of Li-Doping into Li4Ti5O12 Negative Active Material for Li-Ion Cells by New Chemical Method. J. Power Sources 2006, 162, 813–817. [Google Scholar] [CrossRef]
- Veluchamy, A.; Doh, C.H.; Kim, D.H.; Lee, J.H.; Lee, D.J.; Ha, Y.H.; Shin, H.M.; Jin, B.S.; Kim, H.S.; Moon, S.I.; et al. Improvement of Cycle Behaviour of SiO/C Anode Composite by Thermochemically Generated Li4SiO4 Inert Phase for Lithium Batteries. J. Power Sources 2009, 188, 574–577. [Google Scholar] [CrossRef]
- Takezawa, H.; Ito, S.; Yoshizawa, H.; Abe, T. Electrochemical Properties of a SiOx Film Anode Pre-Lithiated by Evaporation of Metallic Li in Li-Ion Batteries. Chem. Lett. 2017, 46, 1365–1367. [Google Scholar] [CrossRef]
- Nayak, P.K.; Penki, T.R.; Markovsky, B.; Aurbach, D. Electrochemical Performance of Li- and Mn-Rich Cathodes in Full Cells with Prelithiated Graphite Negative Electrodes. ACS Energy Lett. 2017, 2, 544–548. [Google Scholar] [CrossRef]
- Sun, Y.G.; Tang, J.; Qin, F.X.; Yuan, J.S.; Zhang, K.; Li, J.; Zhu, D.M.; Qin, L.C. Hybrid Lithium-Ion Capacitors with Asymmetric Graphene Electrodes. J. Mater. Chem. A 2017, 5, 13601–13609. [Google Scholar] [CrossRef]
- Liu, C.Y.; Kim, N.; Rubloff, G.W.; Lee, S.B. High Performance Asymmetric V2O5-SnO2 Nanopore Battery by Atomic Layer Deposition. Nanoscale 2017, 9, 11566–11573. [Google Scholar] [CrossRef] [PubMed]
- Kasnatscheew, J.; Placke, T.; Streipert, B.; Rothermel, S.; Wagner, R.; Meister, P.; Laskovic, I.C.; Winter, M. A Tutorial into Practical Capacity and Mass Balancing of Lithium Ion Batteries. J. Electrochem. Soc. 2017, 64, A2479–A2486. [Google Scholar] [CrossRef]
- Winter, M.; Besenhard, J.O. Lithiated Carbons. In Handbook of Battery Materials; VCH: Weinheim, Germany, 1999; Volume 3, pp. 383–418. [Google Scholar]
- Zhou, H.T.; Wang, X.H.; Chen, D. Li-Metal-Free Prelithiation of Si-Based Negative Electrodes for Full Li-Ion Batteries. ChemSusChem 2015, 8, 2737–2744. [Google Scholar] [CrossRef] [PubMed]
- Grant, R.W.; Sweetland, M.; Acharige, A.M.; Wohl, R. Phased Introduction of Lithium into the Pre-Lithiated Anode of a Lithium Ion Electrochemical Cell. U.S. Patent 9,748,599 B2, 29 August 2017. [Google Scholar]
- Grant, R.W.; Sweetland, M.; Acharige, A.M. Method for Alkaliating Anodes. U.S. Patent US9,598,789 B2, 21 March 2017. [Google Scholar]
- Grant, R.W.; Sweetland, M.; Acharige, A.M. Methods for Alkaliating Roll Anodes. U.S. Patent 20,170,187,030 A1, 29 June 2017. [Google Scholar]
- Shellikeri, A.; Watson, V.G.; Adams, D.L.; Kalu, E.E.; Read, J.A.; Jow, T.R.; Zheng, J.P. Pre-Lithiation of Carbon Anodes Using Different Lithium–Sources. ECS Trans. 2017, 77, 293–303. [Google Scholar] [CrossRef]
- Liu, N.A.; Hu, L.B.; McDowell, M.T.; Jackson, A.; Cui, Y. Prelithiated Silicon Nanowires as an Anode for Lithium Ion Batteries. ACS Nano 2011, 5, 6487–6493. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; He, X.M.; Ren, J.G.; Li, J.J.; Jiang, C.Y.; Wan, C.R. Hard Carbon/Lithium Composite Anode Materials for Li-Ion Batteries. Electrochim. Acta 2007, 52, 4312–4316. [Google Scholar] [CrossRef]
- Fei, L.; Yoo, S.H.; Villamayor, R.A.R.; Williams, B.P.; Gong, S.Y.; Park, S.; Shin, K.; Joo, Y.L. Graphene Oxide Involved Air-Controlled Electrospray for Uniform, Fast, Instantly Dry, and Binder-Free Electrode Fabrication. ACS Appl. Mater. Interfaces 2017, 9, 9738–9746. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xing, G.Z.; Han, Z.J.; Shi, Y.M.; Wong, J.I.; Huang, Z.X.; Ostrikov, K.; Yang, H.Y. Pre-Lithiation of Onion-like Carbon/MoS2 Nano-Urchin Anodes for High-Performance Rechargeable Lithium Ion Batteries. Nanoscale 2014, 6, 8884–8890. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, C.R.; Lain, M.J.; Yakovleva, M.V.; Gao, Y. A Prelithiated Carbon Anode for Lithium-Ion Battery Applications. J. Power Sources 2006, 162, 800–802. [Google Scholar] [CrossRef]
- Jarvis, C.R.; Lain, M.J.; Gao, Y.; Yakovleva, M. A Lithium Ion Cell Containing a Non-Lithiated Cathode. J. Power Sources 2005, 146, 331–334. [Google Scholar] [CrossRef]
- Heine, J.; Kruger, S.; Hartnig, C.; Wietelmann, U.; Winter, M.; Bieker, P. Coated Lithium Powder (CLiP) Electrodes for Lithium-Metal Batteries. Adv. Energy Mater. 2014, 4, 1300815–1300821. [Google Scholar] [CrossRef]
- Heine, J.; Rodehorst, U.; Qi, X.; Badillo, J.P.; Hartnig, C.; Wietelmann, U.; Winter, M.; Bieker, P. Using Polyisobutylene as a Non-Fluorinated Binder for Coated Lithium Powder (CLiP) Electrodes. Electrochim. Acta 2014, 138, 288–293. [Google Scholar] [CrossRef]
- Li, Y.X.; Fitch, B. Effective Enhancement of Lithium-Ion Battery Performance Using SLMP. Electrochem. Commun. 2011, 13, 664–667. [Google Scholar] [CrossRef]
- Xiang, B.; Wang, L.; Liu, G.; Minor, A.M. Electromechanical Probing of Li/Li2CO3 Core/Shell Particles in a TEM. J. Electrochem. Soc. 2013, 160, A415–A419. [Google Scholar] [CrossRef]
- Seong, I.W.; Kim, K.T.; Yoon, W.Y. Electrochemical Behavior of a Lithium-Pre-Doped Carbon-Coated Silicon Monoxide Anode Cell. J. Power Sources 2009, 189, 511–514. [Google Scholar] [CrossRef]
- Pan, Q.R.; Zuo, P.J.; Mu, T.S.; Du, C.Y.; Cheng, X.Q.; Ma, Y.L.; Gao, Y.Z.; Yin, G.P. Improved Electrochemical Performance of Micro-Sized SiO-Based Composite Anode by Prelithiation of Stabilized Lithium Metal Powder. J. Power Sources 2017, 347, 170–177. [Google Scholar] [CrossRef]
- Tahir, M.S.; Weinberger, M.; Balasubramanian, P.; Diemant, T.; Behm, R.J.; Linden, M.; Wohlfahrt-Mehrens, M. Silicon Carboxylate Derived Silicon Oxycarbides as Anodes for Lithium Ion Batteries. J. Mater. Chem. A 2017, 5, 10190–10199. [Google Scholar] [CrossRef]
- Wang, L.; Fu, Y.B.; Battaglia, V.S.; Liu, G. SBR-PVDF Based Binder for the Application of SLMP in Graphite Anodes. RSC Adv. 2013, 3, 15022–15027. [Google Scholar] [CrossRef]
- Ai, G.; Wang, Z.H.; Zhao, H.; Mao, W.F.; Fu, Y.B.; Yi, R.; Gao, Y.; Battaglia, V.; Wang, D.H.; Lopatin, S.; et al. Scalable Process for Application of Stabilized Lithium Metal Powder in Li-Ion Batteries. J. Power Sources 2016, 309, 33–41. [Google Scholar] [CrossRef]
- Mazouzi, D.; Karkar, Z.; Hernandez, C.R.; Manero, P.J.; Guyomard, D.; Roue, L.; Lestriez, B. Critical Roles of Binders and Formulation at Multiscales of Silicon-Based Composite Electrodes. J. Power Sources 2015, 280, 533–549. [Google Scholar] [CrossRef]
- Zhang, H.W.; Sun, X.R.; Huang, X.D.; Zhou, L. Encapsulation of Alpha-Fe2O3 Nanoparticles in Graphitic Carbon Microspheres as High-Performance Anode Materials for Lithium-Ion Batteries. Nanoscale 2015, 7, 3270–3275. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.L.; Han, W.Q.; Chen, H.Y.; Bai, J.M.; Tyson, T.A.; Yu, X.Q.; Wang, X.J.; Yang, X.Q. Amorphous Hierarchical Porous GeOx as High-Capacity Anodes for Li Ion Batteries with Very Long Cycling Life. J. Am. Chem. Soc. 2011, 133, 20692–20695. [Google Scholar] [CrossRef] [PubMed]
- Brutti, S.; Gentili, V.; Reale, P.; Carbone, L.; Panero, S. Mitigation of the Irreversible Capacity and Electrolyte Decomposition in a LiNi0.5Mn1.5O4/Nano-TiO2 Li-Ion Battery. J. Power Sources 2011, 196, 9792–9799. [Google Scholar] [CrossRef]
- Yersak, T.A.; Son, S.B.; Cho, J.S.; Suh, S.S.; Kim, Y.U.; Moon, J.T.; Oh, K.H.; Lee, S.H. An All-Solid-State Li-Ion Battery with a Pre-Lithiated Si-Ti-Ni Alloy Anode. J. Electrochem. Soc. 2013, 160, A1497–A1501. [Google Scholar] [CrossRef]
- Park, H.; Kim, M.; Xu, F.; Jung, C.; Hong, S.M.; Koo, C.M. In Situ Synchrotron Wide-Angle X-ray Scattering Study on Rapid Lithiation of Graphite Anode via Direct Contact Method for Li-Ion Capacitors. J. Power Sources 2015, 283, 68–73. [Google Scholar] [CrossRef]
- Cao, W.J.; Luo, J.F.; Yan, J.; Chen, X.J.; Brandt, W.; Warfield, M.; Lewis, D.; Yturriaga, S.R.; Moye, D.G.; Zheng, J.P. High Performance Li-Ion Capacitor Laminate Cells Based on Hard Carbon/Lithium Stripes Negative Electrodes. J. Electrochem. Soc. 2017, 164, A93–A98. [Google Scholar] [CrossRef]
- Yan, J.; Cao, W.J.; Zheng, J.P. Constructing High Energy and Power Densities Li-Ion Capacitors Using Li Thin Film for Pre-Lithiation. J. Electrochem. Soc. 2017, 164, A2164–A2170. [Google Scholar] [CrossRef]
- Schmuch, R.; Wagner, R.; Hörpel, G.; Placke, T.; Winter, M. Performance and Cost of Materials for Lithium-Based Rechargeable Automotive Batteries. Nat. Energy 2018. accepted. [Google Scholar]
- Shodai, T.; Sakurai, Y.; Suzuki, T. Reaction Mechanisms of Li2.6Co0.4N Anode Material. Solid State Ion. 1999, 122, 85–93. [Google Scholar] [CrossRef]
- Liu, Y.; Horikawa, K.; Fujiyosi, M.; Imanishi, N.; Hirano, A.; Takeda, Y. Layered Lithium Transition Metal Nitrides as Novel Anodes for Lithium Secondary Batteries. Electrochim. Acta 2004, 49, 3487–3496. [Google Scholar] [CrossRef]
- Hanai, K.; Liu, Y.; Matsumura, T.; Imanishi, N.; Hirano, A.; Takeda, Y. Electrochemical Behavior of the Composite Anodes Consisting of Carbonaceous Materials and Lithium Transition-Metal Nitrides for Lithium-Ion Batteries. Solid State Ion. 2008, 179, 1725–1730. [Google Scholar] [CrossRef]
- Liu, D.L.; Du, F.; Pan, W.C.; Chen, G.; Wang, C.Z.; Wei, Y.J. Electrochemical Characterizations of Li2.6Co0.4N/Graphite Anodes for Lithium Ion Batteries. Mater. Lett. 2009, 63, 504–506. [Google Scholar] [CrossRef]
- Sun, H.; He, X.M.; Li, J.J.; Ren, J.G.; Jiang, C.Y.; Wan, C.R. Hard Carbon/Li2.6Co0.4N Composite Anode Materials for Li-Ion Batteries. Solid State Ion. 2006, 177, 1331–1334. [Google Scholar] [CrossRef]
- Liu, Y.; Hanai, K.; Horikawa, K.; Imanishi, N.; Hirano, A.; Takeda, Y. Electrochemical Characterization of a Novel Si-Graphite-Li2.6Co0.4N Composite as Anode Material for Lithium Secondary Batteries. Mater. Chem. Phys. 2005, 89, 80–84. [Google Scholar] [CrossRef]
- Yang, J.; Takeda, Y.; Imanishi, N.; Yamamoto, O. Novel Composite Anodes Based on Nano-Oxides and Li2.6Co0.4N for Lithium Ion Batteries. Electrochim. Acta 2001, 46, 2659–2664. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, J.; Imanishi, N.; Hirano, A.; Takeda, Y.; Yamamoto, O. Composite Anode Containing Nano-SiO1.1 and Li2.6Co0.4N with Solid PEO Electrolytes for Lithium-Ion Batteries. J. Power Sources 2005, 146, 376–379. [Google Scholar] [CrossRef]
- Takeda, Y.; Yang, J. New Composite Anode Systems Combined with Li2.6Co0.4N. J. Power Sources 2001, 97–98, 244–246. [Google Scholar] [CrossRef]
- Yang, J.; Takeda, Y.; Imanishi, N.; Xie, J.Y.; Yamamoto, O. Morphology Modification and Irreversibility Compensation for SnO Anodes. J. Power Sources 2001, 97–98, 216–218. [Google Scholar] [CrossRef]
- Yang, J.; Takeda, Y.; Imanishi, N.; Ichikawa, T.; Yamamoto, O. SnSbx-Based Composite Electrodes for Lithium Ion Cells. Solid State Ion. 2000, 135, 175–180. [Google Scholar] [CrossRef]
- Yang, J.; Takeda, Y.; Capiglia, C.; Liu, X.D.; Imanishi, N.; Yamamoto, O. High-Capacity Composite Anodes with SnSb and Li2.6Co0.4N for Solid Polymer Electrolyte Cells. J. Power Sources 2003, 119, 56–59. [Google Scholar] [CrossRef]
- Yang, J.; Takeda, Y.; Li, Q.; Imanishi, N.; Yamamoto, O. Solid Polymer Electrolyte Cells Using SnSb/Li2.6Co0.4N Composite Anodes. J. Power Sources 2001, 97–98, 779–781. [Google Scholar] [CrossRef]
- Rai, A.K.; Lim, J.; Mathew, V.; Gim, J.; Kang, J.; Paul, B.J.; Kim, D.; Ahn, S.; Kim, S.; Ahn, K.; et al. Highly Reversible Capacity Nanocomposite Anode for Secondary Lithium-Ion Batteries. Electrochem. Commun. 2012, 19, 9–12. [Google Scholar] [CrossRef]
- Liu, Y.; Horikawa, K.; Fujiyoshi, M.; Matsumura, T.; Imanishi, N.; Takeda, Y. Novel Composite Anodes Based on Layered Lithium Transition Metal Nitrides for Lithium Secondary Batteries. Solid State Ion. 2004, 172, 69–72. [Google Scholar] [CrossRef]
- Zhao, J.; Lu, Z.D.; Wang, H.T.; Liu, W.; Lee, H.W.; Yan, K.; Zhuo, D.; Lin, D.C.; Liu, N.; Cui, Y. Artificial Solid Electrolyte Interphase-Protected LixSi Nanoparticles: An Efficient and Stable Prelithiation Reagent for Lithium-Ion Batteries. J. Am. Chem. Soc. 2015, 137, 8372–8375. [Google Scholar] [CrossRef] [PubMed]
- Li, X.M.; Kersey-Bronec, F.E.; Ke, J.; Cloud, J.E.; Wang, Y.L.; Ngo, C.L.; Pylypenko, S.; Yang, Y.G. Study of Lithium Silicide Nanoparticles as Anode Materials for Advanced Lithium Ion Batteries. ACS Appl. Mater. Interfaces 2017, 9, 16071–16080. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Lee, H.W.; Sun, J.; Yan, K.; Liu, Y.Y.; Liu, W.; Lu, Z.D.; Lin, D.C.; Zhou, G.M.; Cui, Y. Metallurgically Lithiated SiOx Anode with High Capacity and Ambient Air Compatibility. Proc. Natl. Acad. Sci. USA 2016, 113, 7408–7413. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Sun, J.; Pei, A.; Zhou, G.; Yan, K.; Liu, Y.; Lin, D.; Cui, Y. A General Prelithiation Approach for Group IV Elements and Corresponding Oxides. Energy Storage Mater. 2018, 10, 275–281. [Google Scholar] [CrossRef]
- Lee, J.H.; Kim, J.M.; Kim, J.H.; Jang, Y.R.; Kim, J.A.; Yeon, S.H.; Lee, S.Y. Toward Ultrahigh-Capacity V2O5 Lithium-Ion Battery Cathodes via One-Pot Synthetic Route from Precursors to Electrode Sheets. Adv. Mater. Interfaces 2016, 3, 1600173. [Google Scholar] [CrossRef]
- Delmas, C.; Brèthes, S.; Ménétrier, M. ω-LixV2O5—A New Electrode Material for Rechargeable Lithium Batteries. J. Power Sources 1991, 34, 113–118. [Google Scholar] [CrossRef]
- Cheah, Y.L.; Aravindan, V.; Madhavi, S. Chemical Lithiation Studies on Combustion Synthesized V2O5 Cathode with Full Cell Application for Lithium Ion Batteries. J. Electrochem. Soc. 2013, 160, A1016–A1024. [Google Scholar] [CrossRef]
- Cheah, Y.L.; Aravindan, V.; Madhavi, S. Synthesis and Enhanced Lithium Storage Properties of Electrospun V2O5 Nanofibers in Full-Cell Assembly with a Spinel Li4Ti5O12 Anode. ACS Appl. Mater. Interfaces 2013, 5, 3475–3480. [Google Scholar] [CrossRef] [PubMed]
- Chernova, N.A.; Roppolo, M.; Dillon, A.C.; Whittingham, M.S. Layered Vanadium and Molybdenum Oxides: Batteries and Electrochromics. J. Mater. Chem. 2009, 19, 2526–2552. [Google Scholar] [CrossRef]
- Garcia, B.; Millet, M.; Pereira-Ramos, J.P.; Baffier, N.; Bloch, D. Electrochemical Behaviour of Chemically Lithiated LixV2O5 phases (0.9 < x < 1.6). J. Power Sources 1999, 81–82, 670–674. [Google Scholar]
- Mai, L.; Xu, L.; Hu, B.; Gu, Y. Improved Cycling Sability of Nanostructured Electrode Materials Enabled by Prelithiation. J. Mater. Res. 2011, 25, 1413–1420. [Google Scholar] [CrossRef]
- Mai, L.Q.; Hu, B.; Chen, W.; Qi, Y.Y.; Lao, C.S.; Yang, R.S.; Dai, Y.; Wang, Z.L. Lithiated MoO3 Nanobelts with Greatly Improved Performance for Lithium Batteries. Adv. Mater. 2007, 19, 3712–3716. [Google Scholar] [CrossRef]
- Sun, Y.; Lee, H.-W.; Seh, Z.W.; Liu, N.; Sun, J.; Li, Y.; Cui, Y. High-Capacity Battery Cathode Prelithiation to Offset Initial Lithium Loss. Nat. Energy 2016, 1, 15008. [Google Scholar] [CrossRef]
- Sun, Y.; Lee, H.-W.; Seh, Z.W.; Zheng, G.; Sun, J.; Li, Y.; Cui, Y. Lithium Sulfide/Metal Nanocomposite as a High-Capacity Cathode Prelithiation Material. Adv. Energy Mater. 2016, 6, 1600154–1600161. [Google Scholar] [CrossRef]
- Zhan, Y.; Yu, H.; Ben, L.; Chen, Y.; Huang, X. Using Li2S to Compensate for the Loss of Active Lithium in Li-ion Batteries. Electrochim. Acta 2017, 255, 212–219. [Google Scholar]
- Park, K.; Yu, B.-C.; Goodenough, J.B. Li3N as a Cathode Additive for High-Energy-Density Lithium-Ion Batteries. Adv. Energy Mater. 2016, 6, 1502534–1502541. [Google Scholar] [CrossRef]
- Sun, Y.; Li, Y.; Sun, J.; Li, Y.; Pei, A.; Cui, Y. Stabilized Li3N for Efficient Battery Cathode Prelithiation. Energy Storage Mater. 2017, 6, 119–124. [Google Scholar] [CrossRef]
- Shanmukaraj, D.; Grugeon, S.; Laruelle, S.; Douglade, G.; Tarascon, J.-M.; Armand, M. Sacrificial Salts: Compensating the Initial Charge Irreversibility in Lithium Batteries. Electrochem. Commun. 2010, 12, 1344–1347. [Google Scholar] [CrossRef]
- Abouimrane, A.; Cui, Y.; Chen, Z.; Belharouak, I.; Yahia, H.B.; Wu, H.; Assary, R.; Curtiss, L.A.; Amine, K. Enabling High Energy Density Li-Ion Batteries through Li2O Activation. Nano Energy 2016, 27, 196–201. [Google Scholar] [CrossRef]
- Bie, Y.; Yang, J.; Wang, J.; Zhou, J.; Nuli, Y. Li2O2 as a Cathode Additive for the Initial Anode Irreversibility Compensation in Lithium-Ion Batteries. Chem. Commun. 2017, 53, 8324–8327. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Lee, H.-W.; Zheng, G.; Seh, Z.W.; Sun, J.; Li, Y.; Cui, Y. In Situ Chemical Synthesis of Lithium Fluoride/Metal Nanocomposite for High Capacity Prelithiation of Cathodes. Nano Lett. 2016, 16, 1497–1501. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Lin, C.; Wang, X.; Maroni, V.A.; Ren, Y.; Johnson, C.S.; Lu, W. A New Strategy to Mitigate the Initial Capacity Loss of Lithium Ion Batteries. J. Power Sources 2016, 324, 150–157. [Google Scholar] [CrossRef]
- Kim, M.G.; Cho, J. Air Stable Al2O3-Coated Li2NiO2 Cathode Additive as a Surplus Current Consumer in a Li-Ion Cell. J. Mater. Chem. 2008, 18, 5880–5887. [Google Scholar] [CrossRef]
- Park, H.; Yoon, T.; Kim, Y.-U.; Ryu, J.H.; Oh, S.M. Li2NiO2 as a Sacrificing Positive Additive for Lithium-Ion Batteries. Electrochim. Acta 2013, 108, 591–595. [Google Scholar] [CrossRef]
- Noh, M.; Cho, J. Role of Li6CoO4 Cathode Additive in Li-Ion Cells Containing Low Coulombic Efficiency Anode Material. J. Electrochem. Soc. 2012, 159, A1329–A1334. [Google Scholar] [CrossRef]
- Park, K.-S.; Im, D.; Benayad, A.; Dylla, A.; Stevenson, K.J.; Goodenough, J.B. LiFeO2-Incorporated Li2MoO3 as a Cathode Additive for Lithium-Ion Battery Safety. Chem. Mater. 2012, 24, 2673–2683. [Google Scholar] [CrossRef]
- Gregory, D.H. Lithium Nitrides as Sustainable Energy Materials. Chem. Record 2008, 8, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Chang, S.-K.; Goh, E.Y.; Jeong, J.-Y.; Lee, J.H.; Kim, H.-J.; Cho, J.-J.; Hong, S.-T. Li2NiO2 as a Novel Cathode Additive for Overdischarge Protection Li-Ion Batteries. Chem. Mater. 2008, 20, 5–7. [Google Scholar] [CrossRef]
- Johnson, C.S.; Kang, S.H.; Vaughey, J.T.; Pol, S.V.; Balasubramanian, M.; Thackeray, M.M. Li2O Removal from Li5FeO4: A Cathode Precursor for Lithium-Ion Batteries. Chem. Mater. 2010, 22, 1263–1270. [Google Scholar] [CrossRef]
- Aravindan, V.; Nan, S.; Keppeler, M.; Madhavi, S. Pre-Lithiated LixMn2O4: A New Approach to Mitigate the Irreversible Capacity Loss in Negative Electrodes for Li-Ion Battery. Electrochim. Acta 2016, 208, 225–230. [Google Scholar] [CrossRef]
- Tarascon, J.M.; Guyomard, D. Li Metal-Free Rechargeable Batteries Based on LixMn2O4 Cathodes and Carbon Anodes. J. Electrochem. Soc. 1991, 138, 2864–2868. [Google Scholar] [CrossRef]
- Peramunage, D. Preparation and Electrochemical Characterization of Overlithiated Spinel LiMn2O4. J. Electrochem. Soc. 1998, 145, 1131–1136. [Google Scholar] [CrossRef]
- Kasnatscheew, J.; Evertz, M.; Streipert, B.; Wagner, R.; Klopsch, R.; Vortmann, B.; Hahn, H.; Nowak, S.; Amereller, M.; Gentschev, A.C.; et al. The Truth about the 1st Cycle Coulombic Efficiency of LiNi1/3Co1/3Mn1/3O2 (NCM) Cathodes. Phys. Chem. Chem. Phys. 2016, 18, 3956–3965. [Google Scholar] [CrossRef] [PubMed]
- Kasnatscheew, J.; Evertz, M.; Streipert, B.; Wagner, R.; Nowak, S.; Laskovic, I.C.; Winter, M. Improving Cycle Life of Layered Lithium Transition Metal Oxide (LiMO2) Based Positive Electrodes for Li Ion Batteries by Smart Selection of the Electrochemical Charge Conditions. J. Power Sources 2017, 359, 458–467. [Google Scholar] [CrossRef]
- Aravindan, V.; Arun, N.; Shubha, N.; Sundaramurthy, J.; Madhavi, S. Overlithiated Li1+xNi0.5Mn1.5O4 in All One Dimensional Architecture with Conversion Type α-Fe2O3: A New Approach to Eliminate Irreversible Capacity Loss. Electrochim. Acta 2016, 215, 647–651. [Google Scholar] [CrossRef]
- Moorhead-Rosenberg, Z.; Allcorn, E.; Manthiram, A. In Situ Mitigation of First-Cycle Anode Irreversibility in a New Spinel/FeSb Lithium-Ion Cell Enabled via a Microwave-Assisted Chemical Lithiation Process. Chem. Mater. 2014, 26, 5905–5913. [Google Scholar] [CrossRef]
- Kasnatscheew, J.; Streipert, B.; Roser, S.; Wagner, R.; Laskovic, I.C.; Winter, M. Determining Oxidative Stability of Battery Electrolytes: Validity of Cmmon Electrochemical Stability Window (ESW) Data and Alternative Strategies. Phys. Chem. Chem. Phys. 2017, 19, 16078–16086. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Blizanac, B.; DuPasquier, A.; Lal, A.; Niehoff, P.; Placke, T.; Oljaca, M.; Li, J.; Winter, M. Influence of Thermal Treated Carbon Black Conductive Additive on the Performance of High Voltage Spinel Cr-Doped LiNi0.5Mn1.5O4 Composite Cathode Electrode. J. Electrochem. Soc. 2015, 162, A339–A343. [Google Scholar] [CrossRef]
- Gabrielli, G.; Marinaro, M.; Mancini, M.; Axmann, P.; Wohlfahrt-Mehrens, M. A New Approach for Compensating the Irreversible Capacity Loss of High-Energy Si/C Vertical Bar LiNi0.5Mn1.5O4 Lithium-Ion Batteries. J. Power Sources 2017, 351, 35–44. [Google Scholar] [CrossRef]
- Mancini, M.; Axmann, P.; Gabrielli, G.; Kinyanjui, M.; Kaiser, U.; Wohlfahrt-Mehrens, M. A High-Voltage and High-Capacity Li1+xNi0.5Mn1.5O4 Cathode Material: From Synthesis to Full Lithium-Ion Cells. ChemSusChem 2016, 9, 1843–1849. [Google Scholar] [CrossRef] [PubMed]
- Simon, P.; Gogotsi, Y. Materials for Electrochemical Capacitors. Nat. Mater. 2008, 7, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Burke, A. R&D Considerations for the Performance and Application of Electrochemical Capacitors. Electrochim. Acta 2007, 53, 1083–1091. [Google Scholar]
- Miller, J.R.; Simon, P. Materials Science–Electrochemical Capacitors for Energy Management. Science 2008, 321, 651–652. [Google Scholar] [CrossRef] [PubMed]
- Winter, M.; Brodd, R.J. What are Batteries, Fuel Cells, and Supercapacitors? Chem. Rev. 2004, 104, 4245–4269. [Google Scholar] [CrossRef] [PubMed]
- Naoi, K.; Ishimoto, S.; Miyamoto, J.; Naoi, W. Second Generation ‘Nanohybrid Supercapacitor’: Evolution of Capacitive Energy Storage Devices. Energy Environ. Sci. 2012, 5, 9363–9373. [Google Scholar] [CrossRef]
- Cericola, D.; Kotz, R. Hybridization of Rechargeable Batteries and Electrochemical Capacitors: Principles and Limits. Electrochim. Acta 2012, 72, 1–17. [Google Scholar] [CrossRef]
- Schroeder, M.; Winter, M.; Passerini, S.; Balducci, A. On the Cycling Stability of Lithium-Ion Capacitors Containing Soft Carbon as Anodic Material. J. Power Sources 2013, 238, 388–394. [Google Scholar] [CrossRef]
- Schroeder, M.; Menne, S.; Segalini, J.; Saurel, D.; Casas-Cabanas, M.; Passerini, S.; Winter, M.; Balducci, A. Considerations about the Influence of the Structural and Electrochemical Properties of Carbonaceous Materials on the Behavior of Lithium-Ion Capacitors. J. Power Sources 2014, 266, 250–258. [Google Scholar] [CrossRef]
- Placke, T.; Fromm, O.; Lux, S.F.; Bieker, P.; Rothermel, S.; Meyer, H.W.; Passerini, S.; Winter, M. Reversible Intercalation of Bis(trifluoromethanesulfonyl)imide Anions from an Ionic Liquid Electrolyte into Graphite for High Performance Dual-Ion Cells. J. Electrochem. Soc. 2012, 159, A1755–A1765. [Google Scholar] [CrossRef]
- Placke, T.; Bieker, P.; Lux, S.F.; Fromm, O.; Meyer, H.W.; Passerini, S.; Winter, M. Dual-Ion Cells Based on Anion Intercalation into Graphite from Ionic Liquid-Based Electrolytes. Z. Phys. Chem. 2012, 226, 391–407. [Google Scholar] [CrossRef]
- Rothermel, S.; Meister, P.; Schmuelling, G.; Fromm, O.; Meyer, H.-W.; Nowak, S.; Winter, M.; Placke, T. Dual-Graphite Cells based on the Reversible Intercalation of Bis(trifluoromethanesulfonyl)imide Anions from an Ionic Liquid Electrolyte. Energy Environ. Sci. 2014, 7, 3412–3423. [Google Scholar] [CrossRef]
- Schmuelling, G.; Placke, T.; Kloepsch, R.; Fromm, O.; Meyer, H.W.; Passerini, S.; Winter, M. X-ray diffraction studies of the electrochemical intercalation of bis(trifluoromethanesulfonyl)imide anions into graphite for dual-ion cells. J. Power Sources 2013, 239, 563–571. [Google Scholar] [CrossRef]
- Amatucci, G.G.; Badway, F.; Du Pasquier, A.; Zheng, T. An Asymmetric Hybrid Nonaqueous Energy Storage Cell. J. Electrochem. Soc. 2001, 148, A930–A939. [Google Scholar] [CrossRef]
- Du Pasquier, A.; Plitz, I.; Menocal, S.; Amatucci, G. A Comparative Study of Li-Ion Battery, Supercapacitor and Nonaqueous Asymmetric Hybrid Devices for Automotive Applications. J. Power Sources 2003, 115, 171–178. [Google Scholar] [CrossRef]
- Zhang, S.S. Eliminating Pre-lithiation Step for Making High Energy Density Hybrid Li-Ion Capacitor. J. Power Sources 2017, 343, 322–328. [Google Scholar] [CrossRef]
- Aida, T.; Yamada, K.; Morita, M. An Advanced Hybrid Electrochemical Capacitor that Uses a Wide Potential Range at the Positive Electrode. Electrochem. Solid State Lett. 2006, 9, A534–A536. [Google Scholar] [CrossRef]
- Xu, N.S.; Sun, X.Z.; Zhao, F.F.; Jin, X.F.; Zhang, X.; Wang, K.; Huang, K.; Ma, Y.W. The Role of Pre-Lithiation in Activated Carbon/Li4Ti5O12 Asymmetric Capacitors. Electrochim. Acta 2017, 236, 443–450. [Google Scholar] [CrossRef]
- Kumagai, S.; Ishikawa, T.; Sawa, N. Cycle Performance of Lithium-Ion Capacitors Using Graphite Negative Electrodes at Different Pre-Lithiation Levels. J. Energy Storage 2015, 2, 1–7. [Google Scholar] [CrossRef]
- Li, J.; Guo, J.Q.; Li, P.Y.; Wang, L.G.; Huang, Y.J. Pre-lithiated Mesocarbon Microbeads Anode and Bifunctional Cathode for High Performance Hybrid Lithium-Ion Capacitors. Int. J. Electrochem. Sci. 2017, 12, 3212–3220. [Google Scholar] [CrossRef]
- Sivakkumar, S.R.; Pandolfo, A.G. Evaluation of Lithium-Ion Capacitors Assembled with Pre-Lithiated Graphite Anode and Activated Carbon Cathode. Electrochim. Acta 2012, 65, 280–287. [Google Scholar] [CrossRef]
- Zhang, S.S. A Cost-Effective Approach for Practically Viable Li-Ion Capacitors by Using Li2S as an in Situ Li-Ion Source Material. J. Mater. Chem. A 2017, 5, 14286–14293. [Google Scholar] [CrossRef]
- Jezowski, P.; Fic, K.; Crosnier, O.; Brousse, T.; Beguin, F. Lithium Rhenium(VII) Oxide as a Novel Material for Graphite Pre-Lithiation in High Performance Lithium Ion Capacitors. J. Mater. Chem. A 2016, 4, 12609–12615. [Google Scholar] [CrossRef]
- Jezowski, P.; Fic, K.; Crosnier, O.; Brousse, T.; Beguin, F. Use of Sacrificial Lithium Nickel Oxide for Loading Graphitic Anode in Li-Ion Capacitors. Electrochim. Acta 2016, 206, 440–445. [Google Scholar] [CrossRef]
- Kim, M.; Xu, F.; Lee, J.H.; Jung, C.; Hong, S.M.; Zhang, Q.M.; Koo, C.M. A Fast and Efficient Pre-Doping Approach to High Energy Density Lithium-Ion Hybrid Capacitors. J. Mater. Chem. A 2014, 2, 10029–10033. [Google Scholar] [CrossRef]
- Cao, W.J.; Zheng, J.P. Li-Ion Capacitors with Carbon Cathode and Hard Carbon/Stabilized Lithium Metal Powder Anode Electrodes. J. Power Sources 2012, 213, 180–185. [Google Scholar] [CrossRef]
- Tsuda, T.; Ando, N.; Mitsuhashi, N.; Tanabe, T.; Itagaki, K.; Soma, N.; Nakamura, S.; Hayashi, N.; Matsumoto, F. Fabrication of Porous Graphite Anodes with Pico-Second Pulse Laser and Enhancement of Pre-Doping of Li+ Ions to Laminated Graphite Anodes with Micrometre-Sized Holes Formed on the Porous Graphite Anodes. ECS Trans. 2017, 77, 1897–1903. [Google Scholar] [CrossRef]
- Tasaki, S.; Ando, N.; Nagai, M.; Shirakami, A.; Matsui, K.; Hato, Y. Lithium Ion Capacitor. U.S. Patent 7,733,629 B2, 8 June 2010. [Google Scholar]
- Tasaki, S.; Nagai, M.; Ando, N. Lithium Metal Foil for Battery or Capacitor. U.S. Patent 8,685,117 B2, 1 April 2014. [Google Scholar]
- Mizukami, M.; Nansaka, K.; Ando, N. Method for Producing Electric Storage Device, and Electric Storage Device. U.S. Patent 9,093,228 B2, 28 July 2015. [Google Scholar]
- Manthiram, A.; Fu, Y.Z.; Chung, S.H.; Zu, C.X.; Su, Y.S. Rechargeable Lithium-Sulfur Batteries. Chem. Rev. 2014, 114, 11751–11787. [Google Scholar] [CrossRef] [PubMed]
- Hagen, M.; Hanselmann, D.; Ahlbrecht, K.; Maca, R.; Gerber, D.; Tubke, J. Lithium-Sulfur Cells: The Gap between the State-of-the-Art and the Requirements for High Energy Battery Cells. Adv. Energy Mater. 2015, 5, 1401986–1401997. [Google Scholar] [CrossRef]
- Tao, T.; Lu, S.; Fan, Y.; Lei, W.; Huang, S.; Chen, Y. Anode Improvement in Rechargeable Lithium-Sulfur Batteries. Adv. Mater. 2017, 29, 1700542. [Google Scholar] [CrossRef] [PubMed]
- Ryou, M.H.; Lee, Y.M.; Lee, Y.J.; Winter, M.; Bieker, P. Mechanical Surface Modification of Lithium Metal: Towards Improved Li Metal Anode Performance by Directed Li Plating. Adv. Funct. Mater. 2015, 25, 834–841. [Google Scholar] [CrossRef]
- Shi, L.; Liu, Y.; Wang, W.; Wang, A.; Jin, Z.; Wu, F.; Yang, Y. High-Safety Lithium-Ion Sulfur Battery with Sulfurized Polyacrylonitrile Cathode, Prelithiated SiOx/C Anode and Carbonate-Based Electrolyte. J. Alloys Compd. 2017, 723, 974–982. [Google Scholar] [CrossRef]
- Agostini, M.; Hassoun, J.; Liu, J.; Jeong, M.; Nara, H.; Momma, T.; Osaka, T.; Sun, Y.-K.; Scrosati, B. A Lithium-Ion Sulfur Battery Based on a Carbon-Coated Lithium-Sulfide Cathode and an Electrodeposited Silicon-Based Anode. ACS Appl. Mater. Interfaces 2014, 6, 10924–10928. [Google Scholar] [CrossRef] [PubMed]
- Agostini, M.; Scrosati, B.; Hassoun, J. An Advanced Lithium-Ion Sulfur Battery for High Energy Storage. Adv. Energy Mater. 2015, 5, 1500481–1500487. [Google Scholar] [CrossRef]
- Fan, K.; Tian, Y.; Zhang, X.; Tan, J. Application of Stabilized Lithium Metal Powder and Hard Carbon in Anode of Lithium–Sulfur Battery. J. Electroanal. Chem. 2016, 760, 80–84. [Google Scholar] [CrossRef]
- Xu, F.; Li, X.; Xiao, F.; Xu, S.; Zhang, X.; He, P.; Zhou, H. A Battery with Sulphur Cathode and Lithiated Graphite Anode Based on Lithium Shuttle Reaction. Mater. Technol. 2016, 31, 517–520. [Google Scholar] [CrossRef]
- Chen, S.; Yu, Z.; Gordin, M.L.; Yi, R.; Song, J.; Wang, D. A Fluorinated Ether Electrolyte Enabled High Performance Prelithiated Graphite/Sulfur Batteries. ACS Appl. Mater. Interfaces 2017, 9, 6959–6966. [Google Scholar] [CrossRef] [PubMed]
- Zeng, P.; Han, Y.; Duan, X.; Jia, G.; Huang, L.; Chen, Y. A Stable Graphite Electrode in Superconcentrated LiTFSI-DME/DOL Electrolyte and its Application in Lithium-Sulfur Full Battery. Mater. Res. Bull. 2017, 95, 61–70. [Google Scholar] [CrossRef]
- Cai, W.; Zhou, J.; Li, G.; Zhang, K.; Liu, X.; Wang, C.; Zhou, H.; Zhu, Y.; Qian, Y. B,N-Co-doped Graphene Supported Sulfur for Superior Stable Li–S Half Cell and Ge–S Full Battery. ACS Appl. Mater. Interfaces 2016, 8, 27679–27687. [Google Scholar] [CrossRef] [PubMed]
- Elazari, R.; Salitra, G.; Gershinsky, G.; Garsuch, A.; Panchenko, A.; Aurbach, D. Rechargeable Lithiated Silicon–Sulfur (SLS) Battery Prototypes. Electrochem. Commun. 2012, 14, 21–24. [Google Scholar] [CrossRef]
- Hagen, M.; Quiroga-González, E.; Dörfler, S.; Fahrer, G.; Tübke, J.; Hoffmann, M.J.; Althues, H.; Speck, R.; Krampfert, M.; Kaskel, S.; et al. Studies on Preventing Li Dendrite Formation in Li–S Batteries by Using Pre-Lithiated Si Microwire Anodes. J. Power Sources 2014, 248, 1058–1066. [Google Scholar] [CrossRef]
- Lee, S.-K.; Oh, S.-M.; Park, E.; Scrosati, B.; Hassoun, J.; Park, M.-S.; Kim, Y.-J.; Kim, H.; Belharouak, I.; Sun, Y.-K. Highly Cyclable Lithium–Sulfur Batteries with a Dual-Type Sulfur Cathode and a Lithiated Si/SiOx Nanosphere Anode. Nano Lett. 2015, 15, 2863–2868. [Google Scholar] [CrossRef] [PubMed]
- Piwko, M.; Kuntze, T.; Winkler, S.; Straach, S.; Härtel, P.; Althues, H.; Kaskel, S. Hierarchical Columnar Silicon Anode Structures for High Energy Density Lithium Sulfur Batteries. J. Power Sources 2017, 351, 183–191. [Google Scholar] [CrossRef]
- Piwko, M.; Thieme, S.; Weller, C.; Althues, H.; Kaskel, S. Enabling Electrolyte Compositions for Columnar Silicon Anodes in High Energy Secondary Batteries. J. Power Sources 2017, 362, 349–357. [Google Scholar] [CrossRef]
- Hassoun, J.; Kim, J.; Lee, D.-J.; Jung, H.-G.; Lee, S.-M.; Sun, Y.-K.; Scrosati, B. A Contribution to the Progress of High Energy Batteries: A Metal-Free, Lithium-Ion, Silicon–Sulfur Battery. J. Power Sources 2012, 202, 308–313. [Google Scholar] [CrossRef]
- Yan, Y.; Yin, Y.-X.; Xin, S.; Su, J.; Guo, Y.-G.; Wan, L.-J. High-Safety Lithium-Sulfur Battery with Prelithiated Si/C Anode and Ionic Liquid Electrolyte. Electrochim. Acta 2013, 91, 58–61. [Google Scholar] [CrossRef]
- Brückner, J.; Thieme, S.; Böttger-Hiller, F.; Bauer, I.; Grossmann, H.T.; Strubel, P.; Althues, H.; Spange, S.; Kaskel, S. Carbon-Based Anodes for Lithium Sulfur Full Cells with High Cycle Stability. Adv. Funct. Mater. 2014, 24, 1284–1289. [Google Scholar] [CrossRef]
- Kang, H.-S.; Park, E.; Hwang, J.-Y.; Kim, H.; Aurbach, D.; Rosenman, A.; Sun, Y.-K. A Scaled-Up Lithium (Ion)-Sulfur Battery: Newly Faced Problems and Solutions. Adv. Mater. Technol. 2016, 1, 1600052–1600059. [Google Scholar] [CrossRef]
- Krause, A.; Dörfler, S.; Piwko, M.; Wisser, F.M.; Jaumann, T.; Ahrens, E.; Giebeler, L.; Althues, H.; Schädlich, S.; Grothe, J.; et al. High Area Capacity Lithium-Sulfur Full-cell Battery with Prelitiathed Silicon Nanowire-Carbon Anodes for Long Cycling Stability. Sci. Rep. 2016, 6, 27982–27994. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Liu, M.; Yun, Q.; Wang, X.; He, Y.-B.; Li, B.; Yang, Q.-H.; Cai, Q.; Kang, F. A Novel Lithiated Silicon-Sulfur Battery Exploiting an Optimized Solid-Like Electrolyte to Enhance Safety and Cycle Life. Small 2017, 13, 1602015–1602023. [Google Scholar] [CrossRef] [PubMed]
- Eom, K.; Lee, J.T.; Oschatz, M.; Wu, F.; Kaskel, S.; Yushin, G.; Fuller, T.F. A Stable Lithiated Silicon–Chalcogen Battery via Synergetic Chemical Coupling between Silicon and Selenium. Nat. Commun. 2017, 8, 13888–13897. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; McDowell, M.T.; Jackson, A.; Cha, J.J.; Hong, S.S.; Cui, Y. New Nanostructured Li2S/Silicon Rechargeable Battery with High Specific Energy. Nano Lett. 2010, 10, 1486–1491. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Momma, T.; Ahn, S.; Yokoshima, T.; Nara, H.; Osaka, T. On-Site Chemical Pre-Lithiation of S Cathode at Room Temperature on a 3D Nano-Structured Current Collector. J. Power Sources 2017, 366, 65–71. [Google Scholar] [CrossRef]
- Zheng, S.Y.; Chen, Y.; Xu, Y.H.; Yi, F.; Zhu, Y.J.; Liu, Y.H.; Yang, J.H.; Wang, C.S. In Situ Formed Lithium Sulfide/Microporous Carbon Cathodes for Lithium-Ion Batteries. ACS Nano 2013, 7, 10995–11003. [Google Scholar] [CrossRef] [PubMed]
- Weinberger, M.; Wohlfahrt-Mehrens, M. Novel Strategies towards the Realization of Larger Lithium Sulfur/Silicon Pouch Cells. Electrochim. Acta 2016, 191, 124–132. [Google Scholar] [CrossRef]
- Wu, Y.; Yokoshima, T.; Nara, H.; Momma, T.; Osaka, T. A Pre-Lithiation Method for Sulfur Cathode Used for Future Lithium Metal Free Full Battery. J. Power Sources 2017, 342, 537–545. [Google Scholar] [CrossRef]
- Hassoun, J.; Scrosati, B. A High-Performance Polymer Tin Sulfur Lithium Ion Battery. Angew. Chem. Int. Ed. 2010, 49, 2371–2374. [Google Scholar] [CrossRef] [PubMed]
- Hassoun, J.; Sun, Y.-K.; Scrosati, B. Rechargeable Lithium Sulfide Electrode for a Polymer Tin/Sulfur Lithium-Ion Battery. J. Power Sources 2011, 196, 343–348. [Google Scholar] [CrossRef]
- Ye, F.; Liu, M.; Zhang, X.; Li, W.; Pan, Z.; Li, H.; Zhang, S.; Zhang, Y. Prelithiation of Nanostructured Sulfur Cathode by an “On-Sheet” Solid-State Reaction. Small 2016, 12, 4966–4972. [Google Scholar] [CrossRef] [PubMed]
- Hassoun, J.; Jung, H.-G.; Lee, D.-J.; Park, J.-B.; Amine, K.; Sun, Y.-K.; Scrosati, B. A Metal-Free, Lithium-Ion Oxygen Battery: A Step Forward to Safety in Lithium-Air Batteries. Nano Lett. 2012, 12, 5775–5779. [Google Scholar] [CrossRef] [PubMed]
- Kwak, W.-J.; Shin, H.-J.; Reiter, J.; Tsiouvaras, N.; Hassoun, J.; Passerini, S.; Scrosati, B.; Sun, Y.-K. Understanding Problems of Lithiated Anodes in Lithium Oxygen Full-Cells. J. Mater. Chem. A 2016, 4, 10467–10471. [Google Scholar] [CrossRef]
- Wu, S.; Zhu, K.; Tang, J.; Liao, K.; Bai, S.; Yi, J.; Yamauchi, Y.; Ishida, M.; Zhou, H. A Long-Life Lithium Ion Oxygen Battery Based on Commercial Silicon Particles as the Anode. Energy Environ. Sci. 2016, 9, 3262–3271. [Google Scholar] [CrossRef]
- Huang, J.; Jin, Z.; Xu, Z.-L.; Qin, L.; Huang, H.; Sadighi, Z.; Yao, S.; Cui, J.; Huang, B.; Kim, J.-K. Porous RuO2 Nanosheet/CNT Electrodes for DMSO-Based Li-O2 and Li Ion O2 Batteries. Energy Storage Mater. 2017, 8, 110–118. [Google Scholar] [CrossRef]
- Elia, G.A.; Bresser, D.; Reiter, J.; Oberhumer, P.; Sun, Y.-K.; Scrosati, B.; Passerini, S.; Hassoun, J. Interphase Evolution of a Lithium-Ion/Oxygen Battery. ACS Appl. Mater. Interfaces 2015, 7, 22638–22643. [Google Scholar] [CrossRef] [PubMed]
- Hirshberg, D.; Sharon, D.; De La Llave, E.; Afri, M.; Frimer, A.A.; Kwak, W.-J.; Sun, Y.-K.; Aurbach, D. Feasibility of Full (Li-Ion)–O2 Cells Comprised of Hard Carbon Anodes. ACS Appl. Mater. Interfaces 2016, 9, 4352–4361. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Dong, X.; Wang, Y.; Xia, Y. A Lithium Air Battery with a Lithiated Al-Carbon Anode. Chem. Commun. 2015, 51, 676–678. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Zhai, D.; Lv, W.; Yang, W.; Huang, J.; Yao, S.; Cui, J.; Chong, W.-G.; Huang, J.-Q.; Kang, F.; et al. A High-Performance Lithium Ion Oxygen Battery Consisting of Li2O2 Cathode and Lithiated Aluminum Anode with Nafion Membrane for Reduced O2 Crossover. Nano Energy 2017, 40, 258–263. [Google Scholar] [CrossRef]



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
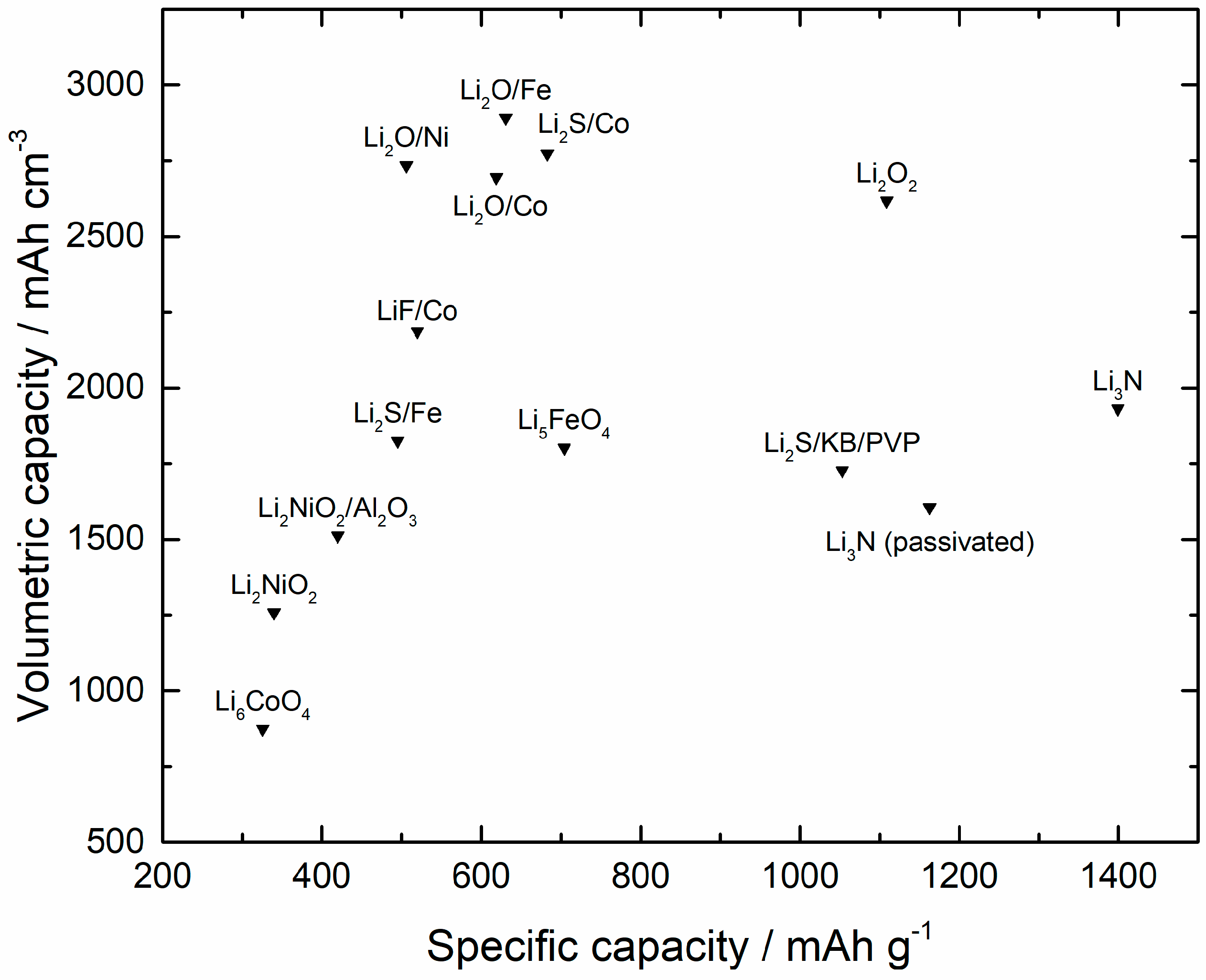{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Active Material | Gravimetric Capacity/mAh g−1 | Stability (Capacity Retention) | Reference |
|---|---|---|---|
| Li2.6Co0.4N | 760 | - | [87] |
| LixSi-Li2O (core-shell) | 1310 | 91% (1 day in dry air) 67% (5 days in dry air) 5% (6 h in ≈40% RH) | [32,105] |
| LixSi (core-shell, artificial SEI) | 2100 | 92% (5 days in dry air) 76% (6 h in 10% RH) | [102] |
| Li4.4Si@LixNySiz (core-shell) | 2808 | - | [103] |
| LixSi/Li2O (composite, based on SiO) | 2120 | 91% (5 days in dry air) 58% (6 h in ≈40% RH) | [104] |
| LixSi/Li2O (composite, based on SiO2) | 1543 | - | [104] |
| LixSn-Li2O (core-shell) | 910 | 93% (5 days in dry air) 45% (6 h in ≈40% RH) | [105] |
| LixSn/Li2O (composite, based on SnO2) | 695 | 56% (6 h in ≈40% RH) | [105] |
| LixGe-Li2O (core-shell) | 1335 | 93% (5 days in dry air) 70% (6 h in ≈40% RH) | [105] |
| LixGe/Li2O (composite, based on GeO2) | 892 | 85% (6 h in ≈40% RH) | [105] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Holtstiege, F.; Bärmann, P.; Nölle, R.; Winter, M.; Placke, T. Pre-Lithiation Strategies for Rechargeable Energy Storage Technologies: Concepts, Promises and Challenges. Batteries 2018, 4, 4. https://doi.org/10.3390/batteries4010004
Holtstiege F, Bärmann P, Nölle R, Winter M, Placke T. Pre-Lithiation Strategies for Rechargeable Energy Storage Technologies: Concepts, Promises and Challenges. Batteries. 2018; 4(1):4. https://doi.org/10.3390/batteries4010004
Chicago/Turabian StyleHoltstiege, Florian, Peer Bärmann, Roman Nölle, Martin Winter, and Tobias Placke. 2018. "Pre-Lithiation Strategies for Rechargeable Energy Storage Technologies: Concepts, Promises and Challenges" Batteries 4, no. 1: 4. https://doi.org/10.3390/batteries4010004
APA StyleHoltstiege, F., Bärmann, P., Nölle, R., Winter, M., & Placke, T. (2018). Pre-Lithiation Strategies for Rechargeable Energy Storage Technologies: Concepts, Promises and Challenges. Batteries, 4(1), 4. https://doi.org/10.3390/batteries4010004