
Designing Strain-Less Electrode Materials: Computational Analysis of Volume Variations in Li-Ion and Na-Ion Batteries


Abstract
1. Introduction
2. Computational Details
3. Results
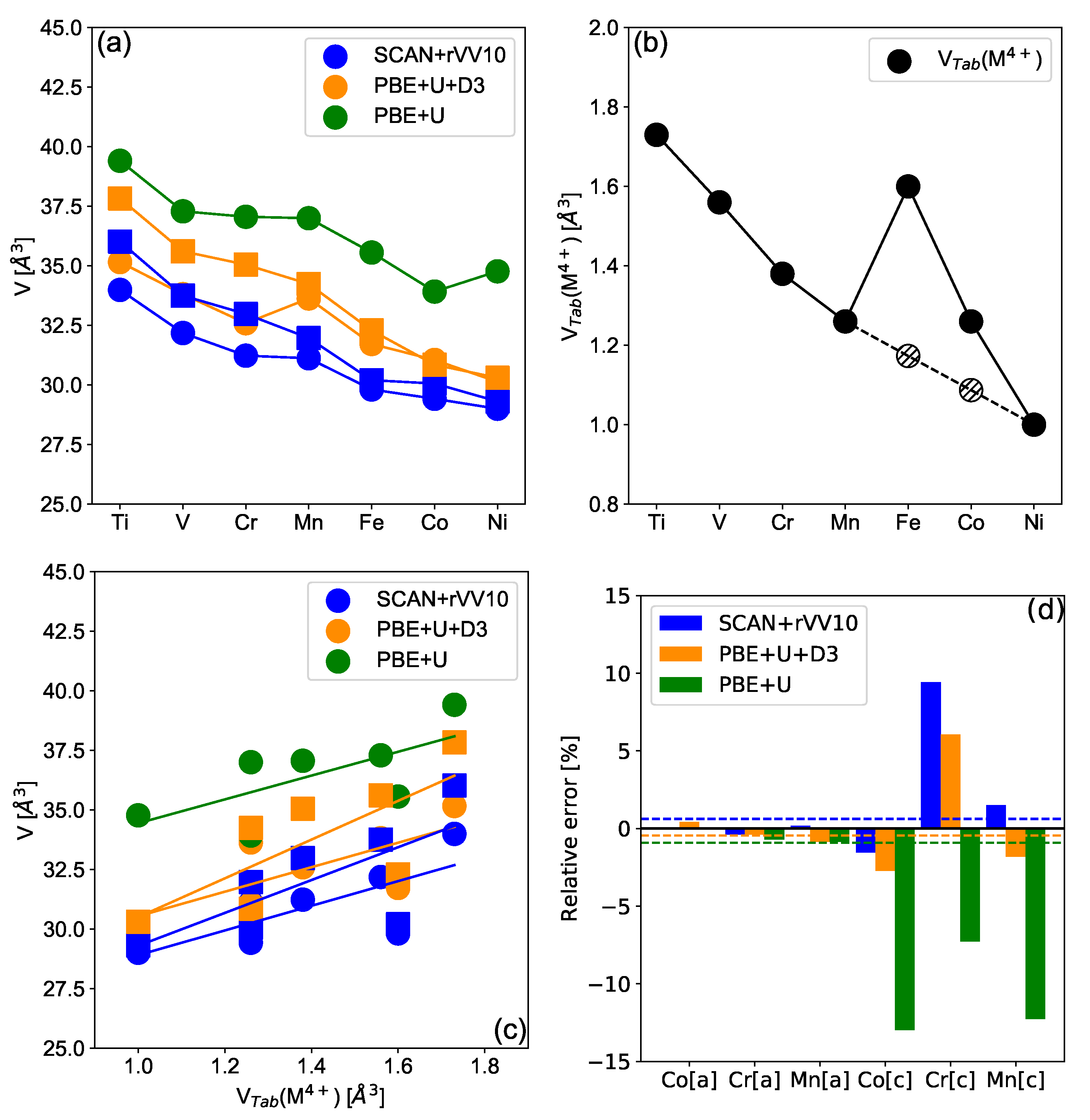
3.1. Volumes and Relation with Ionic Radii
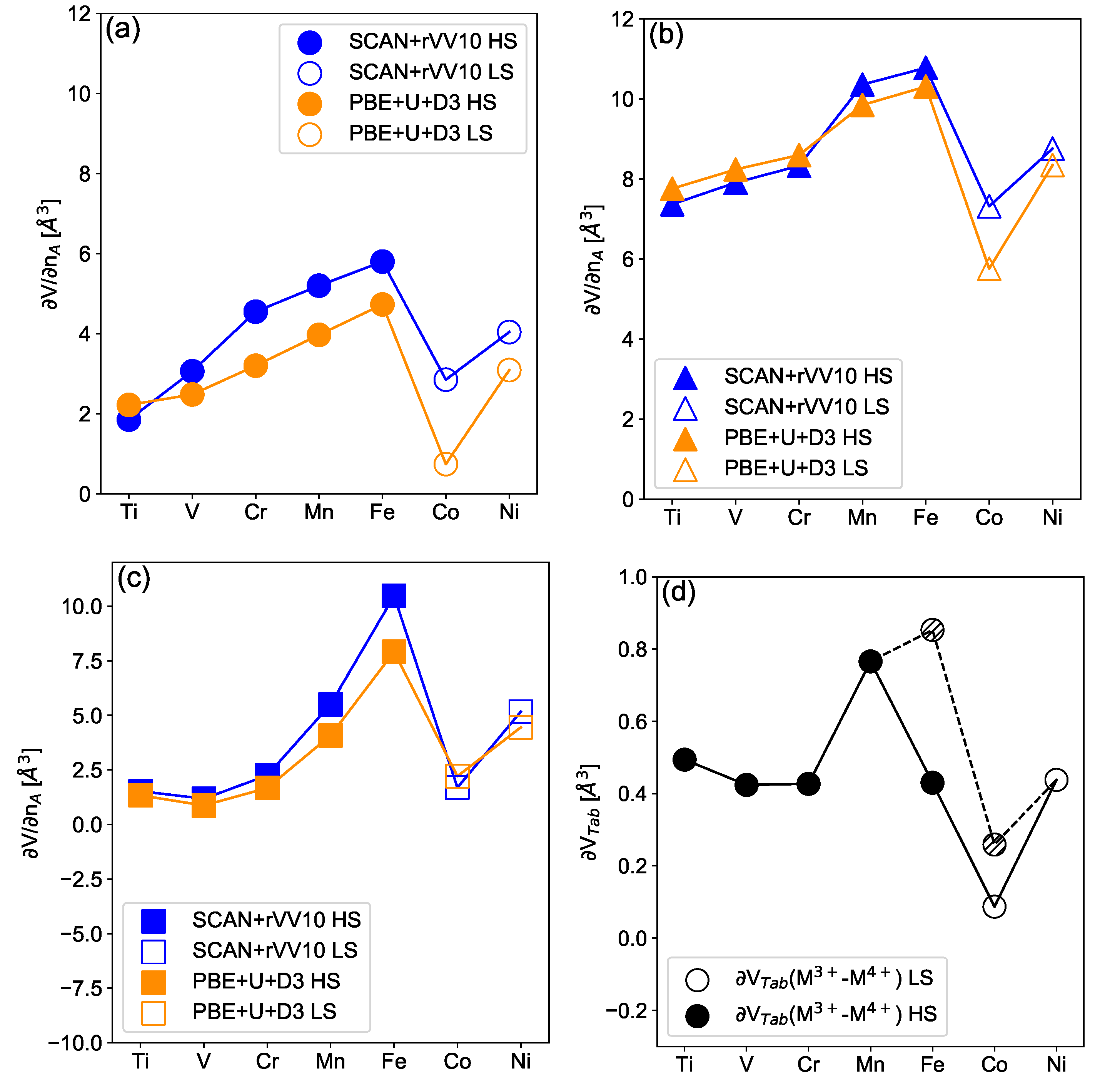
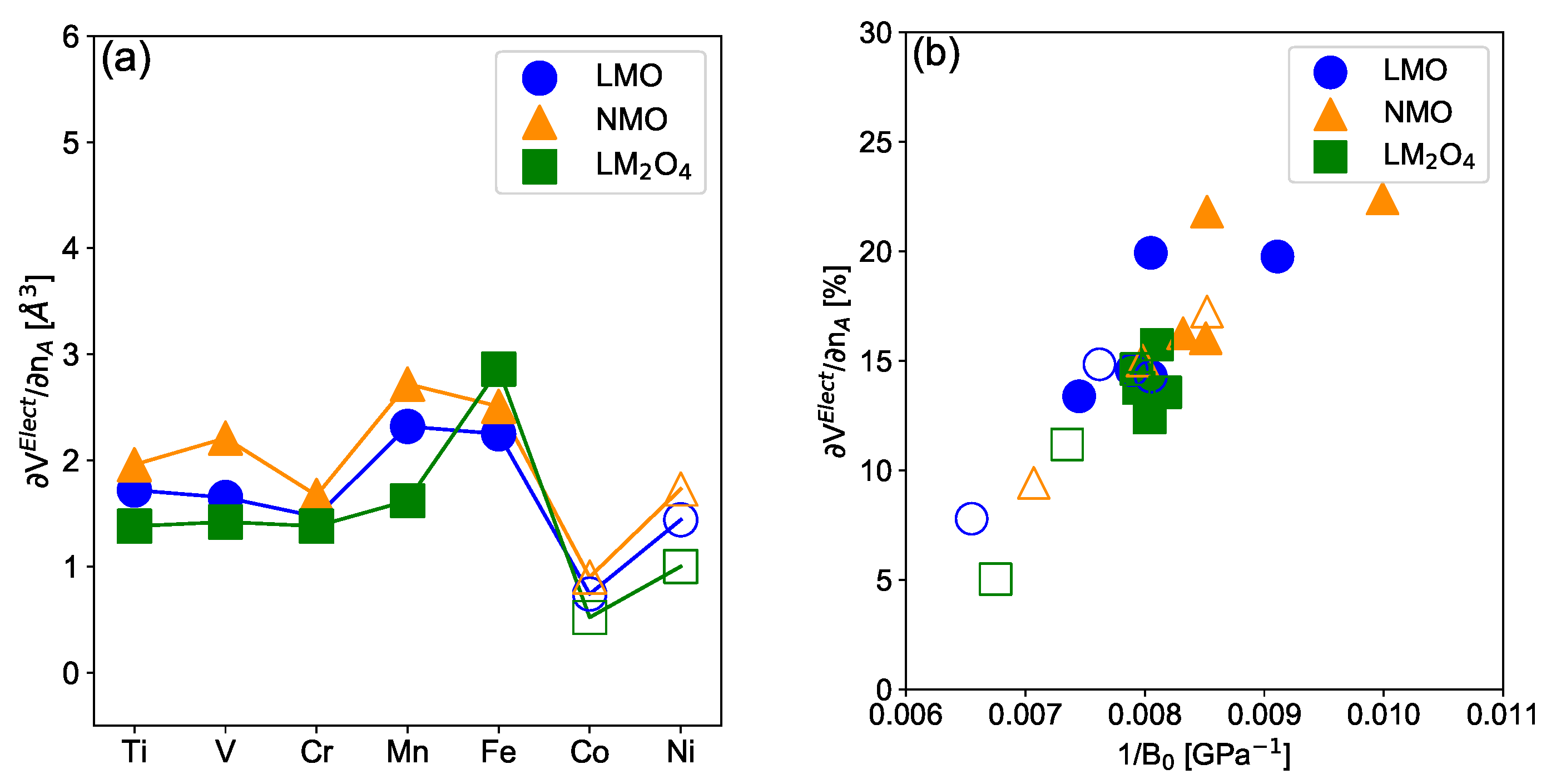
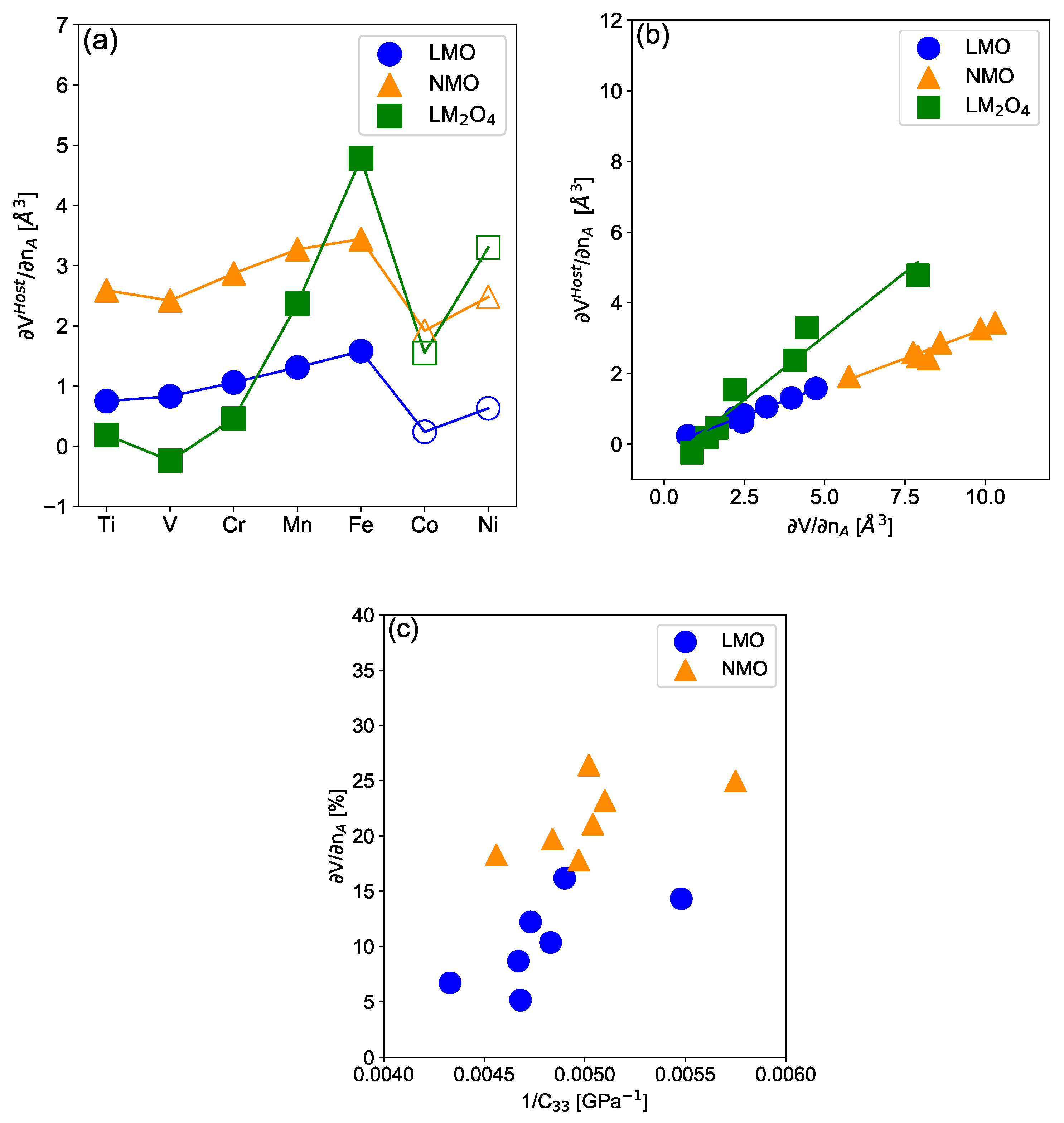
3.2. Electrochemically-Induced Volume Variation
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Berg, H. Neutron Diffraction Study of Electrochemically Delithiated LiMn2O4 Spinel. Solid State Ionics 1999, 126, 227–234. [Google Scholar] [CrossRef]
- Çapraz, Ö.Ö.; Bassett, K.L.; Gewirth, A.A.; Sottos, N.R. Electrochemical Stiffness Changes in Lithium Manganese Oxide Electrodes. Adv. Energy Mater. 2016, 7, 1601778. [Google Scholar] [CrossRef]
- Kondrakov, A.O.; Schmidt, A.; Xu, J.; Geßwein, H.; Mönig, R.; Hartmann, P.; Sommer, H.; Brezesinski, T.; Janek, J. Anisotropic Lattice Strain and Mechanical Degradation of High- and Low-Nickel NCM Cathode Materials for Li-Ion Batteries. J. Phys. Chem. C 2017, 121, 3286–3294. [Google Scholar] [CrossRef]
- Jangid, M.K.; Mukhopadhyay, A. Real-Time Monitoring of Stress Development during Electrochemical Cycling of Electrode Materials for Li-ion Batteries: Overview and Perspectives. J. Mater. Chem. A 2019, 7, 23679–23726. [Google Scholar] [CrossRef]
- Ruess, R.; Schweidler, S.; Hemmelmann, H.; Conforto, G.; Bielefeld, A.; Weber, D.A.; Sann, J.; Elm, M.T.; Janek, J. Influence of NCM Particle Cracking on Kinetics of Lithium-Ion Batteries with Liquid or Solid Electrolyte. J. Electrochem. Soc. 2020, 167, 100532. [Google Scholar] [CrossRef]
- De Biasi, L.; Schwarz, B.; Brezesinski, T.; Hartmann, P.; Janek, J.; Ehrenberg, H. Chemical, Structural, and Electronic Aspects of Formation and Degradation Behavior on Different Length Scales of Ni-Rich NCM and Li-Rich HE-NCM Cathode Materials in Li-Ion Batteries. Adv. Mater. 2019, 31, 1900985. [Google Scholar] [CrossRef] [PubMed]
- Edge, J.S.; O’Kane, S.; Prosser, R.; Kirkaldy, N.D.; Patel, A.N.; Hales, A.; Ghosh, A.; Ai, W.; Chen, J.; Yang, J.; et al. Lithium Ion Battery Degradation: What You Need to Know. Phys. Chem. Chem. Phys. 2021, 23, 8200–8221. [Google Scholar] [CrossRef]
- Stallard, J.C.; Vema, S.; Hall, D.S.; Dennis, A.R.; Penrod, M.E.; Grey, C.P.; Deshpande, V.S.; Fleck, N.A. Effect of Lithiation upon the Shear Strength of NMC811 Single Crystals. J. Electrochem. Soc. 2022, 169, 040511. [Google Scholar] [CrossRef]
- Stallard, J.C.; Wheatcroft, L.; Booth, S.G.; Boston, R.; Corr, S.A.; De Volder, M.F.; Inkson, B.J.; Fleck, N.A. Mechanical Properties of Cathode Materials for Lithium-Ion Batteries. Joule 2022, 6, 984–1007. [Google Scholar] [CrossRef]
- De Vasconcelos, L.S.; Xu, R.; Xu, Z.; Zhang, J.; Sharma, N.; Shah, S.R.; Han, J.; He, X.; Wu, X.; Sun, H.; et al. Chemomechanics of Rechargeable Batteries: Status, Theories, and Perspectives. Chem. Rev. 2022, 122, 13043–13107. [Google Scholar] [CrossRef]
- Park, S.H.; Lee, N.K.; Lee, S.G.; Han, J.H.; Lee, Y.J. Zero-Strain Cathodes for Lithium-Based Rechargeable Batteries: A Comprehensive Review. ACS Appl. Energy Mater. 2023, 6, 12–30. [Google Scholar] [CrossRef]
- Xu, Z.; Rahman, M.M.; Mu, L.; Liu, Y.; Lin, F. Chemomechanical Behaviors of Layered Cathode Materials in Alkali Metal Ion Batteries. J. Mater. Chem. A 2018, 6, 21859–21884. [Google Scholar] [CrossRef]
- Ryu, H.H.; Park, K.J.; Yoon, C.S.; Sun, Y.K. Capacity Fading of Ni-Rich Li[NixCoyMn1−x−y]O2 (0.6 ≤ x ≤ 0.95) Cathodes for High-Energy-Density Lithium-Ion Batteries: Bulk or Surface Degradation? Chem. Mater. 2018, 30, 1155–1163. [Google Scholar] [CrossRef]
- Ryu, H.H.; Namkoong, B.; Kim, J.H.; Belharouak, I.; Yoon, C.S.; Sun, Y.K. Capacity Fading Mechanisms in Ni-Rich Single-Crystal NCM Cathodes. ACS Energy Lett. 2021, 6, 2726–2734. [Google Scholar] [CrossRef]
- Koerver, R.; Aygün, I.; Leichtweiß, T.; Dietrich, C.; Zhang, W.; Binder, J.O.; Hartmann, P.; Zeier, W.G.; Janek, J. Capacity Fade in Solid-State Batteries: Interphase Formation and Chemomechanical Processes in Nickel-Rich Layered Oxide Cathodes and Lithium Thiophosphate Solid Electrolytes. Chem. Mater. 2017, 29, 5574–5582. [Google Scholar] [CrossRef]
- Koerver, R.; Zhang, W.; De Biasi, L.; Schweidler, S.; Kondrakov, A.O.; Kolling, S.; Brezesinski, T.; Hartmann, P.; Zeier, W.G.; Janek, J. Chemo-Mechanical Expansion of Lithium Electrode Materials—On the Route to Mechanically Optimized All-Solid-State Batteries. Energy Environ. Sci. 2018, 11, 2142–2158. [Google Scholar] [CrossRef]
- Shi, T.; Zhang, Y.Q.; Tu, Q.; Wang, Y.; Scott, M.C.; Ceder, G. Characterization of Mechanical Degradation in an All-Solid-State Battery Cathode. J. Mater. Chem. A 2020, 8, 17399–17404. [Google Scholar] [CrossRef]
- Doerrer, C.; Capone, I.; Narayanan, S.; Liu, J.; Grovenor, C.R.M.; Pasta, M.; Grant, P.S. High Energy Density Single-Crystal NMC/Li6PS5Cl Cathodes for All-Solid-State Lithium-Metal Batteries. ACS Appl. Mater. Interfaces 2021, 13, 37809–37815. [Google Scholar] [CrossRef] [PubMed]
- Yoon, K.; Kim, H.; Han, S.; Chan, T.S.; Ko, K.H.; Jo, S.; Park, J.; Kim, S.; Lee, S.; Noh, J.; et al. Detrimental Effect of High-Temperature Storage on Sulfide-Based All-Solid-State Batteries. Appl. Phys. Rev. 2022, 9, 031403. [Google Scholar] [CrossRef]
- Kalnaus, S.; Dudney, N.J.; Westover, A.S.; Herbert, E.; Hackney, S. Solid-State Batteries: The Critical Role of Mechanics. Science 2023, 381, eabg5998. [Google Scholar] [CrossRef]
- Noh, H.J.; Youn, S.; Yoon, C.S.; Sun, Y.K. Comparison of the Structural and Electrochemical Properties of Layered Li[NixCoyMnz]O2 (x = 1/3, 0.5, 0.6, 0.7, 0.8 and 0.85) Cathode Material for Lithium-Ion Batteries. J. Power Sources 2013, 233, 121–130. [Google Scholar] [CrossRef]
- Sun, H.H.; Choi, W.; Lee, J.K.; Oh, I.H.; Jung, H.G. Control of Electrochemical Properties of Nickel-Rich Layered Cathode Materials for Lithium Ion Batteries by Variation of the Manganese to Cobalt Ratio. J. Power Sources 2015, 275, 877–883. [Google Scholar] [CrossRef]
- Yoon, C.S.; Choi, M.H.; Lim, B.B.; Lee, E.J.; Sun, Y.K. Review—High-Capacity Li[Ni1−xCox/2Mnx/2]O2 (x = 0.1, 0.05, 0) Cathodes for Next-Generation Li-Ion Battery. J. Electrochem. Soc. 2015, 162, A2483–A2489. [Google Scholar] [CrossRef]
- Ishidzu, K.; Oka, Y.; Nakamura, T. Lattice Volume Change during Charge/Discharge Reaction and Cycle Performance of Li[NixCoyMnz]O2. Solid State Ionics 2016, 288, 176–179. [Google Scholar] [CrossRef]
- Friedrich, F.; Strehle, B.; Freiberg, A.T.S.; Kleiner, K.; Day, S.J.; Erk, C.; Piana, M.; Gasteiger, H.A. Editors’ Choice—Capacity Fading Mechanisms of NCM-811 Cathodes in Lithium-Ion Batteries Studied by X-ray Diffraction and Other Diagnostics. J. Electrochem. Soc. 2019, 166, A3760–A3774. [Google Scholar] [CrossRef]
- Yuan, K.; Li, N.; Ning, R.; Shen, C.; Hu, N.; Bai, M.; Zhang, K.; Tian, Z.; Shao, L.; Hu, Z.; et al. Stabilizing Surface Chemical and Structural Ni-rich Cathode via a Non-Destructive Surface Reinforcement Strategy. Nano Energy 2020, 78, 105239. [Google Scholar] [CrossRef]
- Liu, T.; Yu, L.; Lu, J.; Zhou, T.; Huang, X.; Cai, Z.; Dai, A.; Gim, J.; Ren, Y.; Xiao, X.; et al. Rational Design of Mechanically Robust Ni-rich Cathode Materials via Concentration Gradient Strategy. Nat. Commun. 2021, 12, 6024. [Google Scholar] [CrossRef] [PubMed]
- Park, N.Y.; Ryu, H.H.; Kuo, L.Y.; Kaghazchi, P.; Yoon, C.S.; Sun, Y.K. High-Energy Cathodes via Precision Microstructure Tailoring for Next-Generation Electric Vehicles. ACS Energy Lett. 2021, 6, 4195–4202. [Google Scholar] [CrossRef]
- Lv, H.; Li, C.; Zhao, Z.; Wu, B.; Mu, D. A Review: Modification Strategies of Nickel-Rich Layer Structure Cathode (Ni ≥ 0.8) Materials for Lithium Ion Power Batteries. J. Energy Chem. 2021, 60, 435–450. [Google Scholar] [CrossRef]
- Lee, J.; Urban, A.; Li, X.; Su, D.; Hautier, G.; Ceder, G. Unlocking the Potential of Cation-Disordered Oxides for Rechargeable Lithium Batteries. Science 2014, 343, 519–522. [Google Scholar] [CrossRef]
- Yabuuchi, N.; Nakayama, M.; Takeuchi, M.; Komaba, S.; Hashimoto, Y.; Mukai, T.; Shiiba, H.; Sato, K.; Kobayashi, Y.; Nakao, A.; et al. Origin of Stabilization and Destabilization in Solid-State Redox Reaction of Oxide Ions for Lithium-Ion Batteries. Nat. Commun. 2016, 7, 13814. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, M.; Yabuuchi, N. Lithium-Excess Cation-Disordered Rocksalt-Type Oxide with Nanoscale Phase Segregation: Li1.25Nb0.25V0.5O2. Chem. Mater. 2017, 29, 6927–6935. [Google Scholar] [CrossRef]
- Zhao, X.; Tian, Y.; Lun, Z.; Cai, Z.; Chen, T.; Ouyang, B.; Ceder, G. Design Principles for Zero-Strain Li-ion Cathodes. Joule 2022, 6, 1654–1671. [Google Scholar] [CrossRef]
- Cho, J.; Kim, Y.J.; Kim, T.J.; Park, B. Zero-Strain Intercalation Cathode for Rechargeable Li-Ion Cell. Angew. Chem. 2001, 113, 3471–3473. [Google Scholar] [CrossRef]
- Kim, J.H.; Kim, H.; Choi, W.; Park, M.S. Bifunctional Surface Coating of LiNbO3 on High-Ni Layered Cathode Materials for Lithium-Ion Batteries. ACS Appl. Mater. Interfaces 2020, 12, 35098–35104. [Google Scholar] [CrossRef]
- Nguyen, A.; Zuo, P.; Jiang, H.; Wang, C.; Wang, D. Dual Protective Mechanism of AlPO4 Coating on High-Nickel Cathode Material for High Energy Density and Long Cycle Life Lithium-Ion Batteries. J. Electrochem. Soc. 2022, 169, 050523. [Google Scholar] [CrossRef]
- Mariyappan, S.; Marchandier, T.; Rabuel, F.; Iadecola, A.; Rousse, G.; Morozov, A.V.; Abakumov, A.M.; Tarascon, J.M. The Role of Divalent (Zn2+/Mg2+/Cu2+) Substituents in Achieving Full Capacity of Sodium Layered Oxides for Na-Ion Battery Applications. Chem. Mater. 2020, 32, 1657–1666. [Google Scholar] [CrossRef]
- Zhang, X.; Qiu, F.; Jiang, K.; He, P.; Han, M.; Guo, S.; Zhou, H. Improving the Structural and Cyclic Stabilities of P2-type Na 0.67 MnO 2 Cathode Material via Cu and Ti Co-Substitution for Sodium Ion Batteries. Chem. Commun. 2020, 56, 6293–6296. [Google Scholar] [CrossRef]
- Xin, F.; Zhou, H.; Zong, Y.; Zuba, M.; Chen, Y.; Chernova, N.A.; Bai, J.; Pei, B.; Goel, A.; Rana, J.; et al. What Is the Role of Nb in Nickel-Rich Layered Oxide Cathodes for Lithium-Ion Batteries? ACS Energy Lett. 2021, 6, 1377–1382. [Google Scholar] [CrossRef]
- Kong, W.; Zhang, J.; Wong, D.; Yang, W.; Yang, J.; Schulz, C.; Liu, X. Tailoring Co3d and O2p Band Centers to Inhibit Oxygen Escape for Stable 4.6 V LiCoO2 Cathodes. Angew. Chem. Int. Ed. 2021, 60, 27102–27112. [Google Scholar] [CrossRef]
- Ou, X.; Liu, T.; Zhong, W.; Fan, X.; Guo, X.; Huang, X.; Cao, L.; Hu, J.; Zhang, B.; Chu, Y.S.; et al. Enabling High Energy Lithium Metal Batteries via Single-Crystal Ni-rich Cathode Material Co-Doping Strategy. Nat. Commun. 2022, 13, 2319. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of Ab-Initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector Augmented-Wave Method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special Points for Brillouin-zone Integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Sun, J.; Ruzsinszky, A.; Perdew, J.P. Strongly Constrained and Appropriately Normed Semilocal Density Functional. Phys. Rev. Lett. 2015, 115, 036402. [Google Scholar] [CrossRef]
- Sun, J.; Remsing, R.C.; Zhang, Y.; Sun, Z.; Ruzsinszky, A.; Peng, H.; Yang, Z.; Paul, A.; Waghmare, U.; Wu, X.; et al. Accurate First-Principles Structures and Energies of Diversely Bonded Systems from an Efficient Density Functional. Nat. Chem. 2016, 8, 831–836. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Sun, J.; Martin, R.M.; Delley, B. Semilocal Density Functionals and Constraint Satisfaction. Int. J .Quantum. Chem. 2016, 116, 847–851. [Google Scholar] [CrossRef]
- Zhang, Y.; Kitchaev, D.A.; Yang, J.; Chen, T.; Dacek, S.T.; Sarmiento-Pérez, R.A.; Marques, M.A.L.; Peng, H.; Ceder, G.; Perdew, J.P.; et al. Efficient First-Principles Prediction of Solid Stability: Towards Chemical Accuracy. NPJ Comput. Mater 2018, 4, 9. [Google Scholar] [CrossRef]
- Vydrov, O.A.; Van Voorhis, T. Nonlocal van Der Waals Density Functional: The Simpler the Better. J. Chem. Phys. 2010, 133, 244103. [Google Scholar] [CrossRef]
- Dudarev, S.L.; Botton, G.A.; Savrasov, S.Y.; Humphreys, C.J.; Sutton, A.P. Electron-Energy-Loss Spectra and the Structural Stability of Nickel Oxide: An LSDA+U Study. Phys. Rev. B 1998, 57, 1505–1509. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Fonseca Guerra, C.; Handgraaf, J.W.; Baerends, E.J.; Bickelhaupt, F.M. Voronoi Deformation Density (VDD) Charges: Assessment of the Mulliken, Bader, Hirshfeld, Weinhold, and VDD Methods for Charge Analysis. J. Comput. Chem. 2004, 25, 189–210. [Google Scholar] [CrossRef]
- Tabuchi, M.; Tsutsui, S.; Masquelier, C.; Kanno, R.; Ado, K.; Matsubara, I.; Nasu, S.; Kageyama, H. Effect of Cation Arrangement on the Magnetic Properties of Lithium Ferrites (LiFeO2) Prepared by Hydrothermal Reaction and Post-annealing Method. J. Solid State Chem. 1998, 140, 159–167. [Google Scholar] [CrossRef]
- Amatucci, G.G.; Tarascon, J.M.; Klein, L.C. CoO2, the End Member of the Lix CoO2 Solid Solution. J. Electrochem. Soc. 1996, 143, 1114. [Google Scholar] [CrossRef]
- Bo, S.H.; Li, X.; Toumar, A.J.; Ceder, G. Layered-to-Rock-Salt Transformation in Desodiated NaxCrO2 (x 0.4). Chem. Mater. 2016, 28, 1419–1429. [Google Scholar] [CrossRef]
- Bruce, P.G.; Armstrong, A.R.; Gitzendanner, R.L. New Intercalation Compounds for Lithium Batteries: Layered LiMnO2. J. Mater. Chem. 1999, 9, 193–198. [Google Scholar] [CrossRef]
- Wang, X.; Li, Y. Synthesis and Formation Mechanism of Manganese Dioxide Nanowires/Nanorods. Chem. Eur. J. 2003, 9, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Sabitov, I.K. Algebraic Methods for Solution of Polyhedra. Russ. Math. Surv. 2011, 66, 445–505. [Google Scholar] [CrossRef]
- Pearce, P.E.; Perez, A.J.; Rousse, G.; Saubanère, M.; Batuk, D.; Foix, D.; McCalla, E.; Abakumov, A.M.; Van Tendeloo, G.; Doublet, M.L.; et al. Evidence for Anionic Redox Activity in a Tridimensional-Ordered Li-rich Positive Electrode β-Li2IrO3. Nat. Mater 2017, 16, 580–586. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| LiMeO (+III) | Ti (d) | V (d) | Cr (d) | Mn (d) | Fe (d) | Co (d) | Ni (d) |
|---|---|---|---|---|---|---|---|
| HS (T2g/Eg) |  |  | 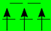 |  |  |  | 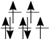 |
| BS (T2g/Eg) | 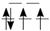 |  | 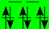 | 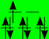 | |||
| MeO (+IV) | Ti (d) | V (d) | Cr (d) | Mn (d) | Fe (d) | Co (d) | Ni (d) |
| HS (T2g/Eg) |  |  |  | 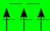 |  |  | 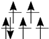 |
| BS (T2g/Eg) |  | 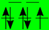 |  |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maréchal, M.; Berthelot, R.; Rozier, P.; Saubanère, M. Designing Strain-Less Electrode Materials: Computational Analysis of Volume Variations in Li-Ion and Na-Ion Batteries. Batteries 2024, 10, 262. https://doi.org/10.3390/batteries10080262
Maréchal M, Berthelot R, Rozier P, Saubanère M. Designing Strain-Less Electrode Materials: Computational Analysis of Volume Variations in Li-Ion and Na-Ion Batteries. Batteries. 2024; 10(8):262. https://doi.org/10.3390/batteries10080262
Chicago/Turabian StyleMaréchal, Maxime, Romain Berthelot, Patrick Rozier, and Matthieu Saubanère. 2024. "Designing Strain-Less Electrode Materials: Computational Analysis of Volume Variations in Li-Ion and Na-Ion Batteries" Batteries 10, no. 8: 262. https://doi.org/10.3390/batteries10080262
APA StyleMaréchal, M., Berthelot, R., Rozier, P., & Saubanère, M. (2024). Designing Strain-Less Electrode Materials: Computational Analysis of Volume Variations in Li-Ion and Na-Ion Batteries. Batteries, 10(8), 262. https://doi.org/10.3390/batteries10080262