
Binuclear Nickel Complexes of a New Di(hydroxyphenyl)imidazolate

 , ,
, ,
Abstract
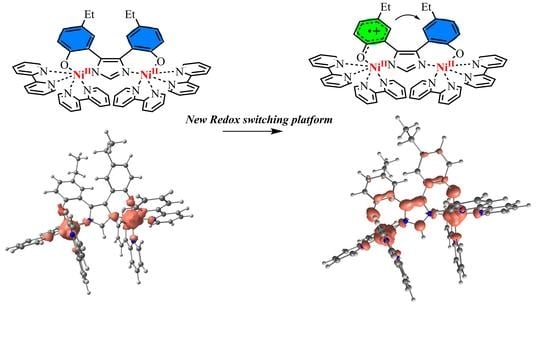
1. Introduction
2. Results and Discussion
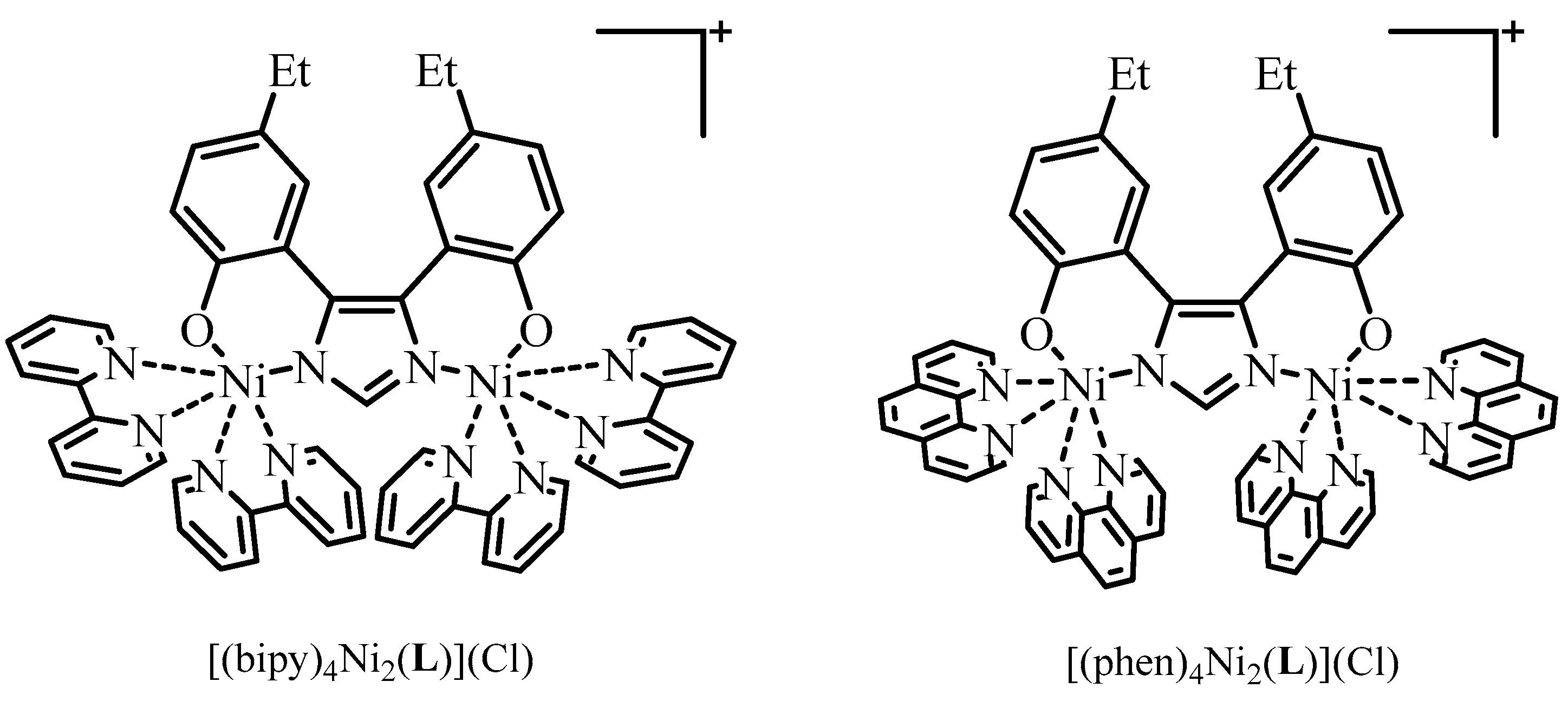
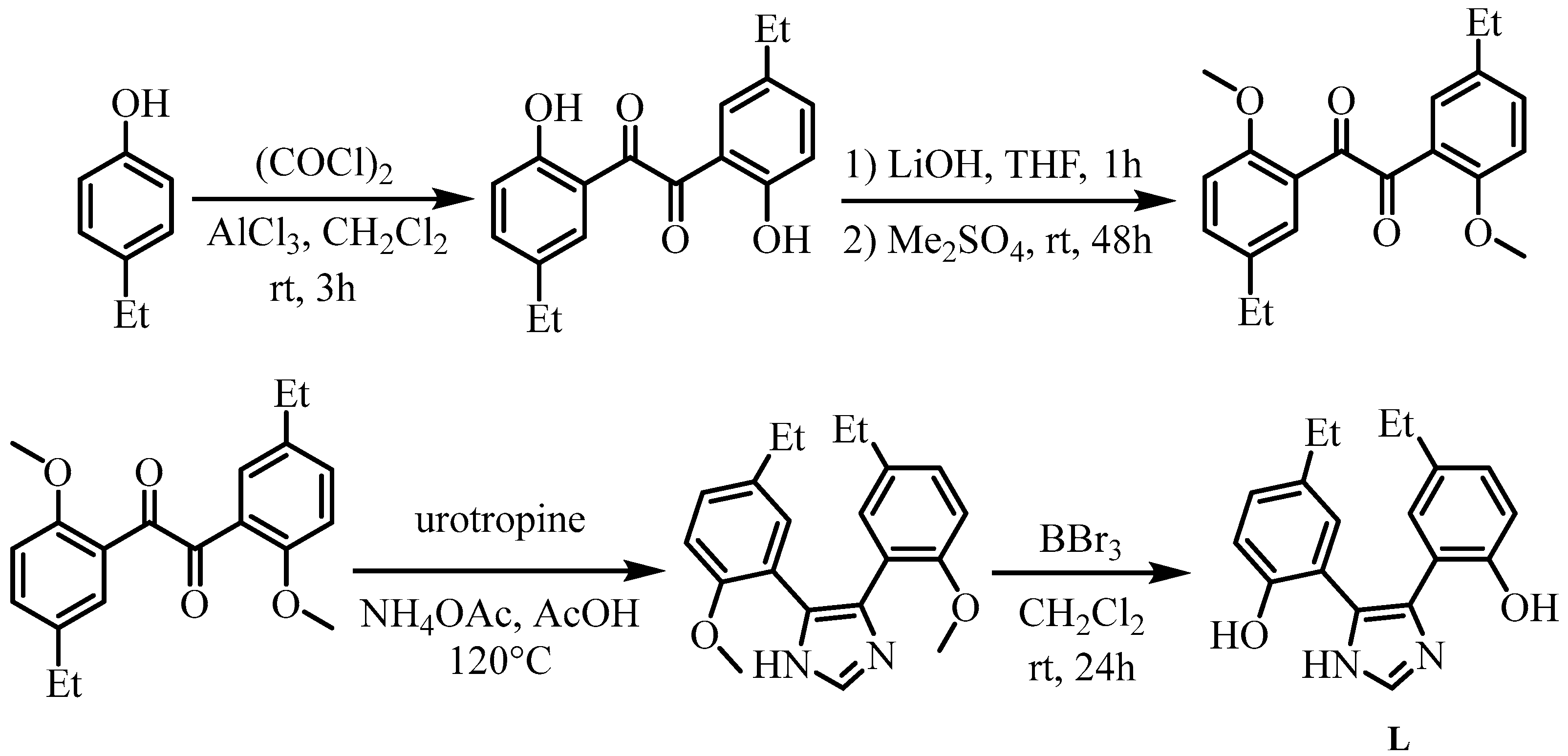
2.1. Synthesis
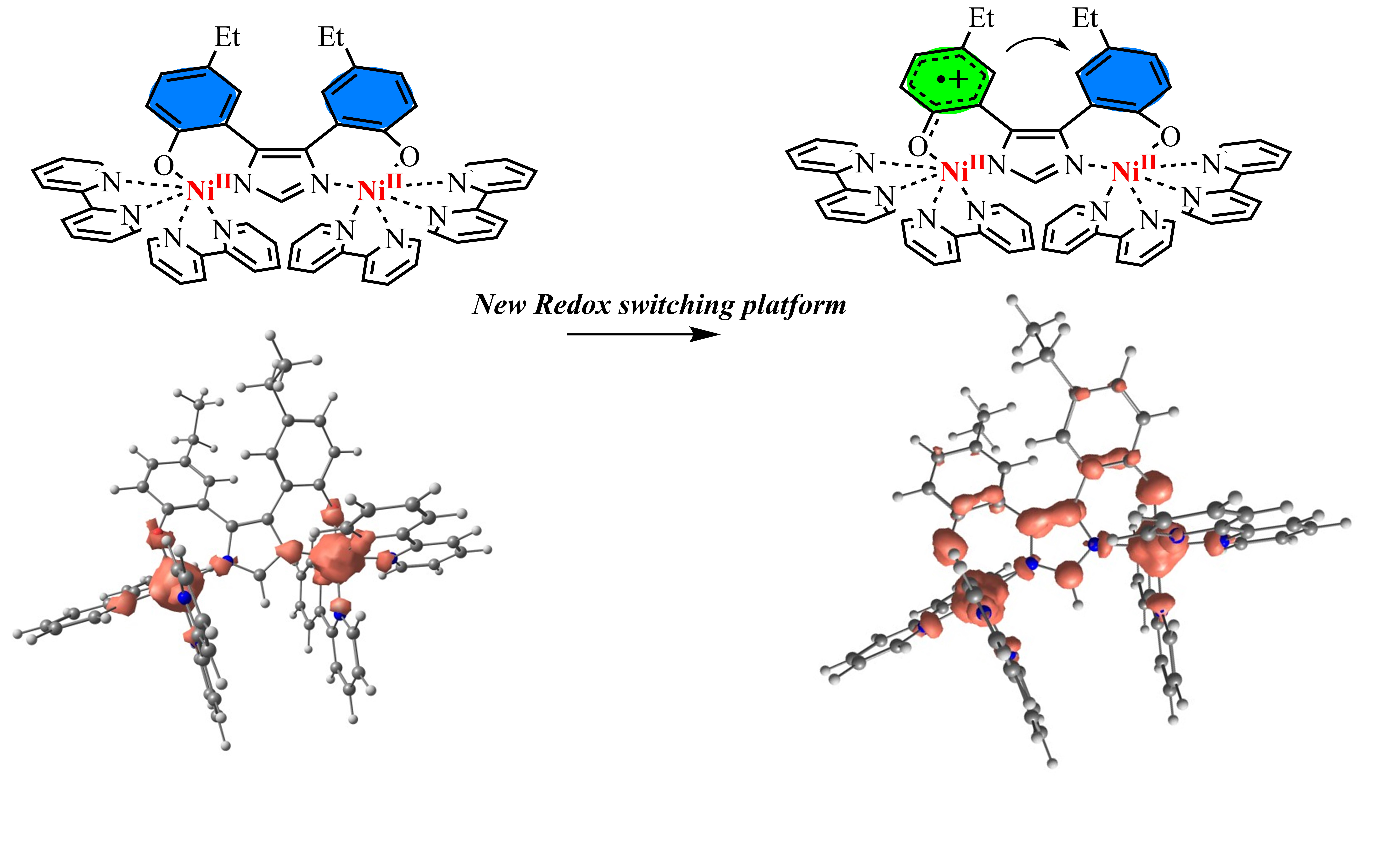
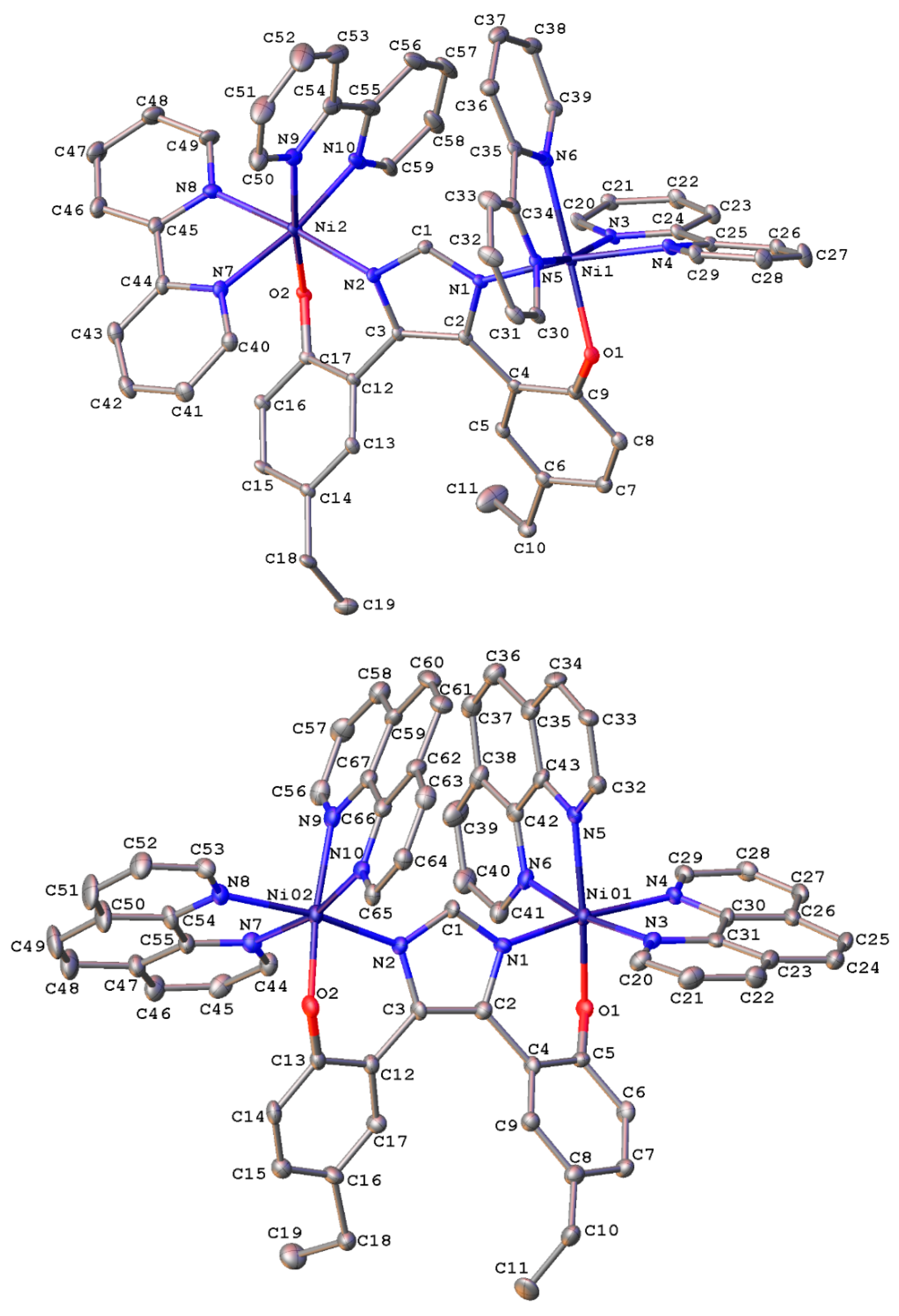
2.2. X-ray Crystallography
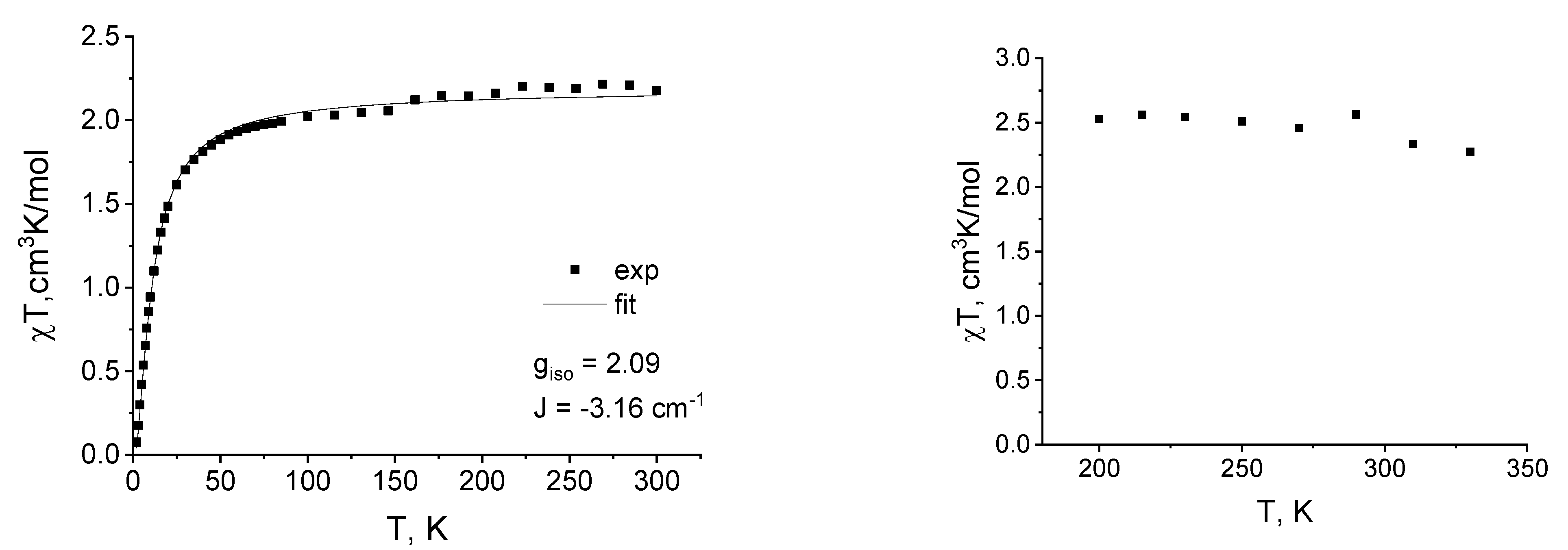
2.3. Magnetic Susceptibility Measurements
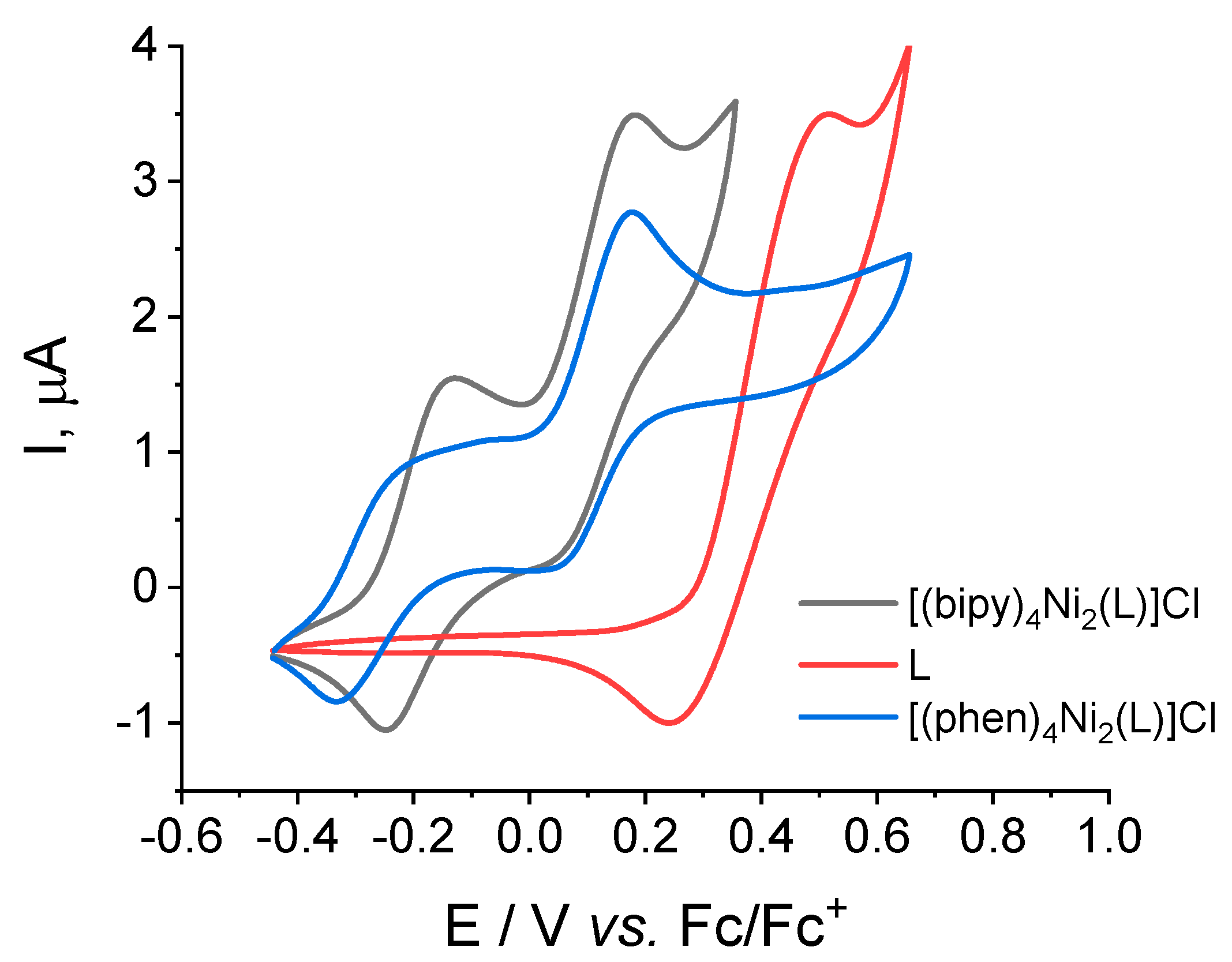
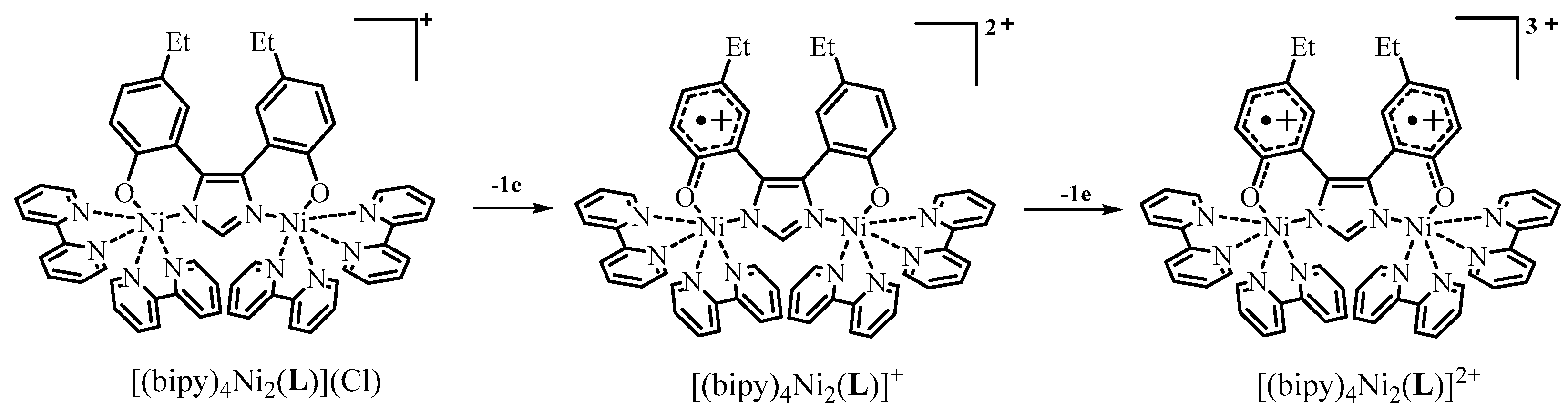
2.4. Redox Properties
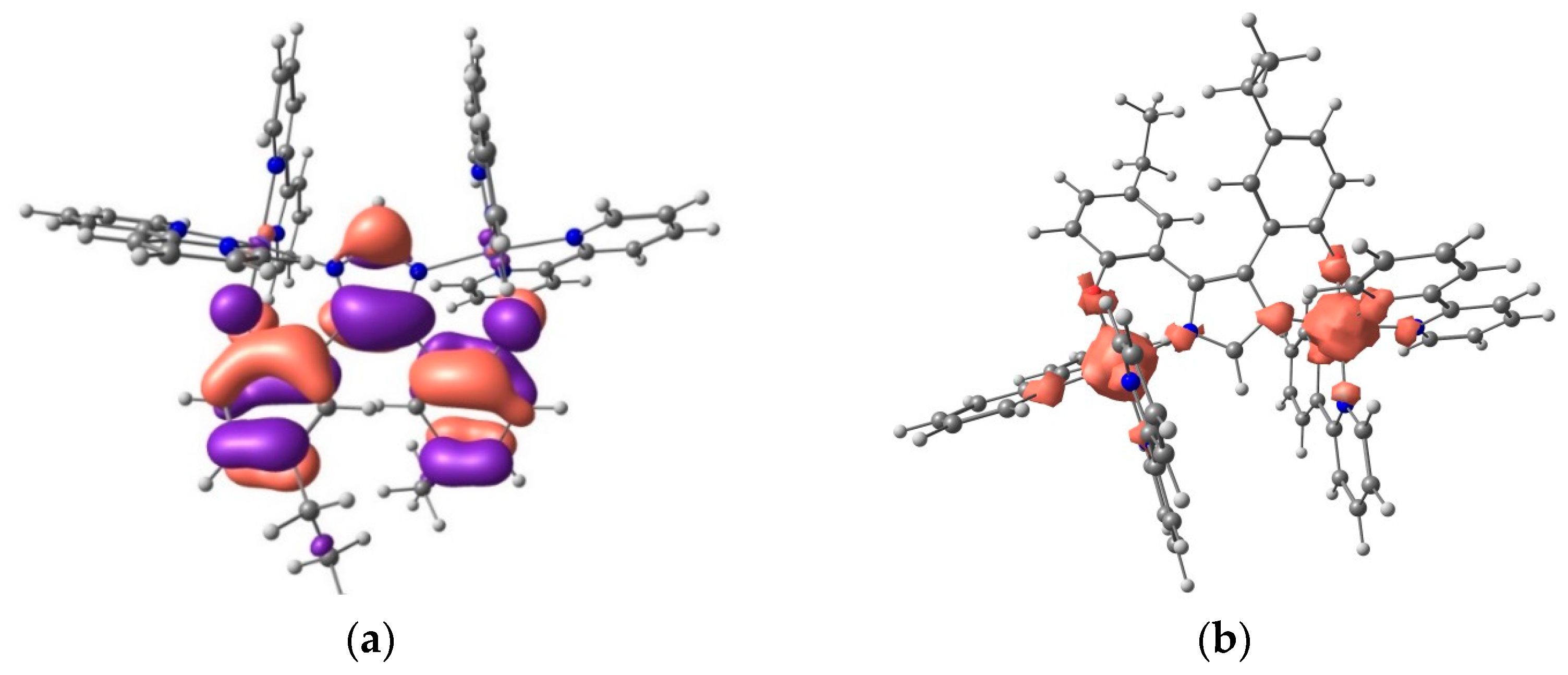
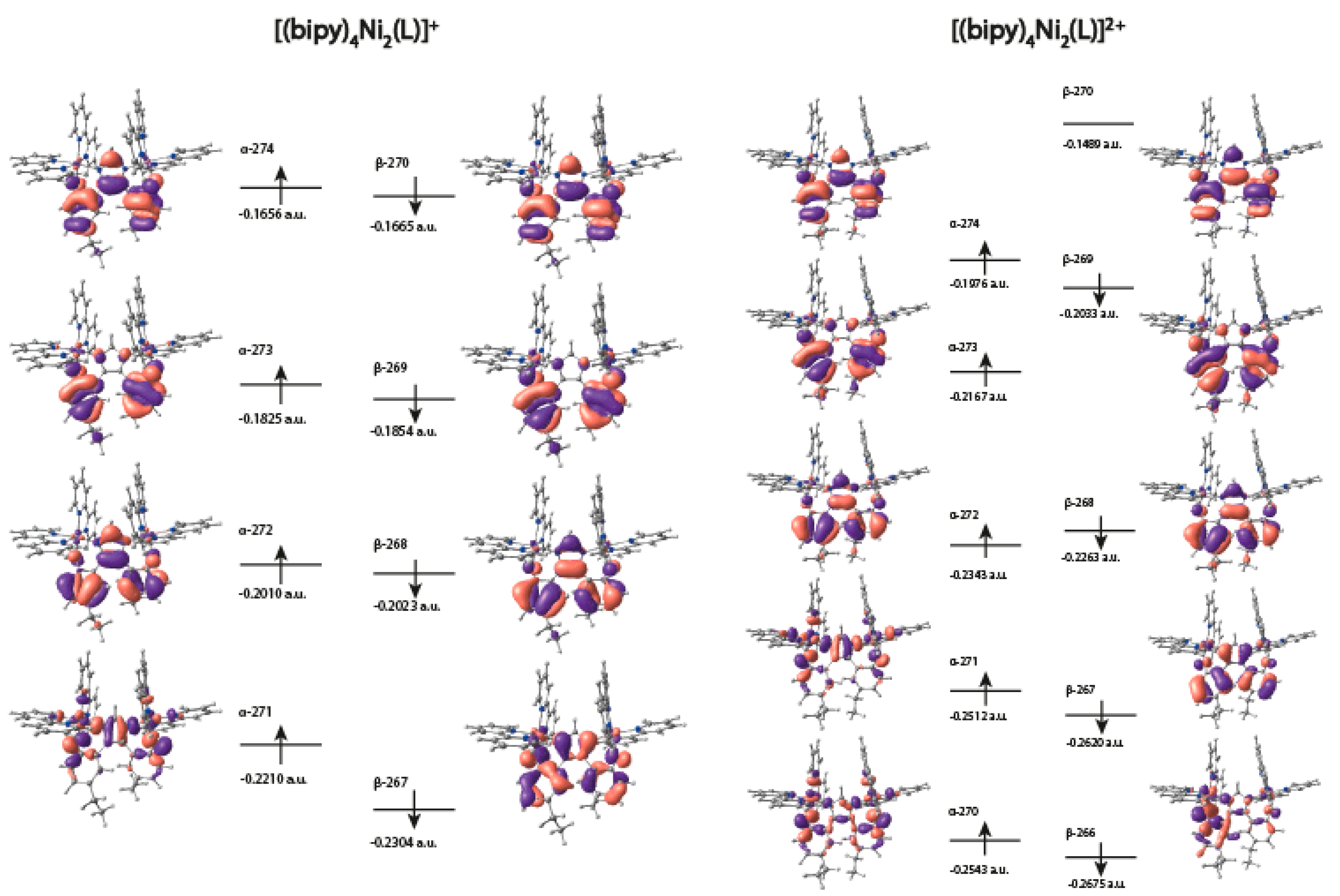
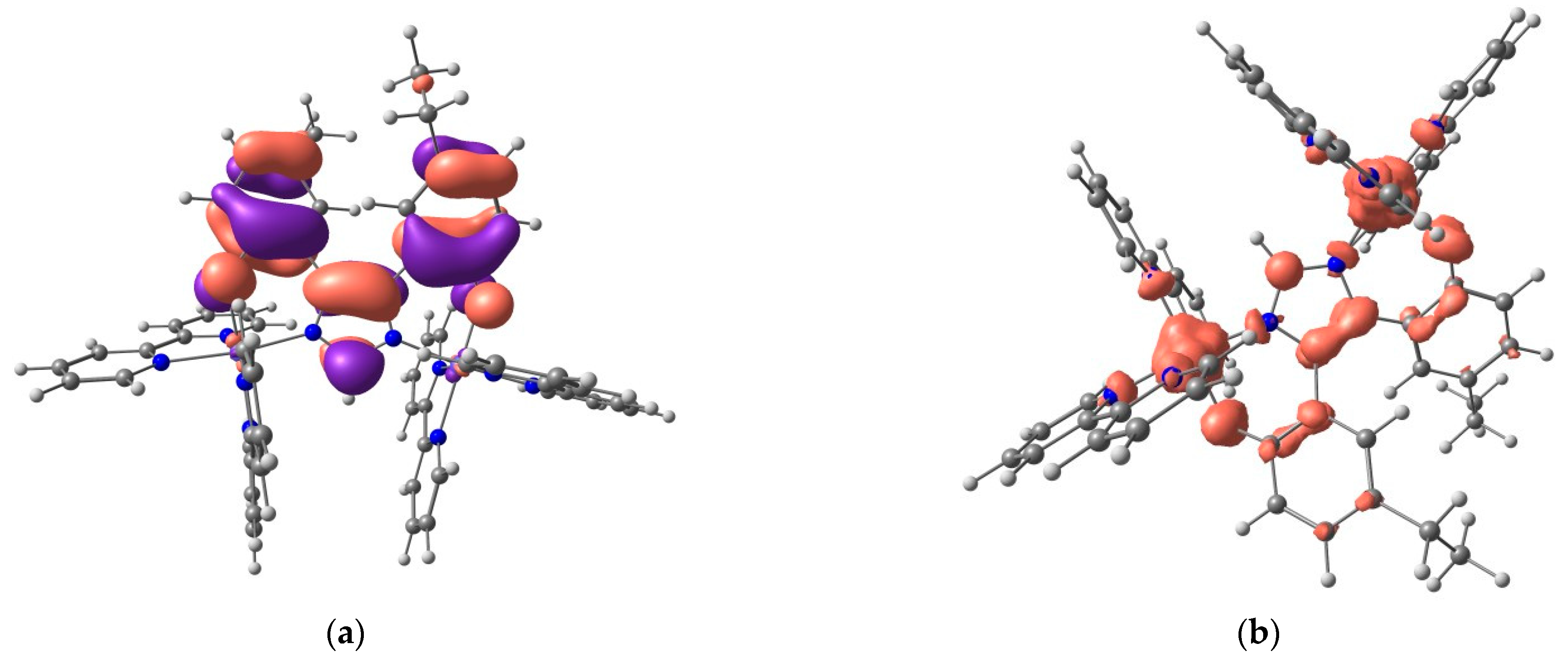
2.5. DFT Calculations
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Senthil Kumar, K.; Ruben, M. Emerging Trends in Spin Crossover (SCO) Based Functional Materials and Devices. Coord. Chem. Rev. 2017, 346, 176–205. [Google Scholar] [CrossRef]
- Nikovskiy, I.; Polezhaev, A.; Novikov, V.; Aleshin, D.; Pavlov, A.; Saffiulina, E.; Aysin, R.; Dorovatovskii, P.; Nodaraki, L.; Tuna, F.; et al. Towards the Molecular Design of Spin-Crossover Complexes of 2,6-Bis(Pyrazol-3-Yl)Pyridines. Chem. —Eur. J. 2020, 26, 5629–5638. [Google Scholar] [CrossRef] [PubMed]
- Tezgerevska, T.; Alley, K.G.; Boskovic, C. Valence Tautomerism in Metal Complexes: Stimulated and Reversible Intramolecular Electron Transfer between Metal Centers and Organic Ligands. Coord. Chem. Rev. 2014, 268, 23–40. [Google Scholar] [CrossRef]
- Palii, A.; Aldoshin, S.; Tsukerblat, B. Mixed-Valence Clusters: Prospects for Single-Molecule Magnetoelectrics. Coord. Chem. Rev. 2021, 426, 213555. [Google Scholar] [CrossRef]
- Poneti, G.; Mannini, M.; Sorace, L.; Sainctavit, P.; Arrio, M.-A.; Otero, E.; Criginski Cezar, J.; Dei, A. Soft-X-ray-Induced Redox Isomerism in a Cobalt Dioxolene Complex. Angew. Chem. Int. Ed. 2010, 49, 1954–1957. [Google Scholar] [CrossRef]
- Gransbury, G.K.; Livesay, B.N.; Janetzki, J.T.; Hay, M.A.; Gable, R.W.; Shores, M.P.; Starikova, A.; Boskovic, C. Understanding the Origin of One- or Two-Step Valence Tautomeric Transitions in Bis(Dioxolene)-Bridged Dinuclear Cobalt Complexes. J. Am. Chem. Soc. 2020, 142, 10692–10704. [Google Scholar] [CrossRef]
- Rajput, A.; Sharma, A.K.; Barman, S.K.; Saha, A.; Mukherjee, R. Valence Tautomerism and Delocalization in Transition Metal Complexes of O-Amidophenolates and Other Redox-Active Ligands. Some Recent Results. Coord. Chem. Rev. 2020, 414, 213240. [Google Scholar] [CrossRef]
- Ohtsu, H.; Tanaka, K. Chemical Control of Valence Tautomerism of Nickel(II) Semiquinone and Nickel(III) Catecholate States. Angew. Chem. Int. Ed. 2004, 43, 6301–6303. [Google Scholar] [CrossRef]
- Evangelio, E.; Ruiz-Molina, D. Valence Tautomerism: New Challenges for Electroactive Ligands. Eur. J. Inorg. Chem. 2005, 2005, 2957–2971. [Google Scholar] [CrossRef]
- Thomas, F. Ligand-Centred Oxidative Chemistry in Sterically Hindered Salen Complexes: An Interesting Case with Nickel. Dalton Trans. 2016, 45, 10866–10877. [Google Scholar] [CrossRef]
- Kurahashi, T. Drastic Redox Shift and Electronic Structural Changes of a Manganese(III)-Salen Oxidation Catalyst upon Reaction with Hydroxide and Cyanide Ion. Inorg. Chem. 2018, 57, 1066–1078. [Google Scholar] [CrossRef] [PubMed]
- Glaser, T.; Heidemeier, M.; Fröhlich, R.; Hildebrandt, P.; Bothe, E.; Bill, E. Trinuclear Nickel Complexes with Triplesalen Ligands: Simultaneous Occurrence of Mixed Valence and Valence Tautomerism in the Oxidized Species. Inorg. Chem. 2005, 44, 5467–5482. [Google Scholar] [CrossRef] [PubMed]
- Ito, N.; Phillips, S.E.V.; Stevens, C.; Ogel, Z.B.; McPherson, M.J.; Keen, J.N.; Yadav, K.D.S.; Knowles, P.F. Novel Thioether Bond Revealed by a 1.7 Å Crystal Structure of Galactose Oxidase. Nature 1991, 350, 87–90. [Google Scholar] [CrossRef]
- Whittaker, M.M.; Kersten, P.J.; Nakamura, N.; Sanders-Loehr, J.; Schweizer, E.S.; Whittaker, J.W. Glyoxal Oxidase from Phanerochaete Chrysosporium Is a New Radical-Copper Oxidase (∗). J. Biol. Chem. 1996, 271, 681–687. [Google Scholar] [CrossRef] [PubMed]
- Launay, J.-P. Electron Transfer in Molecular Binuclear Complexes and Relation with Electron Transport through Nanojunctions. Coord. Chem. Rev. 2013, 257, 1544–1554. [Google Scholar] [CrossRef]
- Inagaki, A.; Yatsuda, S.; Edure, S.; Suzuki, A.; Takahashi, T.; Akita, M. Synthesis of Pd Complexes Combined with Photosensitizing of a Ruthenium(II) Polypyridyl Moiety through a Series of Substituted Bipyrimidine Bridges. Substituent Effect of the Bridging Ligand on the Photocatalytic Dimerization of α-Methylstyrene. Inorg. Chem. 2007, 46, 2432–2445. [Google Scholar] [CrossRef]
- Pourrieux, G.; Fagalde, F.; Katz, N.E.; Parella, T.; Benet-Buchholz, J.; Llobet, A. Synthesis, Spectroscopic and Electrochemical Characterization and Molecular Structure of Polypyridyl Ruthenium Complexes Containing 4,4′-Azobis(Pyridine). Polyhedron 2008, 27, 2990–2996. [Google Scholar] [CrossRef]
- Richardson, C.; Steel, P.J.; D’Alessandro, D.M.; Junk, P.C.; Keene, F.R. Mono- and Di-Nuclear Complexes of the Ligands 3,4-Di(2-Pyridyl)-1,2,5-Oxadiazole and 3,4-Di(2-Pyridyl)-1,2,5-Thiadiazole; New Bridges Allowing Unusually Strong Metal–Metal Interactions. J. Chem. Soc. Dalton Trans. 2002, 2775–2785. [Google Scholar] [CrossRef]
- Slater, J.W.; D’Alessandro, D.M.; Keene, F.R.; Steel, P.J. Metal–Metal Interactions in Dinuclear Ruthenium Complexes Containing Bridging 4,5-Di(2-Pyridyl)Imidazolates and Related Ligands. Dalton Trans. 2006, 1954–1962. [Google Scholar] [CrossRef]
- Rego, R.; Adriaensens, P.J.; Carleer, R.A.; Gelan, J.M. Fully Quantitative Carbon-13 NMR Characterization of Resol Phenol–Formaldehyde Prepolymer Resins. Polymer 2004, 45, 33–38. [Google Scholar] [CrossRef]
- Ebel, K.; Koehler, H.; Gamer, A.O.; Jäckh, R. Imidazole and Derivatives. In Ullmann’s Encyclopedia of Industrial Chemistry; John Wiley & Sons, Ltd: Hoboken, NJ, USA, 2000; ISBN 978-3-527-30673-2. [Google Scholar]
- Yoke, J.T.; Weiss, J.F.; Tollin, G. Reactions of Triethylamine with Copper(I) and Copper(II) Halides. Inorg. Chem. 1963, 2, 1210–1216. [Google Scholar] [CrossRef]
- Demaison, J.; Császár, A.G. Equilibrium CO Bond Lengths. J. Mol. Struct. 2012, 1023, 7–14. [Google Scholar] [CrossRef]
- Alvarez, S. Distortion Pathways of Transition Metal Coordination Polyhedra Induced by Chelating Topology. Chem. Rev. 2015, 115, 13447–13483. [Google Scholar] [CrossRef] [PubMed]
- Galadzhun, I.; Kulmaczewski, R.; Cespedes, O.; Yamada, M.; Yoshinari, N.; Konno, T.; Halcrow, M.A. 2,6-Bis(Pyrazol-1-Yl)Pyridine-4-Carboxylate Esters with Alkyl Chain Substituents and Their Iron(II) Complexes. Inorg. Chem. 2018, 57, 13761–13771. [Google Scholar] [CrossRef]
- Boča, R. Zero-field splitting in metal complexes. Chem. Rev. 2004, 248, 757. [Google Scholar] [CrossRef]
- Piguet, C. Paramagnetic Susceptibility by NMR: The “Solvent Correction” Removed for Large Paramagnetic Molecules. J. Chem. Educ. 1997, 74, 815. [Google Scholar] [CrossRef]
- Koch, F.; Berkefeld, A.; Schubert, H.; Grauer, C. Redox and Acid–Base Properties of Binuclear 4-Terphenyldithiophenolate Complexes of Nickel. Chem. —A Eur. J. 2016, 22, 14640–14647. [Google Scholar] [CrossRef]
- Ikezaki, A.; Nakamura, M.; Cheng, R.-J. Anomalous 13C NMR Chemical Shifts of High-Spin Saddle Shaped Manganese(III) Octaethyltetraphenylporphyrin Complexes. Chem. Lett. 2006, 35, 156–157. [Google Scholar] [CrossRef]
- Arion, V.B.; Rapta, P.; Telser, J.; Shova, S.S.; Breza, M.; Lušpai, K.; Kožišek, J. Syntheses, Electronic Structures, and EPR/UV−Vis−NIR Spectroelectrochemistry of Nickel(II), Copper(II), and Zinc(II) Complexes with a Tetradentate Ligand Based on S-Methylisothiosemicarbazide. Inorg. Chem. 2011, 50, 2918–2931. [Google Scholar] [CrossRef]
- Zheng, B.; Tang, F.; Luo, J.; Schultz, J.W.; Rath, N.P.; Mirica, L.M. Organometallic Nickel(III) Complexes Relevant to Cross-Coupling and Carbon–Heteroatom Bond Formation Reactions. J. Am. Chem. Soc. 2014, 136, 6499–6504. [Google Scholar] [CrossRef]
- Jazdzewski, B.A.; Tolman, W.B. Understanding the Copper–Phenoxyl Radical Array in Galactose Oxidase: Contributions from Synthetic Modeling Studies. Coord. Chem. Rev. 2000, 200–202, 633–685. [Google Scholar] [CrossRef]
- Bühl, M.; Reimann, C.; Pantazis, D.A.; Bredow, T.; Neese, F. Geometries of Third-Row Transition-Metal Complexes from Density-Functional Theory. J. Chem. Theory Comput. 2008, 4, 1449–1459. [Google Scholar] [CrossRef] [PubMed]
- Radical Localization in a Series of Symmetric NiII Complexes with Oxidized Salen Ligands—Chiang—2012—Chemistry—A European Journal—Wiley Online Library. Available online: https://chemistry-europe.onlinelibrary.wiley.com/doi/abs/10.1002/chem.201201410 (accessed on 16 September 2022).
- Yamaguchi, K.; Takahara, Y.; Fueno, T. Ab-Initio Molecular Orbital Studies of Structure and Reactivity of Transition Metal-OXO Compounds. In Proceedings of the Applied Quantum Chemistry; Smith, V.H., Schaefer, H.F., Morokuma, K., Eds.; Springer: Dordrecht, The Netherlands, 1986; pp. 155–184. [Google Scholar]
- Noodleman, L.; Davidson, E.R. Ligand Spin Polarization and Antiferromagnetic Coupling in Transition Metal Dimers. Chem. Phys. 1986, 109, 131–143. [Google Scholar] [CrossRef]
- Bencini, A.; Gatteschi, D. X.Alpha.-SW Calculations of the Electronic Structure and Magnetic Properties of Weakly Coupled Transition-Metal Clusters. The [Cu2Cl6]2− Dimers. J. Am. Chem. Soc. 1986, 108, 5763–5771. [Google Scholar] [CrossRef] [PubMed]
- Fontaine, F.G. Cis-Bis(2,2-Bi pyridine) di chloro nickel(II) Methanol Solvate. Acta Cryst. E 2001, 57, m270–m271. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.a.K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated Space-Group and Crystal-Structure Determination. Acta Cryst. A 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A Short History of SHELX. Acta Cryst A 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Neese, F. Orca 4.2.1. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 73. [Google Scholar] [CrossRef]
- Sinnecker, S.; Neese, F.; Noodleman, L.; Lubitz, W. Calculating the Electron Paramagnetic Resonance Parameters of Exchange Coupled Transition Metal Complexes Using Broken Symmetry Density Functional Theory: Application to a MnIII/MnIV Model Compound. J. Am. Chem. Soc. 2004, 126, 2613–2622. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | [(bipy)4Ni2(L)]Cl•2CH3OH•0.75Et2O | [(phen)4Ni2(L)]Cl•3CH3OH | ||
|---|---|---|---|---|
| Ni(1) | Ni(2) | Ni(1) | Ni(2) | |
| Ni–NPy, Å | 2.074(5)–2.132(2) | 2.061(5)–2.095(5) | - | - |
| Ni–NPhen, Å | - | - | 2.076(5)–2.146(6) | 2.080(5)–2.135(6) |
| Ni–NL, Å | 2.040(4) | 2.050(4) | 2.036(6) | 2.033(5) |
| Ni–OL, Å | 2.014(4) | 2.050(4) | 2.028(4) | 2.036(5) |
| S(TP-6) | 12.997 | 15.215 | 13.850 | 12.366 |
| S(OC-6) | 0.958 | 0.733 | 0.658 | 0.885 |
| [(bipy)4Ni2(L)]Cl•2CH3OH•0.75Et2O | [(phen)4Ni2(L)]Cl•3CH3OH | |
|---|---|---|
| Empirical formula | C64H64.5ClN10Ni2O4.75 | C70H61ClN10Ni2O5 |
| Formula weight | 1202.62 | 1275.15 |
| T, K | 120 | 100 |
| Crystal system | triclinic | triclinic |
| Space group | P-1 | P-1 |
| Z | 2 | 2 |
| a, Å | 13.555(2) | 12.2327(3) |
| b, Å | 15.389(3) | 13.0491(3) |
| c, Å | 17.007(3) | 20.7742(5) |
| α, ° | 77.732(3) | 72.9350(10) |
| β, ° | 70.231(3) | 77.2100(10) |
| γ, ° | 81.830(3) | 89.9820(10) |
| V, Å3 | 3253.0(9) | 3084.00(13) |
| Dcalc, (g cm−1) | 1.228 | 1.373 |
| μ, cm−1 | 6.73 | 7.15 |
| F(000) | 1259 | 1328 |
| 2θmax, ° | 54 | 50 |
| Reflections measured | 45,304 | 41,247 |
| Independent reflections | 14,150 | 10,859 |
| Observed reflections [I > 2σ(I)] | 6921 | 5967 |
| Parameters | 756 | 811 |
| R1 | 0.0794 | 0.0811 |
| wR2 | 0.2188 | 0.2702 |
| GOF | 0.955 | 1.100 |
| Δρmax/Δρmin (e Å−3) | 0.955/−0.694 | 2.109/−0.415 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nikovskiy, I.A.; Karnaukh, K.M.; Aleshin, D.Y.; Spiridonov, K.A.; Danshina, A.A.; Nelyubina, Y.V.; Polezhaev, A.V.; Novikov, V.V. Binuclear Nickel Complexes of a New Di(hydroxyphenyl)imidazolate. Magnetochemistry 2022, 8, 132. https://doi.org/10.3390/magnetochemistry8100132
Nikovskiy IA, Karnaukh KM, Aleshin DY, Spiridonov KA, Danshina AA, Nelyubina YV, Polezhaev AV, Novikov VV. Binuclear Nickel Complexes of a New Di(hydroxyphenyl)imidazolate. Magnetochemistry. 2022; 8(10):132. https://doi.org/10.3390/magnetochemistry8100132
Chicago/Turabian StyleNikovskiy, Igor A., Kseniia M. Karnaukh, Dmitry Yu. Aleshin, Kirill A. Spiridonov, Anastasia A. Danshina, Yulia V. Nelyubina, Alexander V. Polezhaev, and Valentin V. Novikov. 2022. "Binuclear Nickel Complexes of a New Di(hydroxyphenyl)imidazolate" Magnetochemistry 8, no. 10: 132. https://doi.org/10.3390/magnetochemistry8100132
APA StyleNikovskiy, I. A., Karnaukh, K. M., Aleshin, D. Y., Spiridonov, K. A., Danshina, A. A., Nelyubina, Y. V., Polezhaev, A. V., & Novikov, V. V. (2022). Binuclear Nickel Complexes of a New Di(hydroxyphenyl)imidazolate. Magnetochemistry, 8(10), 132. https://doi.org/10.3390/magnetochemistry8100132