
Pyridyl-Thioethers as Capping Ligands for the Design of Heteroleptic Fe(II) Complexes with Spin-Crossover Behavior

 ,
,  and
and 
Abstract
:1. Introduction
2. Results
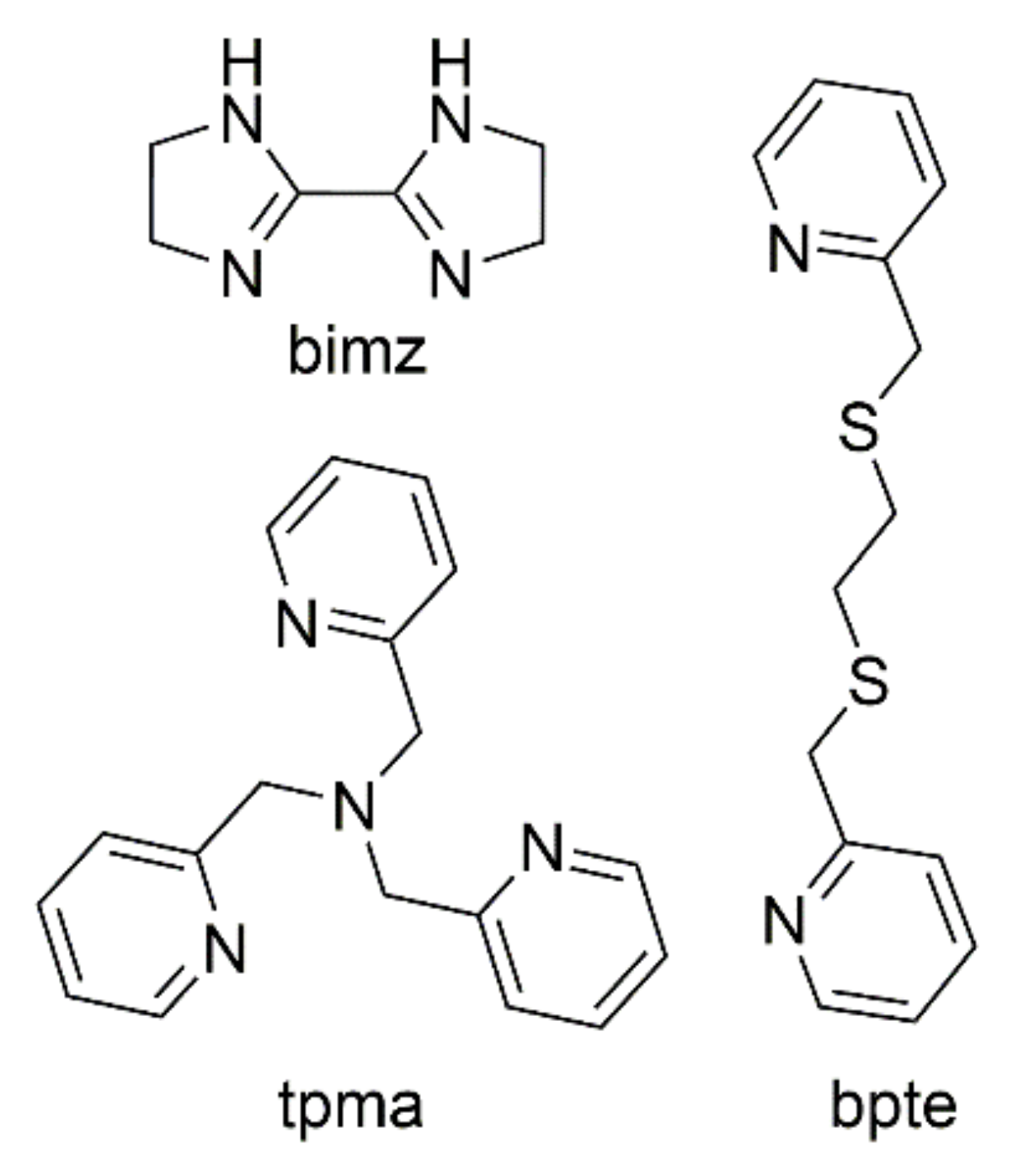
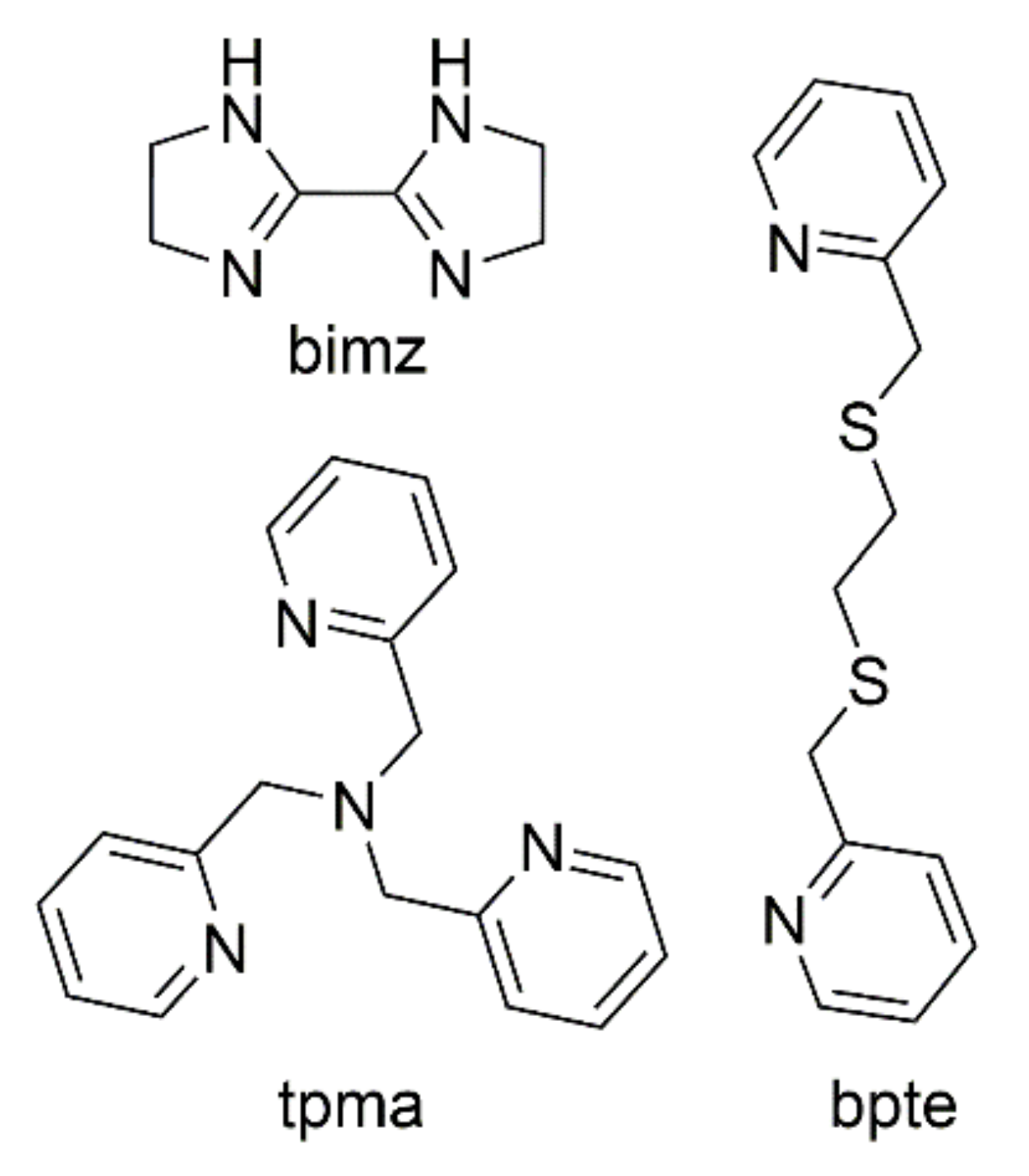
2.1. Synthesis
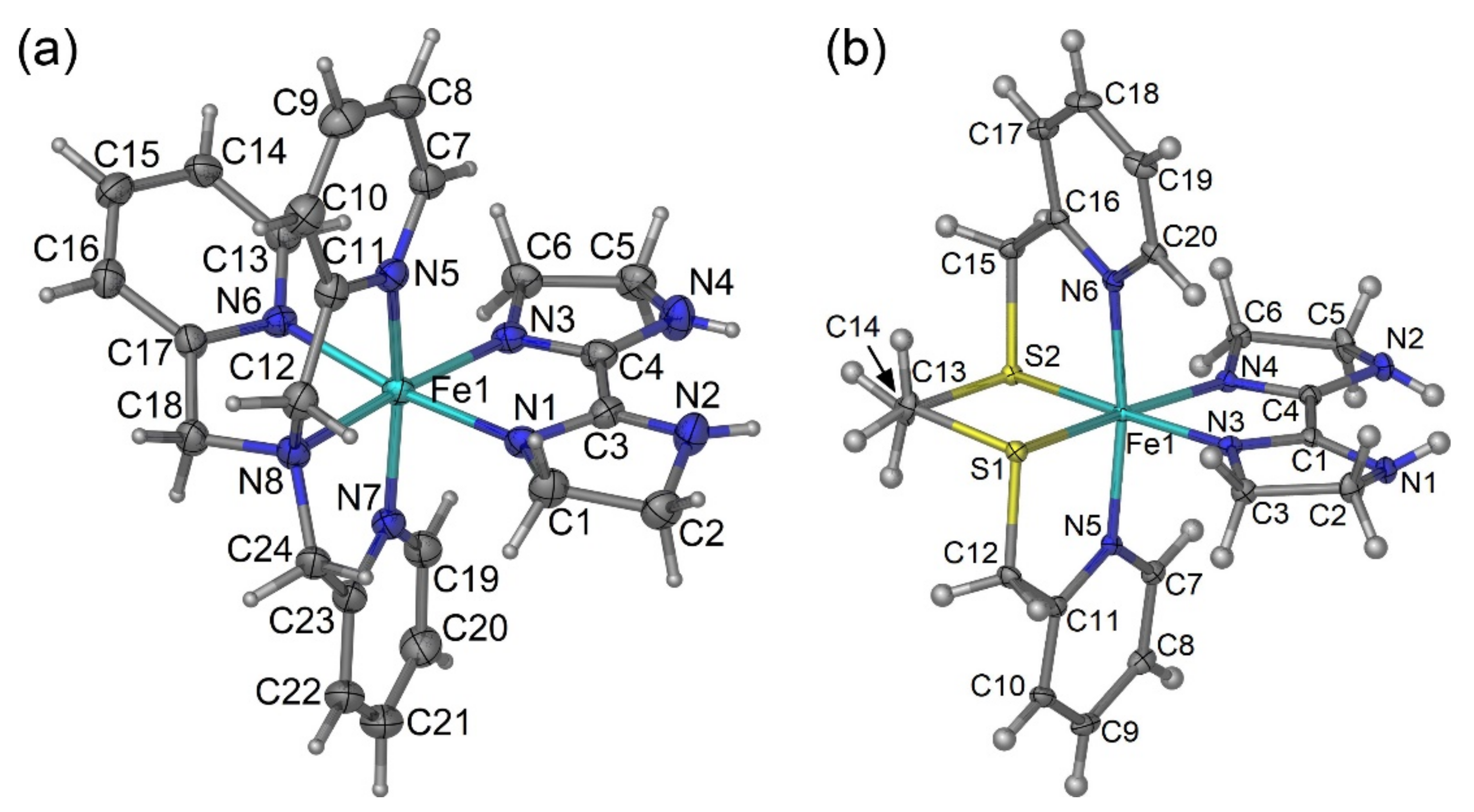
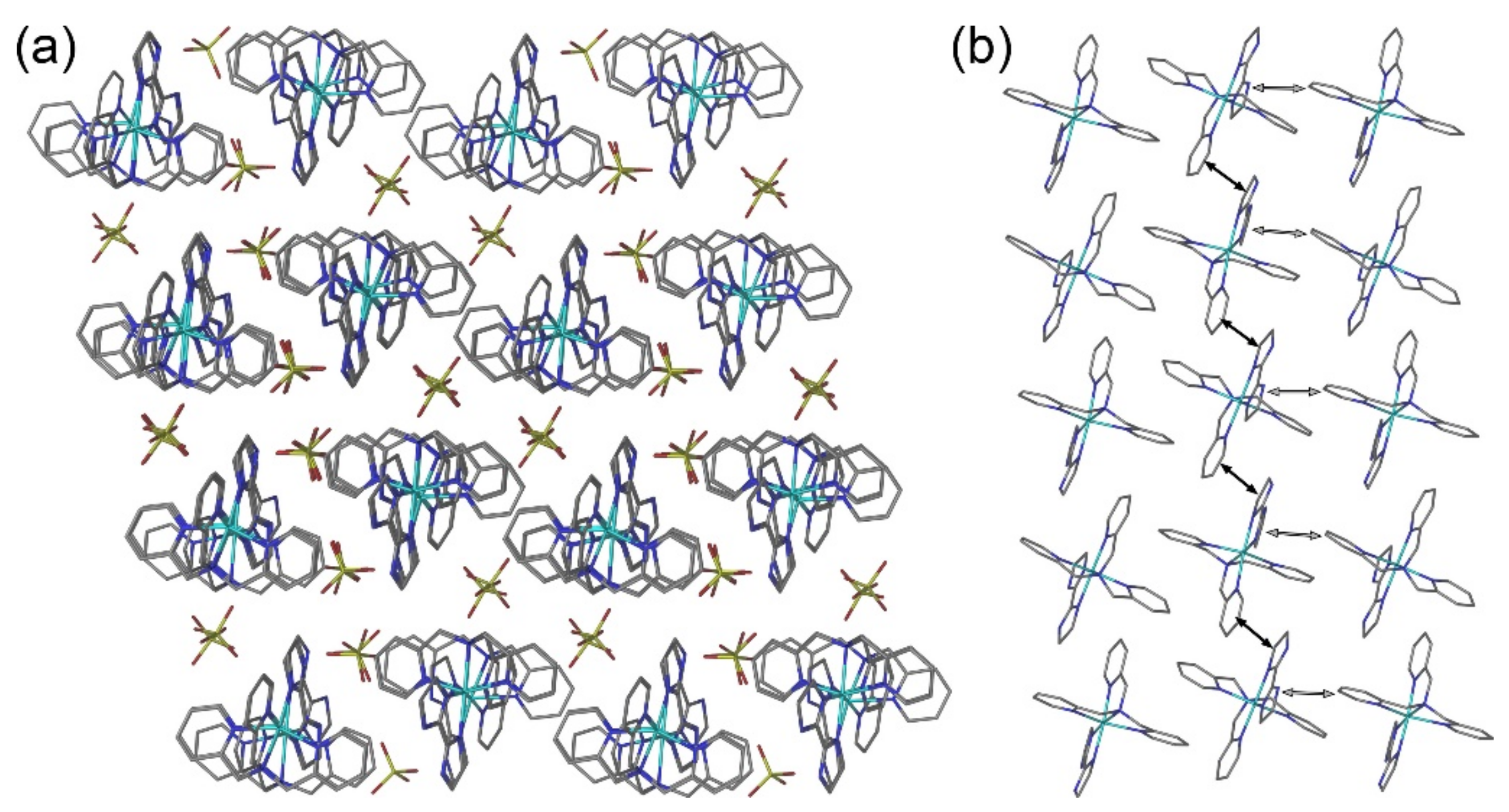
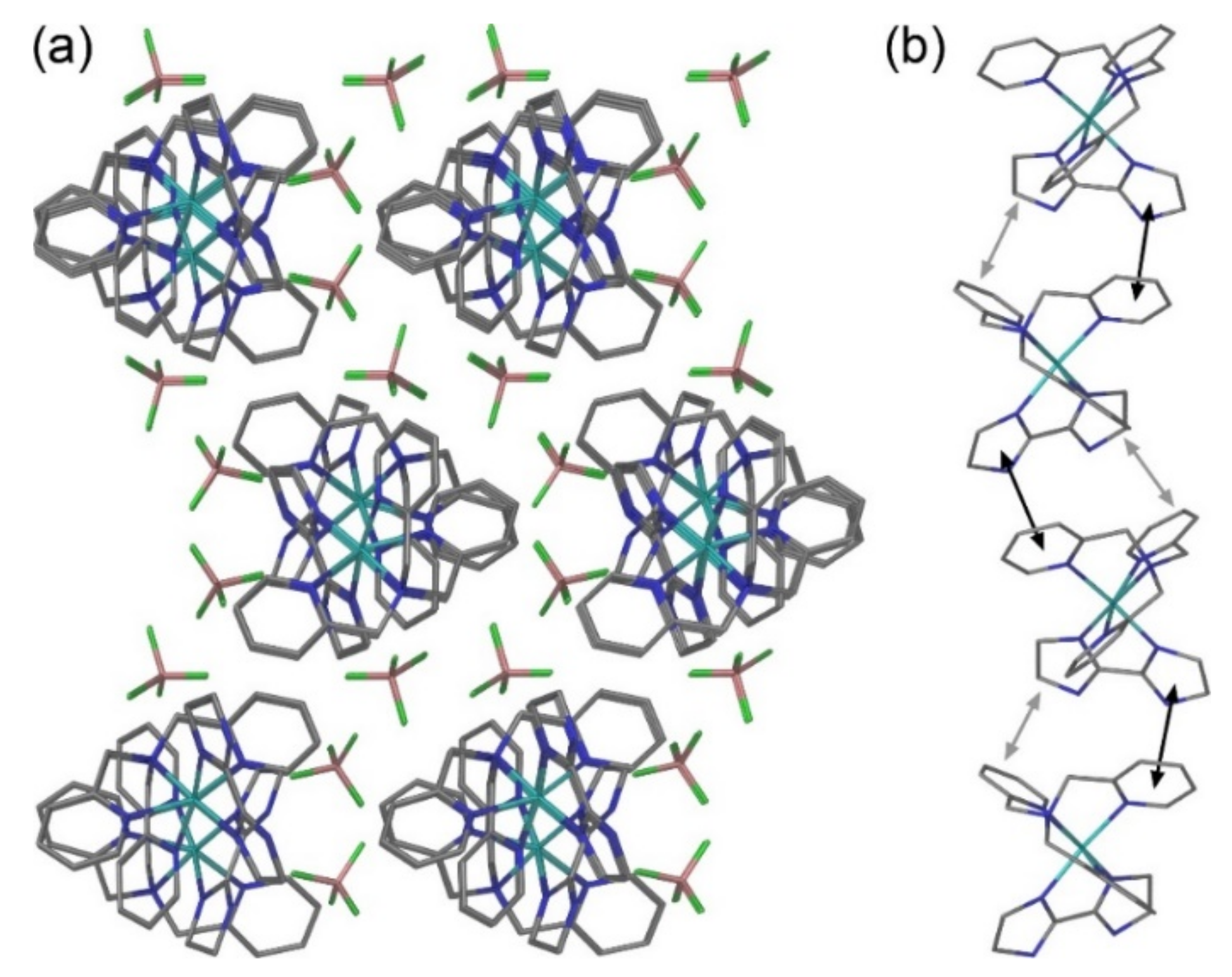
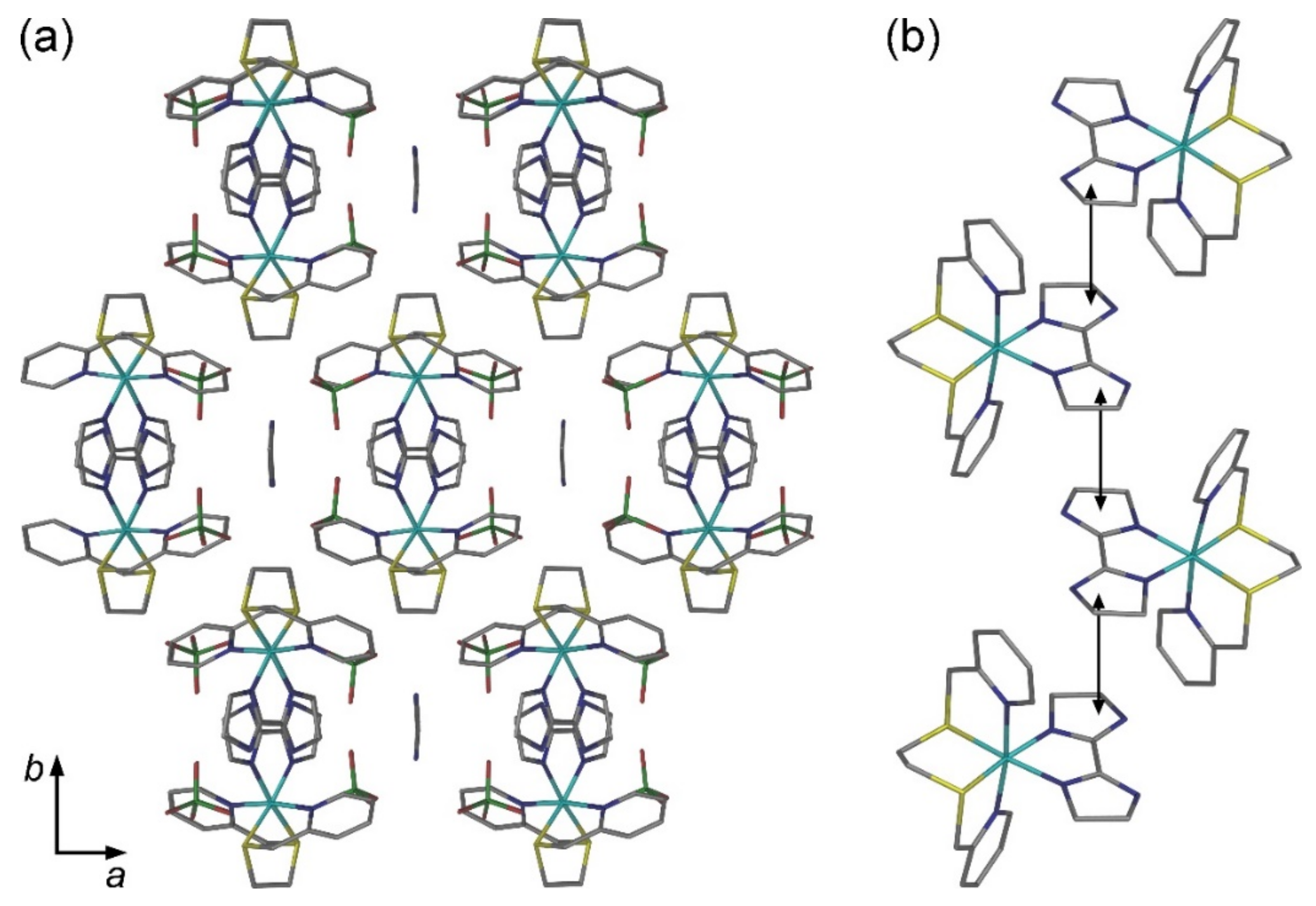
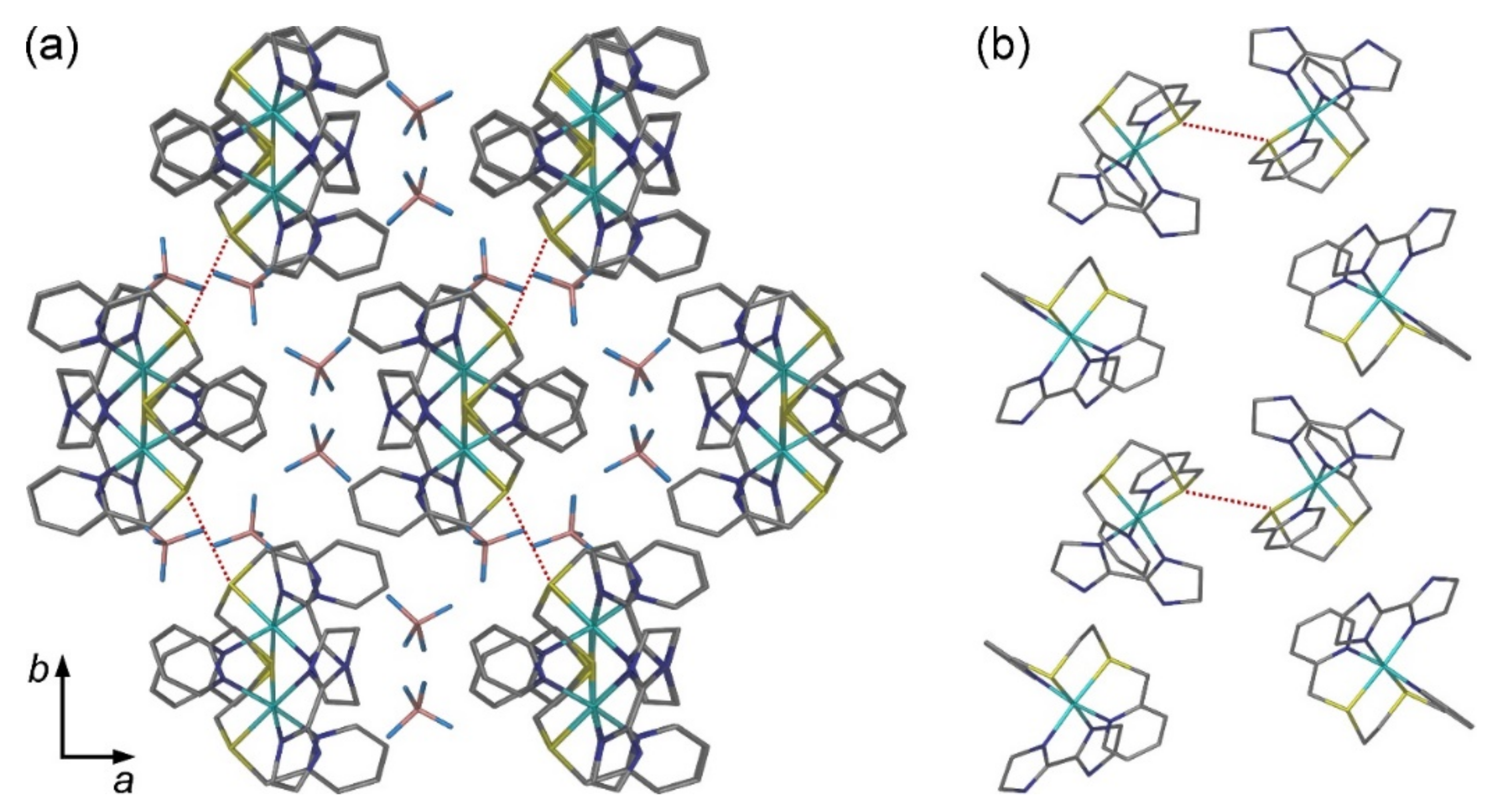
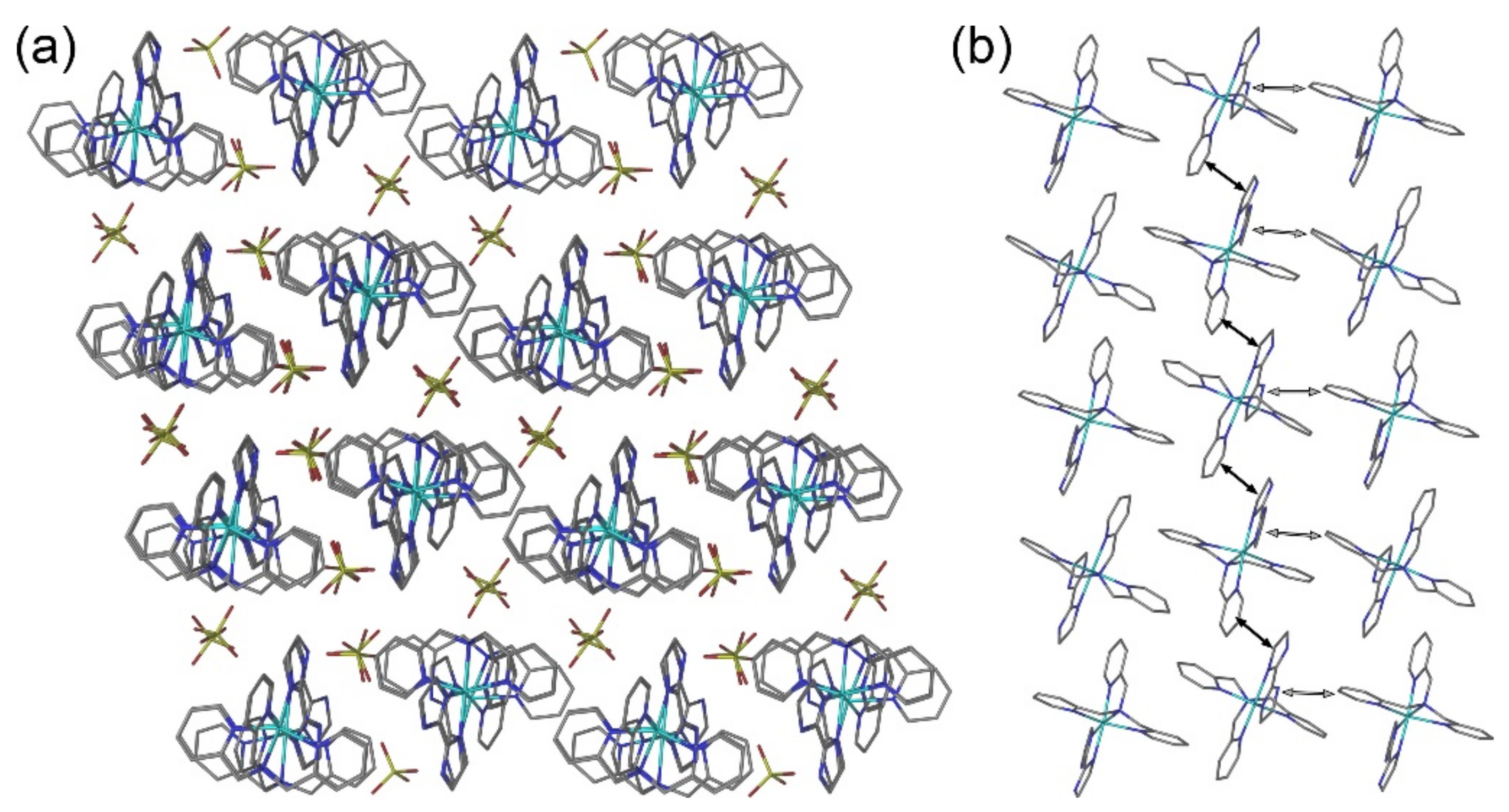
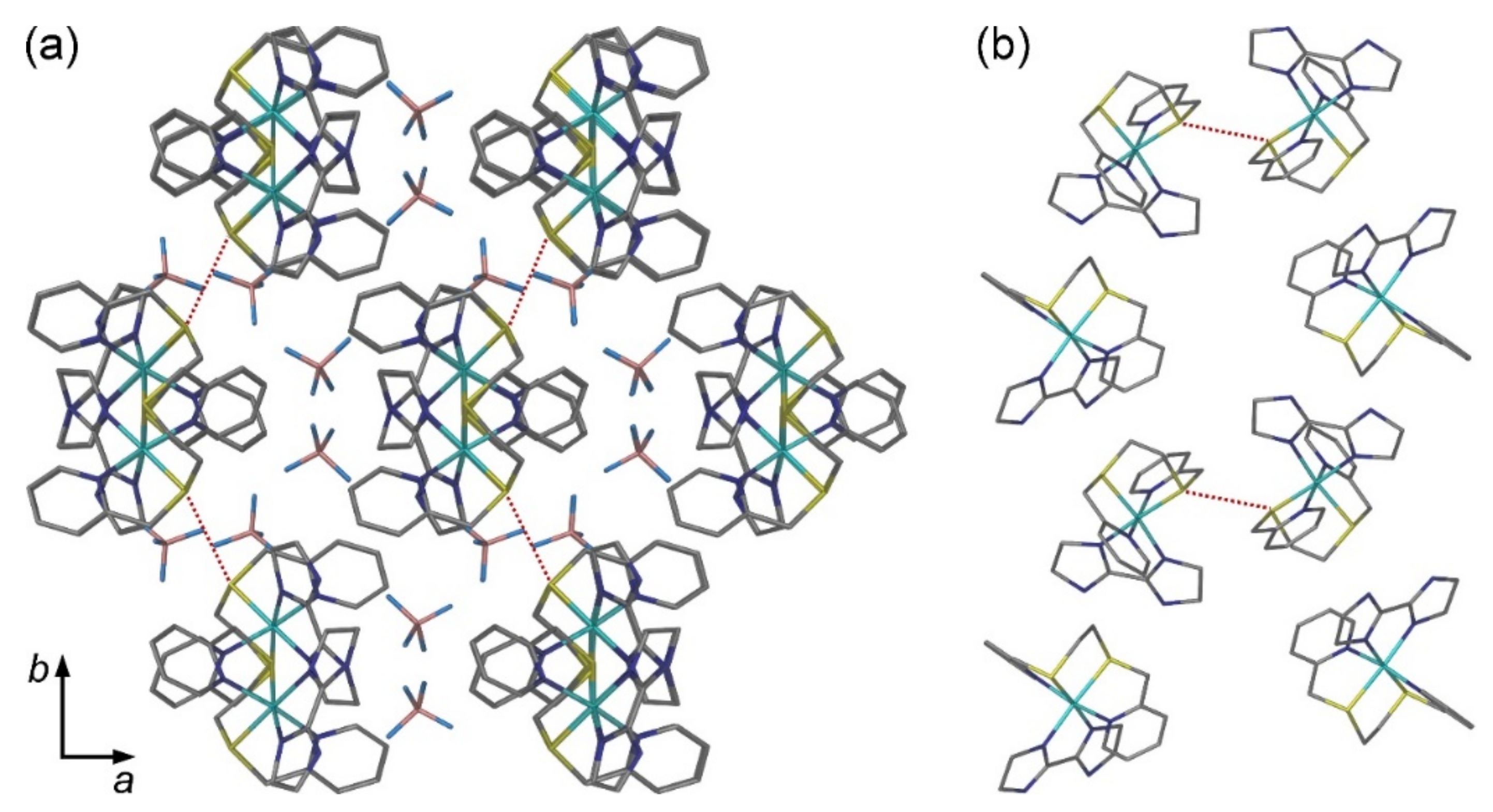
2.2. Crystal Structures
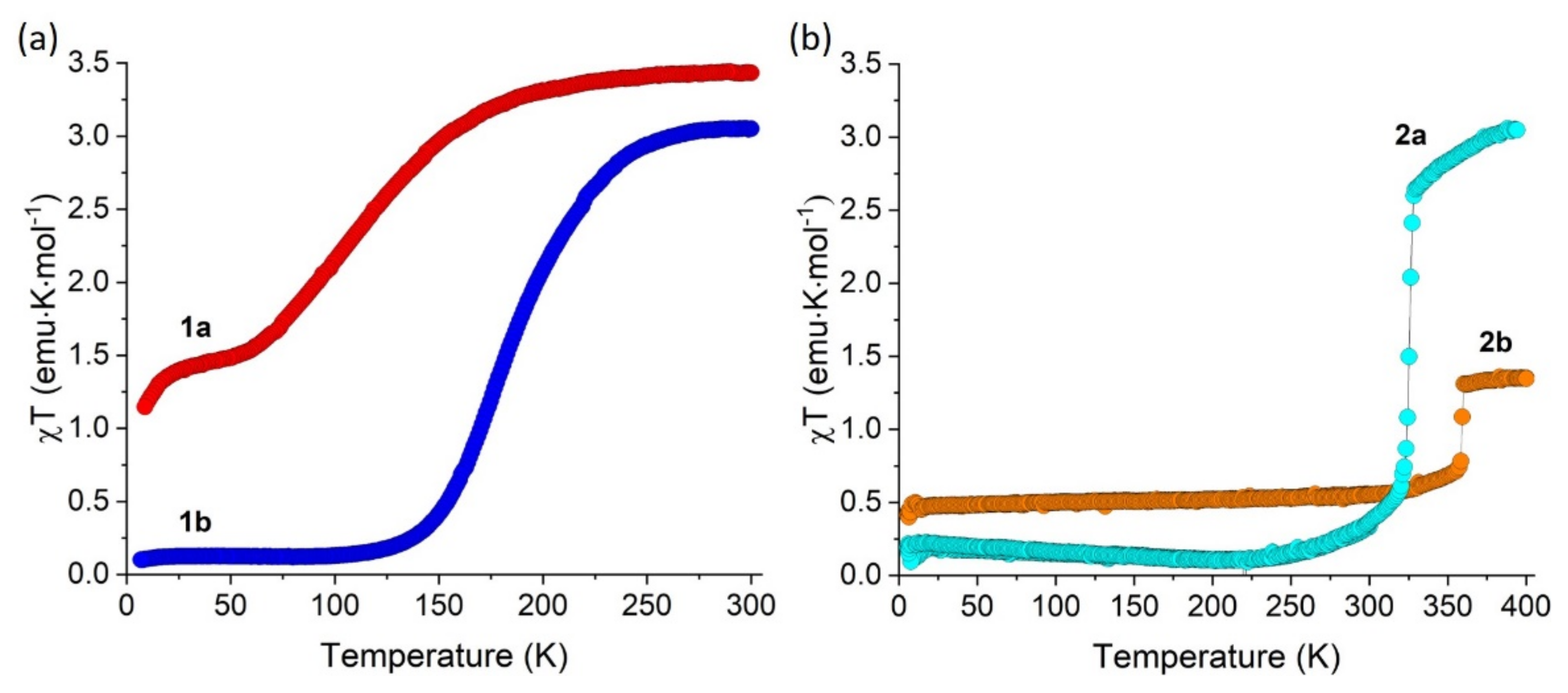
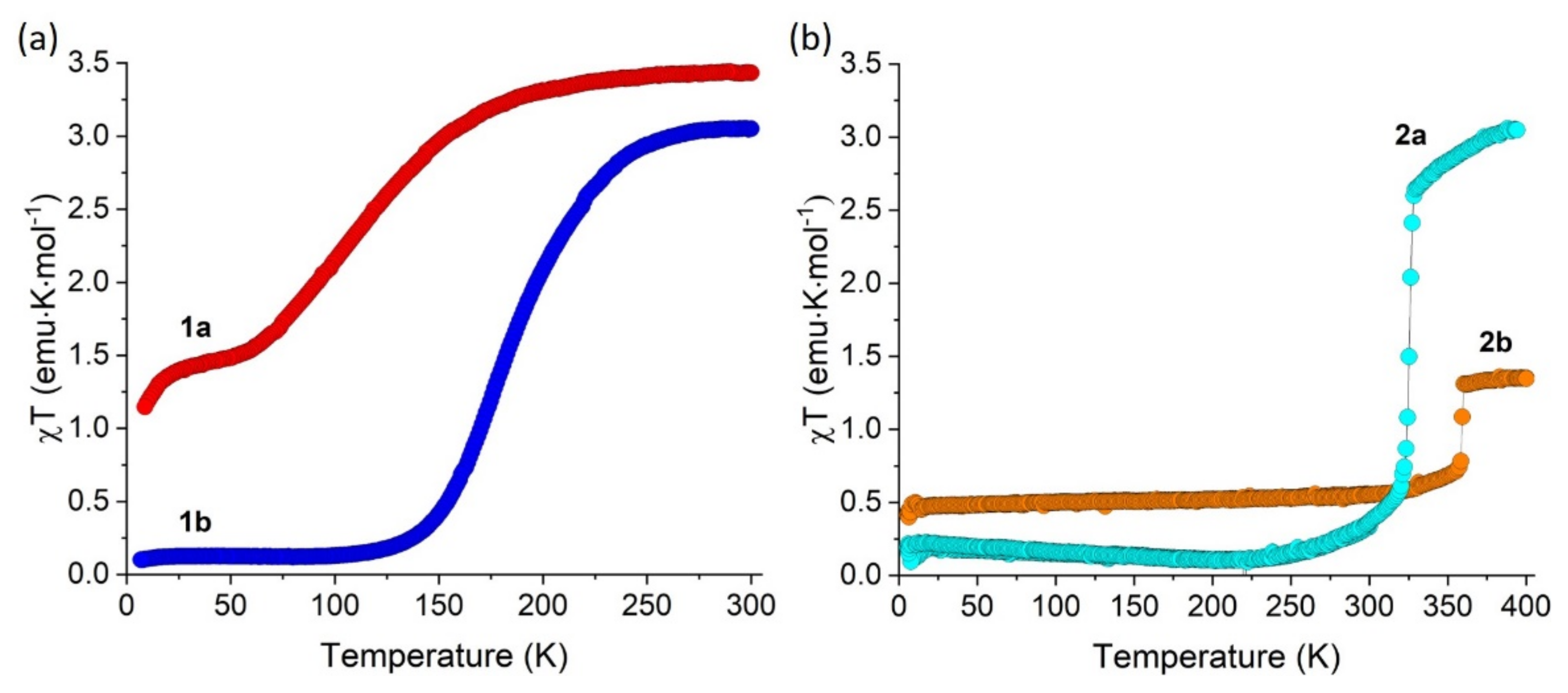
2.3. Magnetic Properties
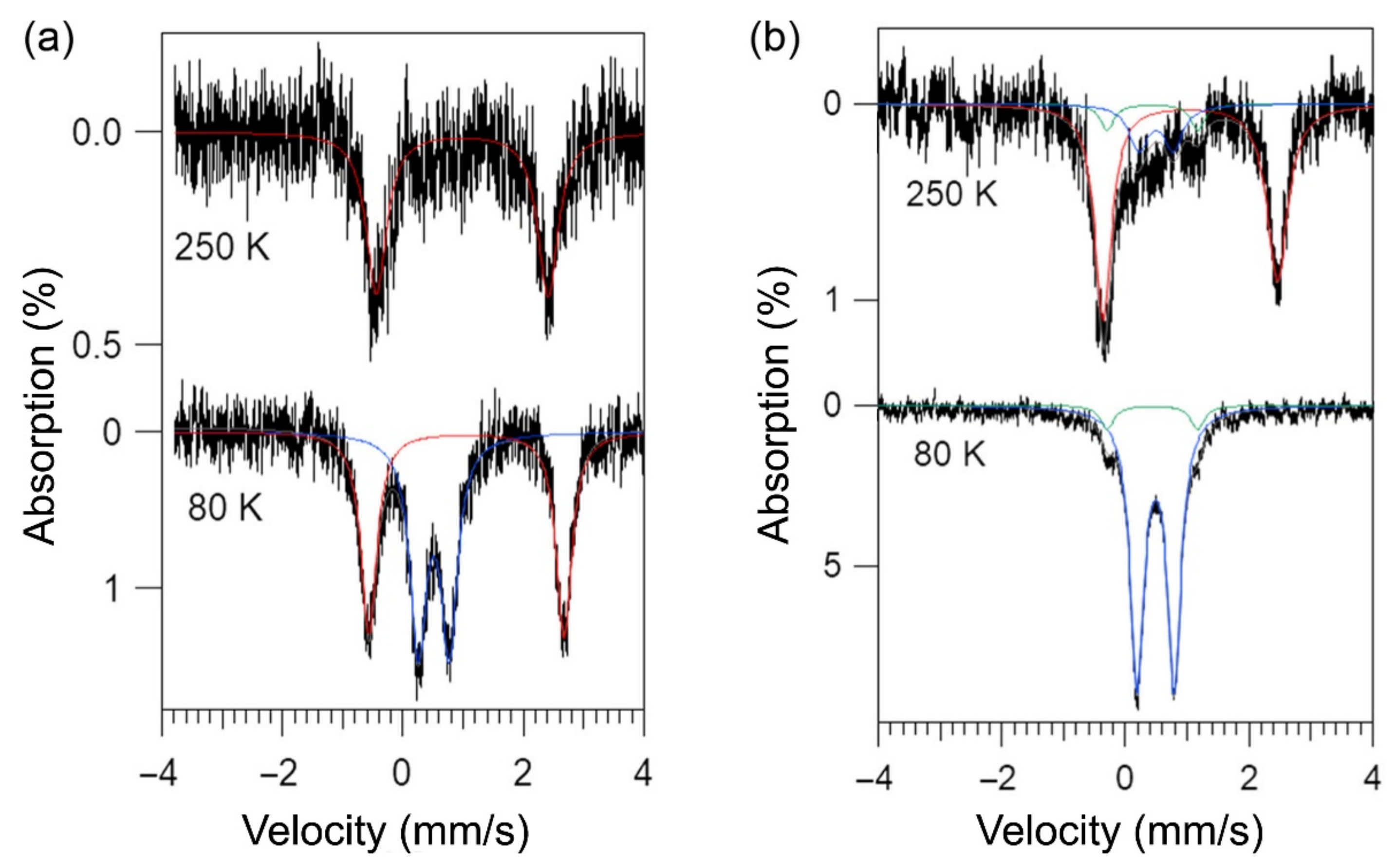
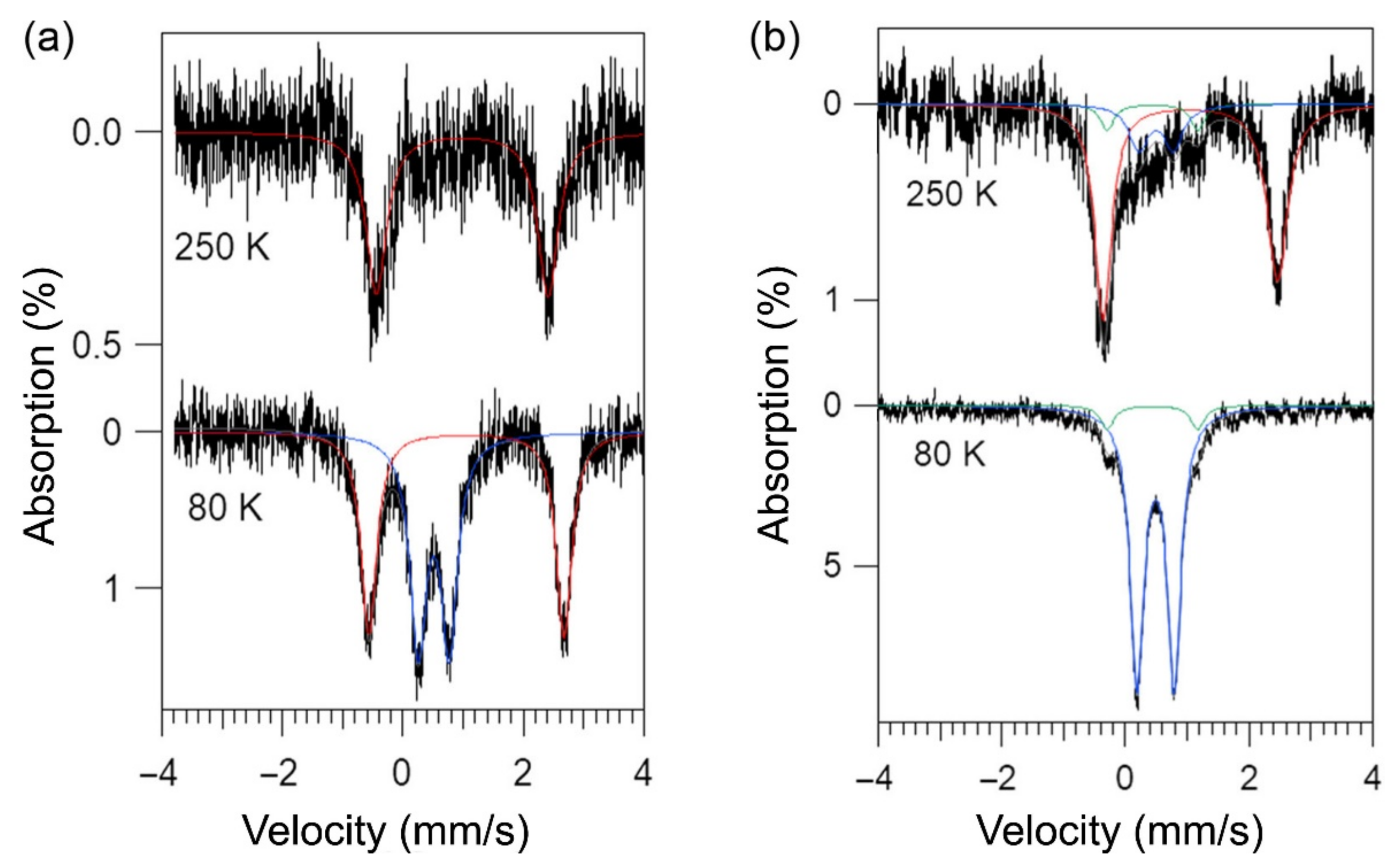
2.4. Mössbauer Spectroscopy
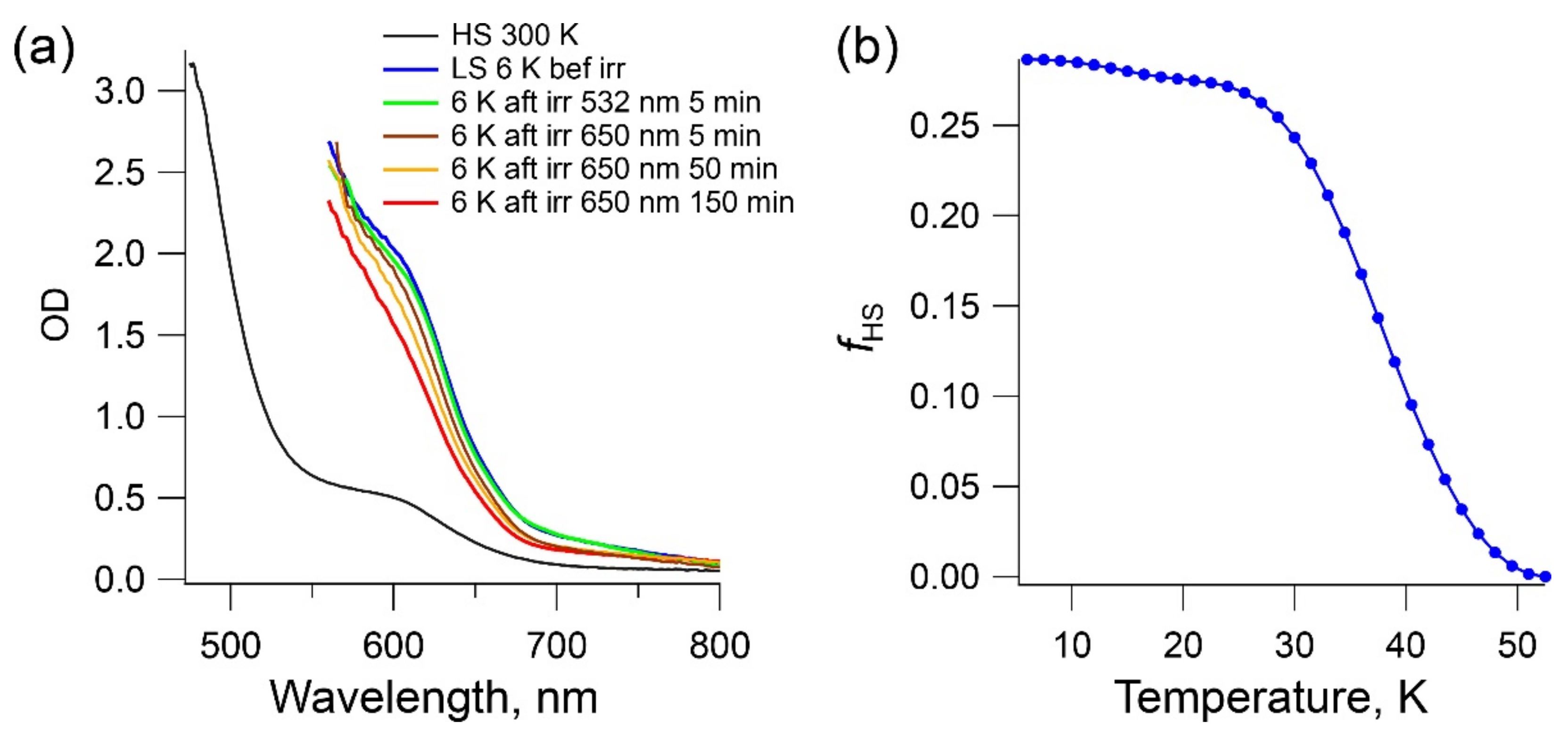
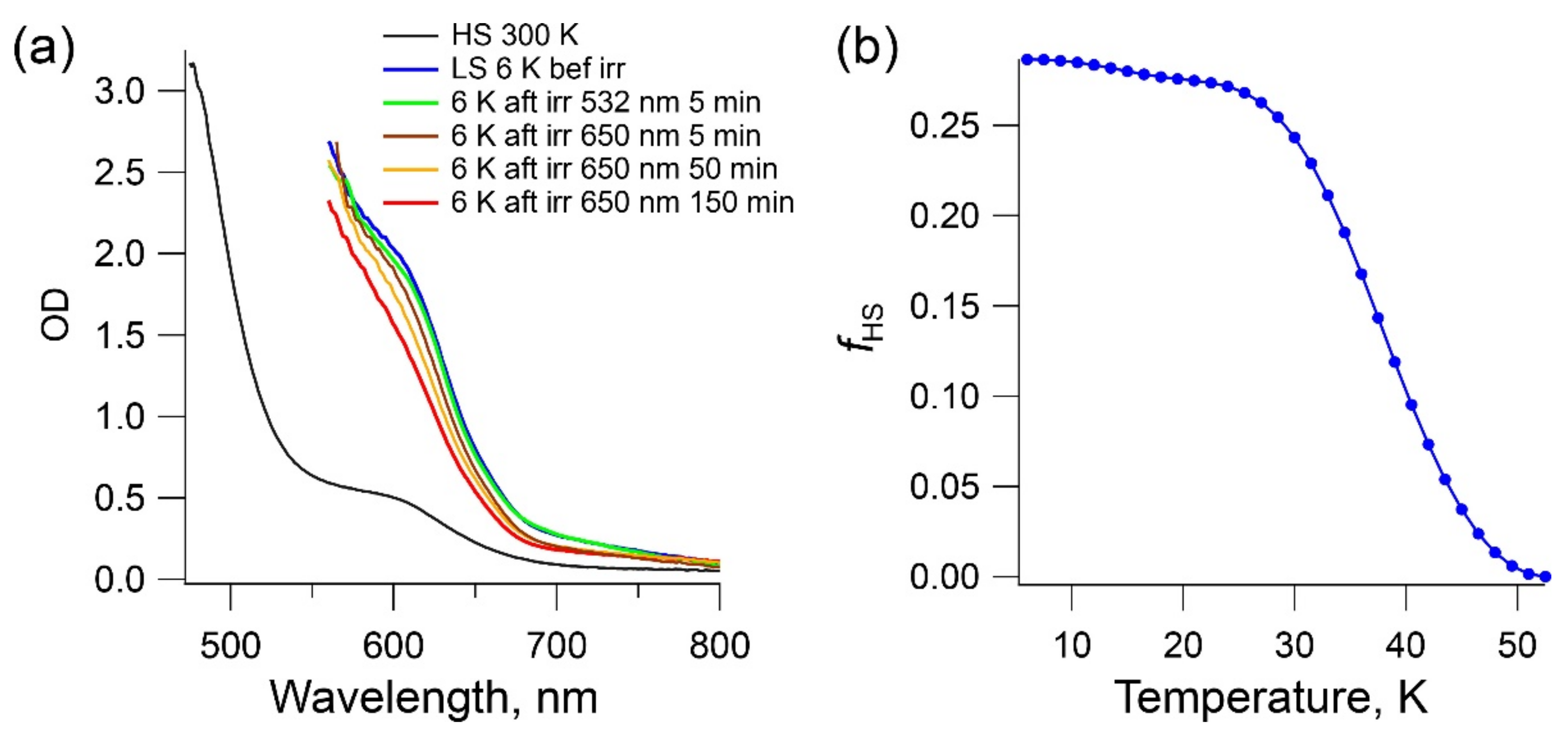
2.5. LIESST Effect in Complex 1b
3. Conclusions
4. Materials and Methods
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formula | FeCl2O8N8C24H28 (1a) | FeF8N8C24B2H28 (1b) | FeCl2O8N7C22H29S2 (2a·CH3CN) | FeSF8N6C20B2H26S2 (2b) | ||||
|---|---|---|---|---|---|---|---|---|
| T, K | 90(1) | 230(1) | 100(1) | 230(1) | 100(1) | 230(1) | 100(1) | 230(1) |
| CCDC number | 1965486 | 1965490 | 1965488 | 1965489 | 1965484 | 1965491 | 1965485 | 1965487 |
| Space group | P | P | P21/c | P21/c | P21/c | P21/c | P21/c | P21/c |
| a, Å | 12.2975(3) | 12.4579(7) | 12.836(4) | 13.348(3) | 11.460(1) | 11.594(3) | 13.134(2) | 13.135(2) |
| b, Å | 13.6406(4) | 13.8651(8) | 17.474(6) | 17.562(4) | 20.491(2) | 20.691(5) | 17.169(3) | 17.171(3) |
| c, Å | 17.3808(5) | 17.512(1) | 15.887(4) | 16.243(4) | 12.484(1) | 12.529(3) | 14.335(2) | 14.336(2) |
| α, deg | 81.828(2) | 82.266(5) | ||||||
| β, deg | 77.321(5) | 77.320(5) | 129.56(2) | 130.85(1) | 108.122(1) | 108.091(3) | 125.200(9) | 125.199(2) |
| γ, deg | 84.175(2) | 84.248(5) | ||||||
| V, Å3 | 2809.3(1) | 2916.4(3) | 2747(2) | 2880(1) | 2786.1(5) | 2857(1) | 2641.5(7) | 2642.2(7) |
| Z | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 |
| Crystal color | light yellow | light yellow | dark orange | orange | red | red | red | red |
| Crystal size, mm3 | 0.50 × 0.30 × 0.14 | 0.48 × 0.32 × 0.12 | 0.13 × 0.13 × 0.11 | 0.19 × 0.18 × 0.16 | 0.53 × 0.47 × 0.19 | 0.29 × 0.12 × 0.03 | 0.13 × 0.13 × 0.11 | 0.17 × 0.13 × 0.08 |
| dcalc, g/cm3 | 1.616 | 1.556 | 1.591 | 1.518 | 1.694 | 1.652 | 1.620 | 1.619 |
| μ, mm−1 | 0.791 | 0. 762 | 0.636 | 0.606 | 0.944 | 0.921 | 0.809 | 0.809 |
| λ, Å | 0.71073 | 0.71073 | 0.71073 | 0.71073 | 0.71073 | 0.71073 | 0.71073 | 0.71073 |
| 2θmax, deg | 27.48 | 27.48 | 26.37 | 28.51 | 28.52 | 28.53 | 28.59 | 28.58 |
| Total refls. (Rint) | 39132 (0.052) | 33276 (0.047) | 26980 (0.148) | 33221 (0.036) | 31167 (0.019) | 26075 (0.022) | 28863 (0.030) | 28869 (0.029) |
| Unique refls. | 12690 | 13208 | 5568 | 6920 | 6667 | 6745 | 6309 | 6309 |
| Parameters/Restraints | 787/0 | 787/2 | 456/32 | 454/10 | 402/0 | 386/0 | 433/10 | 423/10 |
| R1, wR2 [I > 2σ(I)] a | 0.069, 0.174 | 0.076, 0.206 | 0.072, 0.200 | 0.042, 0.120 | 0.037, 0.097 | 0.038, 0.106 | 0.051, 0.139 | 0.040, 0.101 |
| R1, wR2 (all data) | 0.086, 0.186 | 0.108, 0.228 | 0.077, 0.204 | 0.064, 0.129 | 0.041, 0.098 | 0.050, 0.115 | 0.067, 0.151 | 0.056, 0.111 |
| Goodness of fit b | 1.031 | 1.023 | 1.095 | 1.041 | 1.130 | 1.032 | 1.067 | 1.084 |
| Diff. peak/hole, e/Å3 | 1.60, −0.87 | 1.58, −0.50 | 1.59, −1.10 | 0.58, −0.49 | 0.51, −0.61 | 1.07, −0.50 | 0.95, −0.63 | 0.52, −0.42 |
References
- Gütlich, P.; Goodwin, H.A. Spin crossover—An overall perspective. Top. Curr. Chem. 2004, 233, 1–47. [Google Scholar] [CrossRef]
- Halcrow, M.A. The spin-states and spin-transitions of mononuclear iron(II) complexes of nitrogen-donor ligands. Polyhedron 2007, 26, 3523–3576. [Google Scholar] [CrossRef]
- Lennartson, A.; Bond, A.D.; Piligkos, S.; McKenzie, C.J. Four-site cooperative spin crossover in a mononuclear FeII Complex. Angew. Chem. Int. Ed. 2012, 51, 11049–11052. [Google Scholar] [CrossRef] [Green Version]
- Lennartson, A.; Southon, P.; Sciortino, N.F.; Kepert, C.J.; Frandsen, C.; Morup, S.; Piligkos, S.; McKenzie, C.J. Reversible guest binding in a non-porous FeII coordination polymer host toggles spin crossover. Chem. Eur. J. 2015, 21, 16066–16072. [Google Scholar] [CrossRef]
- Arroyave, A.; Lennartson, A.; Dragulescu-Andrasi, A.; Pedersen, K.S.; Piligkos, S.; Stoian, S.A.; Greer, S.M.; Pak, C.; Hietsoi, O.; Phan, H.; et al. Spin crossover in Fe(II) complexes with N4S2 coordination. Inorg. Chem. 2016, 55, 5904–5913. [Google Scholar] [CrossRef]
- Hogue, R.W.; Feltham, H.L.C.; Miller, R.G.; Brooker, S. Spin crossover in dinuclear N4S2 iron(II) thioether-triazole complexes: Access to [HS-HS], [HS-LS], and [LS-LS] states. Inorg. Chem. 2016, 55, 4152–4165. [Google Scholar] [CrossRef]
- Yergeshbayeva, S.; Hrudka, J.J.; Lengyel, J.; Erkasov, R.; Stoian, S.A.; Dragulescu-Andrasi, A.; Shatruk, M. Heteroleptic Fe(II) complexes with N4S2 coordination as a platform for designing spin-crossover materials. Inorg. Chem. 2017, 56, 11096–11103. [Google Scholar] [CrossRef]
- Dragulescu-Andrasi, A.; Hietsoi, O.; Üngör, Ö.; Dunk, P.W.; Stubbs, V.; Arroyave, A.; Kovnir, K.; Shatruk, M. Dicyanometalates as building blocks for multinuclear iron(II) spin-crossover complexes. Inorg. Chem. 2019, 58, 11920–11926. [Google Scholar] [CrossRef]
- Klokishner, S.; Reu, O. Modeling of spin crossover in iron(II) complexes with N4S2 coordination. J. Phys. Chem. C 2019, 123, 19984–19990. [Google Scholar] [CrossRef]
- Kawamura, A.; Xie, J.; Boyn, J.-N.; Jesse, K.A.; McNeece, A.J.; Hill, E.A.; Collins, K.A.; Valdez-Moreira, J.A.; Filatov, A.S.; Kurutz, J.W.; et al. Reversible switching of organic diradical character via iron-based spin-crossover. J. Am. Chem. Soc. 2020, 142, 17670–17680. [Google Scholar] [CrossRef]
- Plaza-Lozano, D.; Conde-Gallardo, A.; Olguín, J. Spin crossover vs. high-spin iron(II) complexes in N4S2 coordination sphere containing picolyl-thioether ligands and NCE (E=S, Se and BH3) co-ligands. Eur. J. Inorg. Chem. 2021, 2021, 2846–2856. [Google Scholar] [CrossRef]
- Guionneau, P.; Marchivie, M.; Bravic, G.; Létard, J.F.; Chasseau, D. Structural aspects of spin crossover. Example of the [FeIILn(NCS)2] complexes. Top. Curr. Chem. 2004, 234, 97–128. [Google Scholar] [CrossRef]
- Phan, H.; Hrudka, J.J.; Igimbayeva, D.; Lawson Daku, L.M.; Shatruk, M. A simple approach for predicting the spin state of homoleptic Fe(II) tris-diimine complexes. J. Am. Chem. Soc. 2017, 139, 6437–6447. [Google Scholar] [CrossRef]
- Matouzenko, G.S.; Bousseksou, A.; Lecocq, S.; van Koningsbruggen, P.J.; Perrin, M.; Kahn, O.; Collet, A. Spin transition in [Fe(DPEA)(NCS)2], a compound with the new tetradentate ligand (2-aminoethyl)bis(2-pyridylmethyl)amine (DPEA): Crystal structure, magnetic properties, and Mössbauer spectroscopy. Inorg. Chem. 1997, 36, 2975–2981. [Google Scholar] [CrossRef]
- Phan, H.V.; Chakraborty, P.; Chen, M.M.; Calm, Y.M.; Kovnir, K.; Keniley, L.K.; Hoyt, J.M.; Knowles, E.S.; Besnard, C.; Meisel, M.W.; et al. Heteroleptic FeII complexes of 2,2′-biimidazole and its alkylated derivatives: Spin-crossover and photomagnetic behavior. Chem. Eur. J. 2012, 18, 15805–15815. [Google Scholar] [CrossRef]
- Li, B.; Wei, R.J.; Tao, J.; Huang, R.B.; Zheng, L.S.; Zheng, Z. Solvent-induced transformation of single crystals of a spin-crossover (SCO) compound to single crystals with two distinct SCO centers. J. Am. Chem. Soc. 2010, 132, 1558–1566. [Google Scholar] [CrossRef] [PubMed]
- Wei, R.-J.; Tao, J.; Huang, R.-B.; Zheng, L.-S. Reversible and irreversible vapor-induced guest molecule exchange in spin-crossover compounds. Inorg. Chem. 2011, 50, 8553–8564. [Google Scholar] [CrossRef] [PubMed]
- Abushamleh, A.S.; Goodwin, H.A. Coordination of 2,2′-biimidazole with iron, cobalt, nickel and copper. Aus. J. Chem. 1979, 32, 513–518. [Google Scholar] [CrossRef]
- König, E.; Ritter, G.; Kulshreshtha, S.K.; Nelson, S.M. The high-spin (5T2) <-> low-spin (1A1) transition in solid tris(2,2′-bi-2-imidazoline)iron(II) diperchlorate. Hysteresis effects, simultaneous change of spin and lattice characteristics, and order-disorder phenomena of the perchlorate anion. Inorg. Chem. 1982, 21, 3022–3029. [Google Scholar] [CrossRef]
- Shatruk, M.; Dragulescu-Andrasi, A.; Chambers, K.E.; Stoian, S.A.; Bominaar, E.L.; Achim, C.; Dunbar, K.R. Properties of Prussian blue materials manifested in molecular complexes: Observation of cyanide linkage isomerism and spin-crossover behavior in pentanuclear cyanide clusters. J. Am. Chem. Soc. 2007, 129, 6104–6116. [Google Scholar] [CrossRef]
- Chakraborty, P.; Enachescu, C.; Walder, C.; Bronisz, R.; Hauser, A. Thermal and light-induced spin switching dynamics in the 2D coordination network of {[Zn1-xFex(bbtr)3](ClO4)2}∞: The role of cooperative effects. Inorg. Chem. 2012, 51, 9714–9722. [Google Scholar] [CrossRef] [PubMed]
- Shatruk, M.; Phan, H.; Chrisostomo, B.A.; Suleimenova, A. Symmetry-breaking structural phase transitions in spin crossover complexes. Coord. Chem. Rev. 2015, 289, 62–73. [Google Scholar] [CrossRef]
- Gütlich, P.; Bill, E.; Trautwein, A.X. Mössbauer Spectroscopy and Transition Metal Chemistry Fundamentals and Applications; Springer: Berlin/Heidelberg, Gemany, 2011. [Google Scholar]
- Hauser, A. Light-induced spin crossover and the high-spin → low-spin relaxation. Top. Curr. Chem. 2004, 234, 155–198. [Google Scholar] [CrossRef]
- Létard, J.F.; Asthana, S.; Shepherd, H.J.; Guionneau, P.; Goeta, A.E.; Suemura, N.; Ishikawa, R.; Kaizaki, S. Photomagnetism of a sym-cis-dithiocyanato iron(II) complex with a tetradentate N,N′-bis(2-pyridylmethyl)1,2-ethanediamine ligand. Chem. Eur. J. 2012, 18, 5924–5934. [Google Scholar] [CrossRef]
- Kojima, T.; Leising, R.A.; Yan, S.; Que, L., Jr. Alkane functionalization at nonheme iron centers. Stoichiometric transfer of metal-bound ligands to alkane. J. Am. Chem. Soc. 1993, 115, 11328–11335. [Google Scholar] [CrossRef]
- Lennartson, A.; McKenzie, C.J. 1,2-Bis[(pyridin-2-ylmethyl)sulfanyl]ethane and its dimorphic hydrochloride salt. Acta Crystallogr. Sect. C 2011, 67, 354–358. [Google Scholar] [CrossRef] [Green Version]
- Forssel, G. Über die Einwirkung des Aethylendiamins auf Thioamide. I. Ber. Deutsch. Chem. Gesell. 1892, 25, 2132–2142. [Google Scholar] [CrossRef] [Green Version]
- Bain, G.A.; Berry, J.F. Diamagnetic corrections and Pascal’s constants. J. Chem. Educ. 2008, 85, 532–536. [Google Scholar] [CrossRef]
- Prescher, C.; McCammon, C.; Dubrovinsky, L. MossA. A program for analyzing energy-domain Mössbauer spectra from conventional and synchrotron sources. J. Appl. Crystallogr. 2012, 45, 329–331. [Google Scholar] [CrossRef]
- SMART and SAINT; Bruker AXS Inc.: Madison, WI, USA, 2007.
- CrysAlis; Oxford Diffraction Ltd.: Abingdon, UK, 2006.
- Sheldrick, G.M. SADABS; University of Göttingen: Göttingen, Germany, 1996. [Google Scholar]
- Sheldrick, G.M. XPREP. Space Group Determination and Reciprocal Space Plots; Siemens Analytical X-ray Instruments: Madison, WI, USA, 1991. [Google Scholar]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef]



| Complex | 1a a | 1b | 2a·CH3CN | 2b | ||||
|---|---|---|---|---|---|---|---|---|
| Temperature, K | 90 | 230 | 100 | 230 | 100 | 230 | 100 | 230 |
| Bond lengths, Å | ||||||||
| Fe‒Ntpma(amine) | Fe1: 2.262(3) | 2.269(3) | 2.004(4) | 2.189(2) | – | – | – | – |
| Fe2: 2.240(3) | 2.256(3) | |||||||
| Fe–Ntpma(pyridyl) b | Fe1: 2.178(3) | 2.187(3) | 1.989(4) | 2.149(2) | – | – | – | – |
| Fe2: 2.184(3) | 2.198(3) | |||||||
| Fe–Nbimz(1) b | Fe1: 2.159(3) | 2.165(3) | 2.034(4) | 2.138(2) | 1.983(2) | 1.989(2) | 2.008(3) | 2.009(2) |
| Fe2: 2.145(3) | 2.155(3) | |||||||
| Fe‒Nbpte b | – | – | – | – | 2.006(2) | 2.007(2) | 2.032(2) | 2.033(2) |
| Fe‒S(1) a | – | – | – | – | 2.2402(7) | 2.2408(6) | 2.2668(9) | 2.2678(7) |
| Σ90(cis-N/S-Fe-N/S), deg | Fe1: 113.5(1) | Fe1: 111.1(1) | 79.5(5) | 114.4(2) | 44.5(2) | 45.2(2) | 54.4(3) | 54.4(2) |
| Fe2: 108.8(1) | Fe2: 110.0(1) | |||||||
| Complex | T (K) | Site | δ (mm/s) | ΔEQ (mm/s) | ΓL/R (mm/s) | Area (%) |
|---|---|---|---|---|---|---|
| 1a | 80 | LS-FeII | 0.52(1) | 0.51(2) | 0.33(1) | 53(3) |
| HS-FeII | 1.07(2) | 3.24(2) | 0.31 | 47(3) | ||
| 250 | HS-FeII | 1.00(4) | 2.84(6) | 0.41 | 100 | |
| 1b | 80 | LS-FeII | 0.509(5) | 0.598(5) | 0.27 | 93(2) |
| HS-FeIII | 0.46(2) | 1.47(2) | 0.27 | 7(2) | ||
| 250 | LS-FeII | 0.51(8) | 0.56(8) | 0.37 | 18(5) | |
| HS-FeII | 1.07(2) | 2.82(2) | 0.32/0.40 | 75(5) | ||
| HS-FeIII | 0.46(8) | 1.47(8) | 0.27 | 7(5) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Üngör, Ö.; Igimbayeva, D.; Dragulescu-Andrasi, A.; Yergeshbayeva, S.; Delgado, T.; Greer, S.M.; Donalson, G.; Jo, M.; Erkasov, R.; Shatruk, M. Pyridyl-Thioethers as Capping Ligands for the Design of Heteroleptic Fe(II) Complexes with Spin-Crossover Behavior. Magnetochemistry 2021, 7, 134. https://doi.org/10.3390/magnetochemistry7100134
Üngör Ö, Igimbayeva D, Dragulescu-Andrasi A, Yergeshbayeva S, Delgado T, Greer SM, Donalson G, Jo M, Erkasov R, Shatruk M. Pyridyl-Thioethers as Capping Ligands for the Design of Heteroleptic Fe(II) Complexes with Spin-Crossover Behavior. Magnetochemistry. 2021; 7(10):134. https://doi.org/10.3390/magnetochemistry7100134
Chicago/Turabian StyleÜngör, Ökten, Dilyara Igimbayeva, Alina Dragulescu-Andrasi, Sandugash Yergeshbayeva, Teresa Delgado, Samuel M. Greer, Gabrielle Donalson, Minyoung Jo, Rakhmetulla Erkasov, and Michael Shatruk. 2021. "Pyridyl-Thioethers as Capping Ligands for the Design of Heteroleptic Fe(II) Complexes with Spin-Crossover Behavior" Magnetochemistry 7, no. 10: 134. https://doi.org/10.3390/magnetochemistry7100134
APA StyleÜngör, Ö., Igimbayeva, D., Dragulescu-Andrasi, A., Yergeshbayeva, S., Delgado, T., Greer, S. M., Donalson, G., Jo, M., Erkasov, R., & Shatruk, M. (2021). Pyridyl-Thioethers as Capping Ligands for the Design of Heteroleptic Fe(II) Complexes with Spin-Crossover Behavior. Magnetochemistry, 7(10), 134. https://doi.org/10.3390/magnetochemistry7100134