Synthesis, NMR Characterization, and Antileukemic Activity of N-Nonanoylpiperazinyl-5α-Androstane-3α,17β-Diol A-Ring Derivatives



Abstract
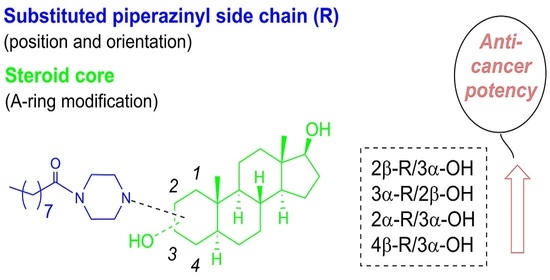
1. Introduction
2. Results and Discussion
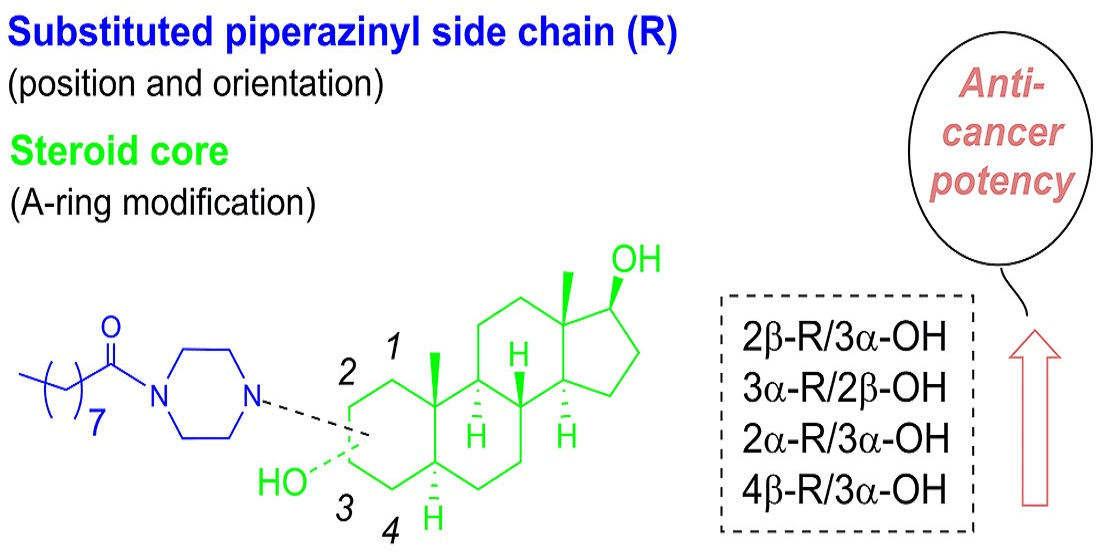
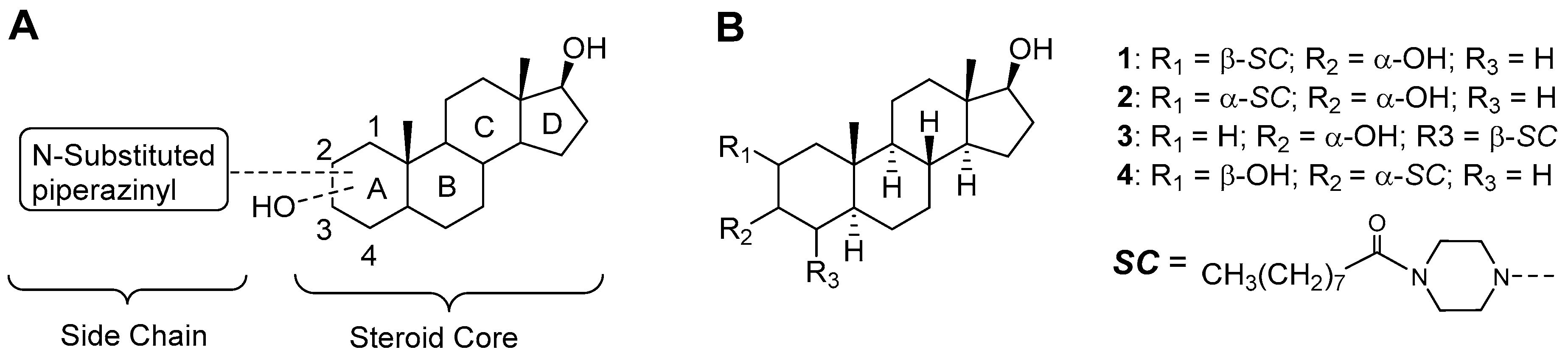
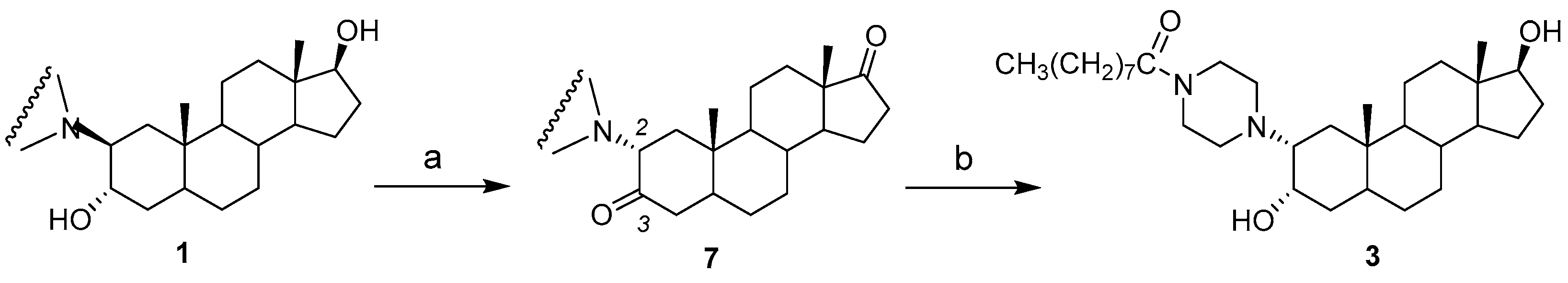
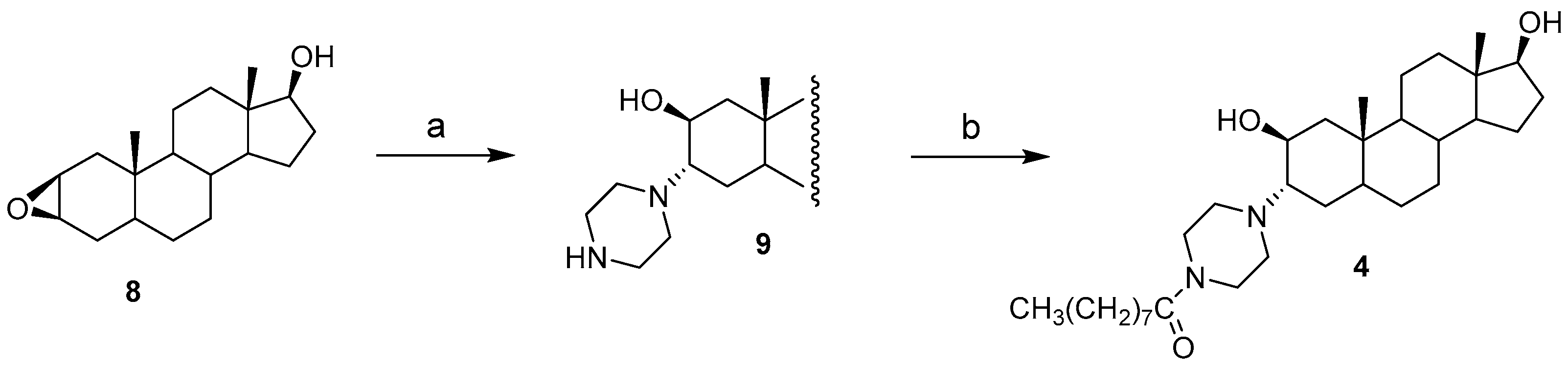
2.1. Chemical Synthesis of Compounds 1–4
2.2. Nuclear Magnetic Resonance (NMR) Characterization
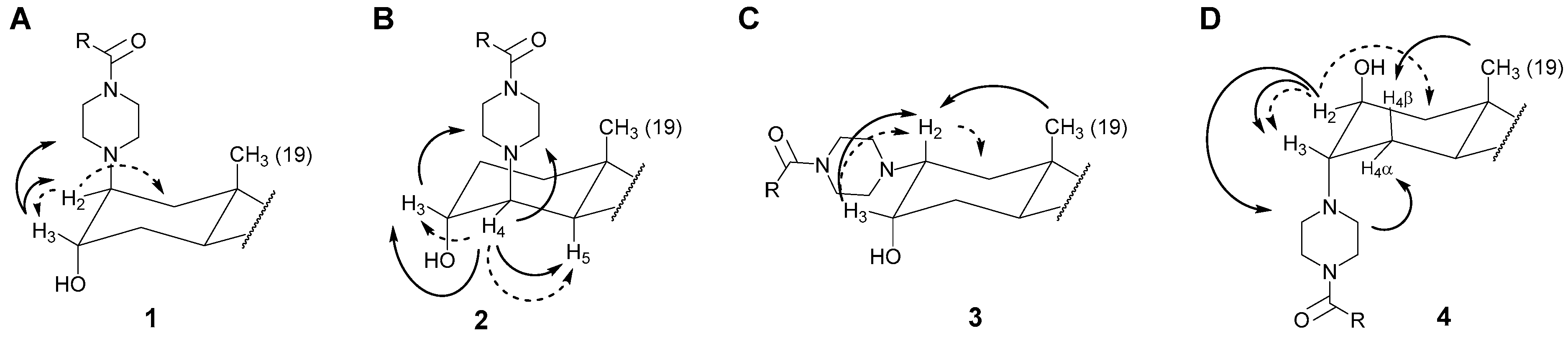
2.2.1. Assignments of Carbons and Protons
2.2.2. Positioning and Orientation of A-Ring Sidechain and OH
2.3. Inhibition of HL-60 Cell Proliferation
3. Materials and Methods
3.1. General
3.2. Synthesis of Compounds 1 and 2
3.3. Synthesis of Compound 3
3.4. Synthesis of Compound 4
3.5. Cell Proliferation Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- He, Q.; Na, X. The effects and mechanisms of a novel 2-aminosteroid on murine WEHI-3B leukemia cells in vitro and in vivo. Leukemia Res. 2001, 25, 455–461. [Google Scholar] [CrossRef]
- Thibeault, D.; Roy, J.; DeRoy, P.; Poirier, D. Chemical synthesis of 2β-amino-5α-androstane-3α,17β-diol N-derivatives and their antiproliferative effect on HL-60 human leukemia cells. Bioorg. Med. Chem. 2008, 16, 5062–5077. [Google Scholar] [CrossRef] [PubMed]
- Roy, J.; Maltais, R.; Jegham, H.; Poirier, D. Libraries of 2β-(N-substituted piperazino)-5α-androstane-3α,17β-diols: Chemical synthesis and cytotoxic effects on human leukemia HL-60 cells and on normal lymphocytes. Mol. Divers. 2011, 15, 317–339. [Google Scholar] [CrossRef] [PubMed]
- Ayan, D.; Maltais, R.; Hospital, A.; Poirier, D. Chemical synthesis, cytotoxicity, selectivity and bioavailability of 5alpha-androstane-3alpha,17beta-diol derivatives. Bioorg. Med. Chem. 2014, 22, 5847–5859. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Kumar, B.S.; Negi, A.S. Current status on development of steroids as anticancer agents. J. Steroid Biochem. Mol. Biol. 2013, 137, 242–270. [Google Scholar] [CrossRef] [PubMed]
- Jegham, H.; Maltais, R.; Roy, J.; Doillon, C.; Poirier, D. Biological evaluation of a new family of aminosteroids that display a selective toxicity for various malignant cell lines. Anti-Cancer Drugs 2012, 23, 803–814. [Google Scholar] [CrossRef] [PubMed]
- Kenmogne, L.C.; Ayan, D.; Roy, J.; Maltais, R.; Poirier, D. The aminosteroid derivative RM-133 shows in vitro and in vivo antitumor activity in human ovarian and pancreatic cancers. PLoS ONE 2015, 10, e0144890. [Google Scholar] [CrossRef] [PubMed]
- Maltais, R.; Hospital, A.; Delhomme, A.; Roy, J.; Poirier, D. Chemical synthesis, NMR analysis and evaluation on a cancer xenograft model (HL-60) of the aminosteroid derivative RM-133. Steroids 2014, 82, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Roy, J.; DeRoy, P.; Poirier, D. 2β-(N-substituted piperazino)-5α-androstane-3α,17β-diols: Parallel solid-phase synthesis and antiproliferative activity on human leukemia HL-60 mcells. J. Comb. Chem. 2007, 9, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Thibeault, D.; Poirier, D. An efficient method for the regioselective aminolysis of 2,3α-steroidal epoxyde. Synlett 2003, 8, 1192–1194. [Google Scholar]
- Barton, D.H.R. The stereochemistry of cyclohexane derivatives. J. Chem. Soc. 1953, 1027–1040. [Google Scholar] [CrossRef]
- Matthews, G.J.; Hassner, A. Organic Reactions in Organic Chemistry; Fried, J., Edwards, J.A., Eds.; Van Nostrand Reinhold Company: New York, NY, USA, 1972; Volume 2, pp. 1–53. [Google Scholar]
- Anderson, A.; Boyd, A.C.; Byford, A.; Campbell, A.C.; Gemmell, D.K.; Hamilton, N.M.; Hill, D.R.; Hill-Venning, C.; Lambert, J.J.; Maidment, M.S.; et al. Anestetic activity of novel water-soluble 2β-morpholinyl steroids and their modulatory effects at GABAA receptors. J. Med. Chem. 1997, 40, 1668–1681. [Google Scholar] [CrossRef]
- He, Q.; Xu, Y.H. Synthesis of 2β-(4’-methyl-1’-piperazino)-3α-hydroxyl-16,17-substituted-5α-androstanes. Acta Pharm Sin. 1992, 27, 101–106. [Google Scholar]
- Lewett, C.L.; Savage, D.S. Amino-steroids. Part III.1 2- and 3-Amino-5α-androstanes. J. Chem. Soc. 1968, 1134–1140. [Google Scholar] [CrossRef]
- Mancuso, A.J.; Huang, S.L.; Swern, D. Oxidation of long-chain and related alcohols to carbonyls by dimethylsulfoxide “activated” by oxalyl chloride. J. Org. Chem. 1978, 43, 2480–2482. [Google Scholar] [CrossRef]
- Cadot, C.; Laplante, Y.; Kamal, F.; Luu-The, V.; Poirier, D. C6-(N,N-butyl-methyl-heptanamide) derivatives of estrone and estradiol as inhibitors of type 1 17β-hydroxysteroid dehydrogenase: Chemical synthesis and biological evaluation. Bioorg. Med. Chem. 2007, 15, 714–726. [Google Scholar] [CrossRef] [PubMed]
- Blunt, J.W.; Stothers, J.B. 13C N.m.r. spectra of steroids—A survey and commentary. J. Magn. Reson. 1977, 9, 439–464. [Google Scholar] [CrossRef]
- Poirier, D.; Maltais, R. NMR-assisted structure elucidation of an anticancer steroid-β-enaminone derivative. Magnetochemistry 2017, 3, 37. [Google Scholar] [CrossRef]
- Tchédam-Ngatcha, B.; Trottier, M.C.; Poirier, D. 13C Nuclear magnetic resonance spectroscopy data of a variety of androsterone and epi-androsterone derivatives substituted at position 3beta or/and 3alpha. Curr. Top. Steroid Res. 2011, 8, 35–46. [Google Scholar]
- Fielding, L. 1H and 13C NMR studies of some steroidal neuromuscular blocking drugs: Solution conformations and dynamics. Magn. Reson. Chem. 1998, 36, 387–397. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| C and H Assignment | 1 (1H) | 1 (13C) | 2 (1H) | 2 (13C) | 3 (1H) | 3 (13C) | 4 (1H) | 4 (13C) |
|---|---|---|---|---|---|---|---|---|
| 2β-Chain- | -3α-OH | 4β-Chain- | -3α-OH | 2α-Chain- | -3α-OH | 3α-Chain- | -2β-OH | |
| CH2-1 | 1.40/1.82 | 34.5 | 1.40/1.52 | 34.0 | 1.30/1.73 | 36.9 | 1.54/1.68 | 42.4 |
| CH-2 or CH2-2 | 2.40 | 66.5 | 1.90 | 26.9 | 2.24 | 63.0 | 4.09 | 68.5 |
| CH-3 | 4.10 | 66.6 | 4.10 | 66.6 | 4.15 | 66.8 | 2.25 | 65.8 |
| CH-4 or CH2-4 | 1.35 | 34.2 | 2.34 | 70.5 | 1.55 | 36.5 | 1.42/1.82 | 26.6 |
| CH-5 | 1.66 | 40.6 | 1.77 | 45.7 | 1.60 | 40.0 | 1.48 | 41.1 |
| CH2-6 | 1.30 | 29.0 | 1.40 | 29.2 | 1.30 | 28.9 | 1.28 | 29.2 |
| CH2-7 | 0.92/1.71 | 32.7 | 0.91/1.80 | 34.0 | 0.95/1.88 | 32.8 | 0.92/1.71 | 32.8 |
| CH-8 | 1.42 | 36.6 | 1.40 | 37.0 | 1.45 | 36.6 | 1.46 | 36.5 |
| CH-9 | 0.75 | 56.9 | 0.73 | 57.4 | 0.82 | 56.3 | 0.72 | 57.1 |
| C-10 | - | 37.3 | - | 37.2 | - | 37.7 | - | 37.1 |
| CH2-11 | 1.38/1.62 | 21.8 | 1.33/1.58 | 21.1 | 1.38/1.70 | 21.6 | 1.33/1.60 | 21.7 |
| CH2-12 | 1.05/1.84 | 38.1 | 1.04/1.84 | 38.1 | 1.10/1.85 | 38.0 | 1.05/1.84 | 38.1 |
| C-13 | - | 44.2 | - | 44.0 | - | 44.1 | - | 44.2 |
| CH-14 | 0.95 | 52.4 | 0.93 | 52.4 | 1.00 | 52.4 | 0.96 | 52.4 |
| CH2-15 | 1.25/1.60 | 24.3 | 1.27/1.62 | 24.3 | 1.30/1.62 | 24.3 | 1.26/1.60 | 24.3 |
| CH2-16 | 1.46/1.98 | 30.6 | 1.45/1.98 | 30.6 | 1.50/2.00 | 30.6 | 1.47/1.98 | 30.6 |
| CH-17 | 3.57 | 82.5 | 3.57 | 82.5 | 3.57 | 82.5 | 3.57 | 82.5 |
| CH3-18 | 0.74 | 11.7 | 0.74 | 11.6 | 0.75 | 11.7 | 0.74 | 11.7 |
| CH3-19 | 1.04 | 14.6 | 1.11 | 14.5 | 0.86 | 12.9 | 1.05 | 15.6 |
| CH2-2′/CH2-6′ | 2.53/2.63 | 51.4/51.9 | 2.63/2.68 | 53.8/54.4 | 2.65/2.70 | 50.9/51.3 | 2.50/2.56 | 51.4/51.9 |
| CH2-3′/CH2-5′ | 3.57 | 43.1/47.2 | 3.50/3.57 | 43.6/47.7 | 3.58 | 43.0/47.1 | 3.57 | 43.1/47.2 |
| C-1″ | - | 174.1 | - | 174.1 | - | 174.1 | - | 174.0 |
| CH2-2″ | 2.40 | 34.0 | 2.38 | 34.0 | 2.40 | 34.0 | 2.40 | 34.0 |
| CH2-3″ | 1.61 | 26.6 | 1.61 | 26.6 | 1.62 | 26.6 | 1.61 | 26.6 |
| CH2-4″ | 1.34 | 30.3 | 1.34 | 30.3 | 1.36 | 30.3 | 1.35 | 30.3 |
| CH2-5″ | 1.34 | 30.5 | 1.34 | 30.5 | 1.36 | 30.5 | 1.35 | 30.5 |
| CH2-6″ | 1.34 | 30.5 | 1.34 | 30.4 | 1.36 | 30.4 | 1.35 | 30.4 |
| CH2-7″ | 1.34 | 33.0 | 1.34 | 33.0 | 1.36 | 33.0 | 1.33 | 33.0 |
| CH2-8″ | 1.34 | 23.7 | 1.34 | 23.7 | 1.35 | 23.7 | 1.33 | 23.7 |
| CH2-9″ | 0.92 | 14.4 | 0.92 | 14.4 | 0.92 | 14.4 | 0.92 | 14.5 |
| Compound | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| Structure |  |  |  |  |
| Substitution | 2β-R (axial) 3α-OH (axial) | 4β-R (axial) 3α-OH (axial) | 2α-R (equatorial) 3α-OH (axial) | 3α-R (axial) 2β-OH (axial) |
| AA (%)a | 82 | 1 | 42 | 51 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poirier, D.; Raad, I.; Roy, J.; Maltais, R. Synthesis, NMR Characterization, and Antileukemic Activity of N-Nonanoylpiperazinyl-5α-Androstane-3α,17β-Diol A-Ring Derivatives. Magnetochemistry 2021, 7, 3. https://doi.org/10.3390/magnetochemistry7010003
Poirier D, Raad I, Roy J, Maltais R. Synthesis, NMR Characterization, and Antileukemic Activity of N-Nonanoylpiperazinyl-5α-Androstane-3α,17β-Diol A-Ring Derivatives. Magnetochemistry. 2021; 7(1):3. https://doi.org/10.3390/magnetochemistry7010003
Chicago/Turabian StylePoirier, Donald, Imad Raad, Jenny Roy, and René Maltais. 2021. "Synthesis, NMR Characterization, and Antileukemic Activity of N-Nonanoylpiperazinyl-5α-Androstane-3α,17β-Diol A-Ring Derivatives" Magnetochemistry 7, no. 1: 3. https://doi.org/10.3390/magnetochemistry7010003
APA StylePoirier, D., Raad, I., Roy, J., & Maltais, R. (2021). Synthesis, NMR Characterization, and Antileukemic Activity of N-Nonanoylpiperazinyl-5α-Androstane-3α,17β-Diol A-Ring Derivatives. Magnetochemistry, 7(1), 3. https://doi.org/10.3390/magnetochemistry7010003