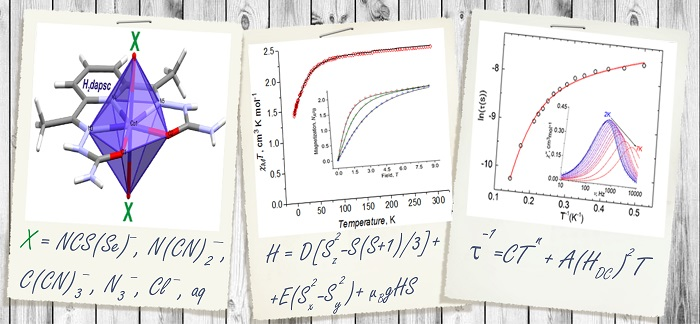
A Series of Field-Induced Single-Ion Magnets Based on the Seven-Coordinate Co(II) Complexes with the Pentadentate (N3O2) H2dapsc Ligand

 , ,
, ,
Abstract

1. Introduction
2. Results and Discussion
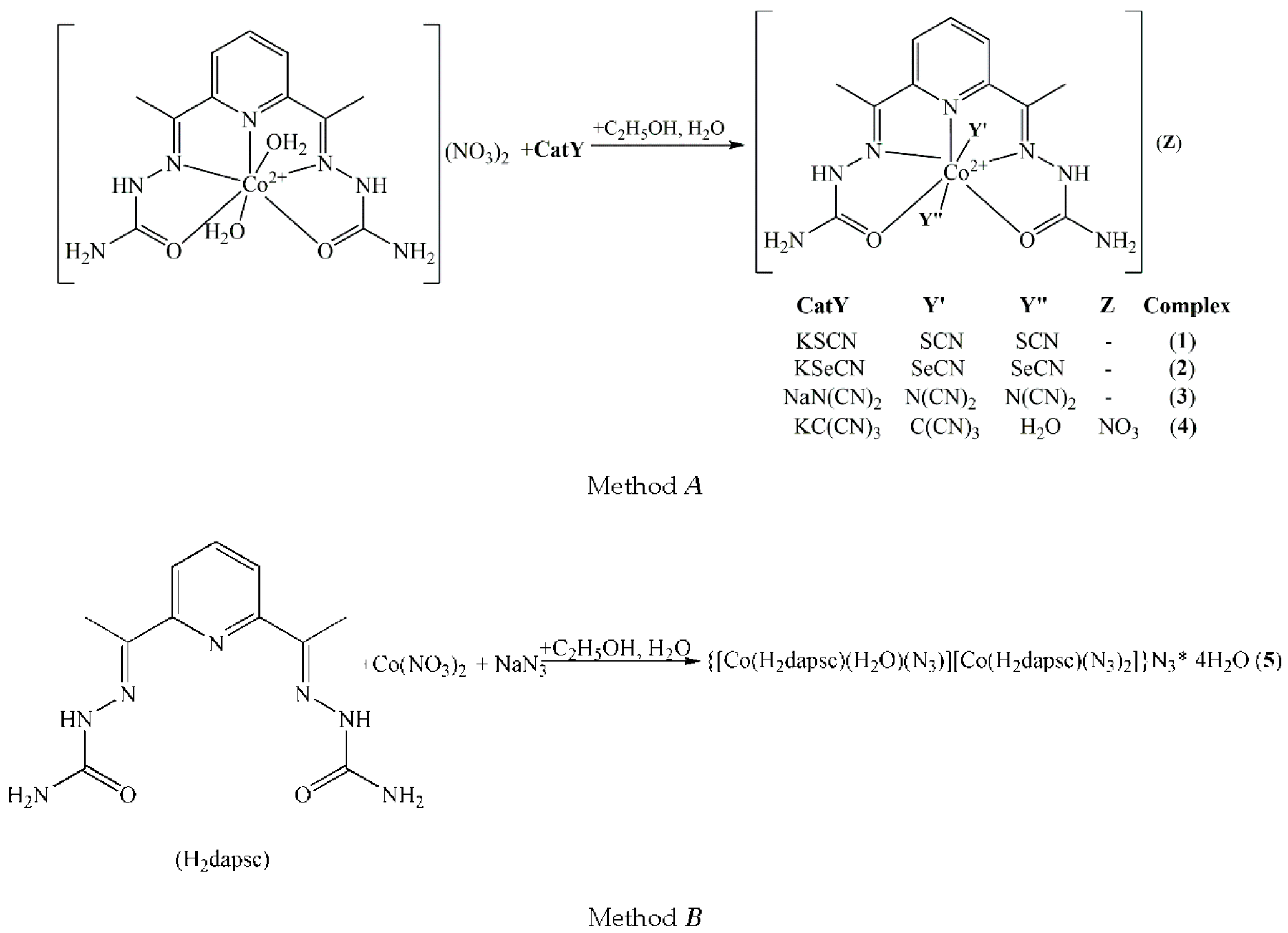
2.1. Synthesis and Characterization
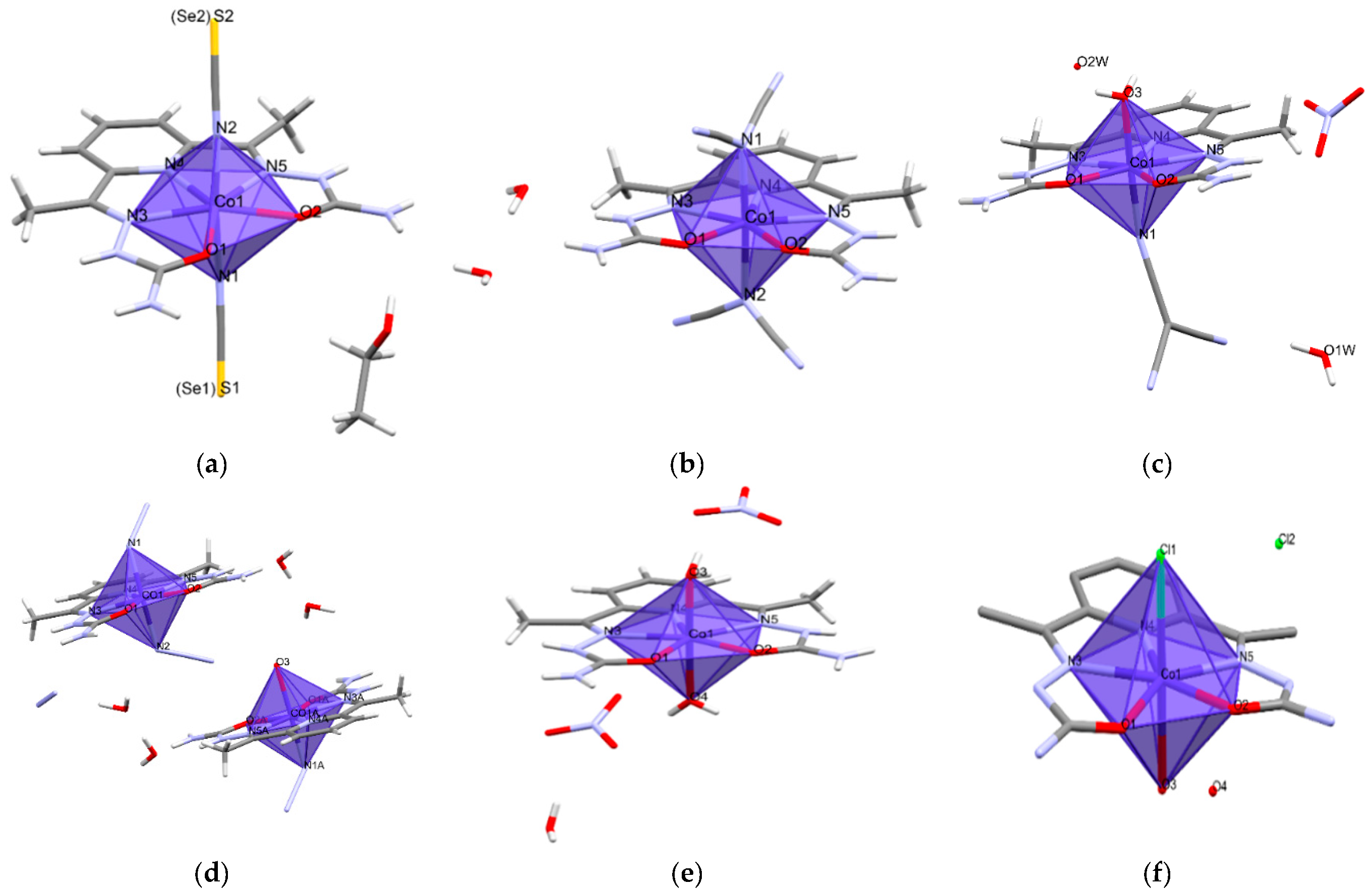
2.2. Description of the Structure
2.3. Magnetic Properties
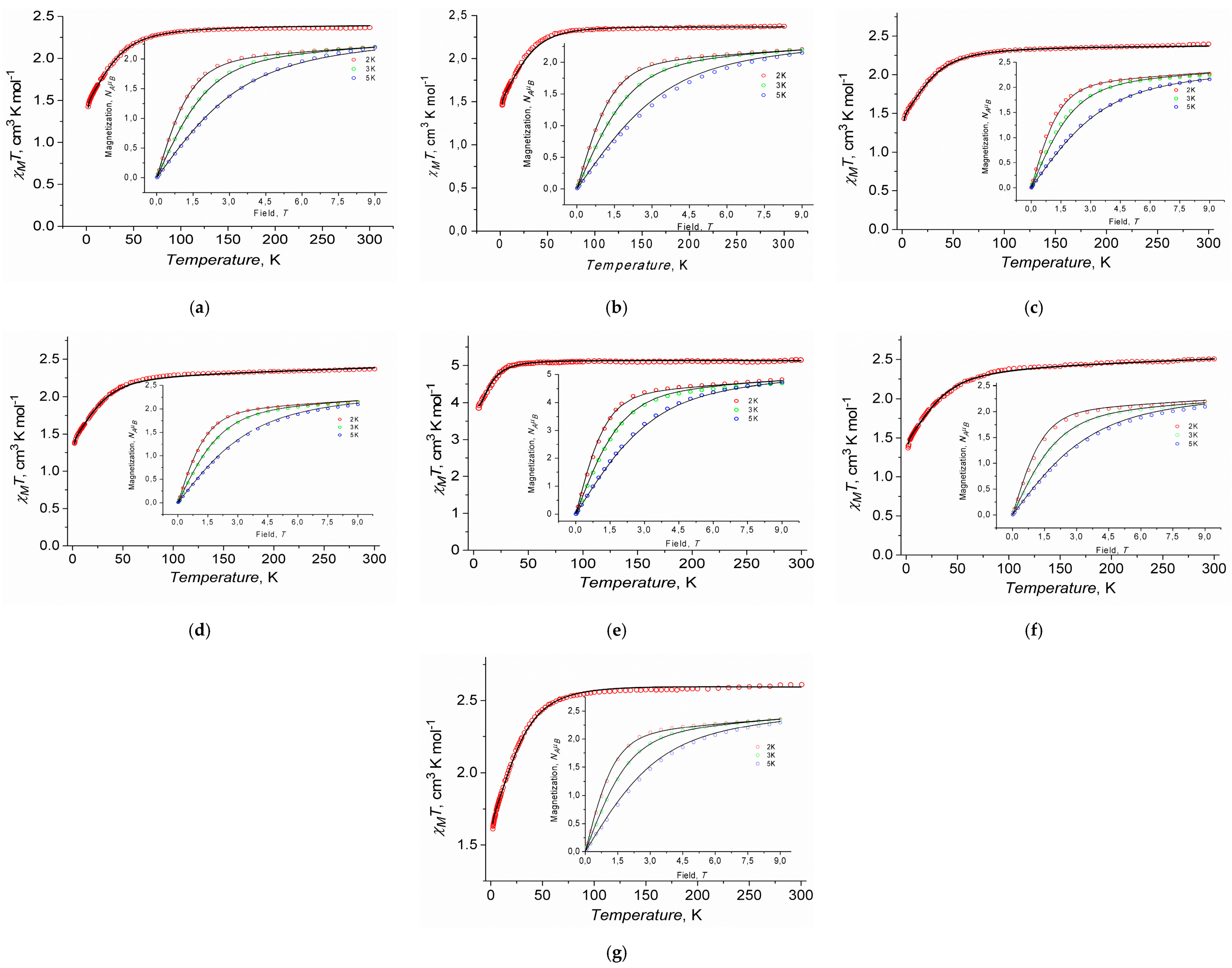
2.3.1. Static Magnetic Measurements
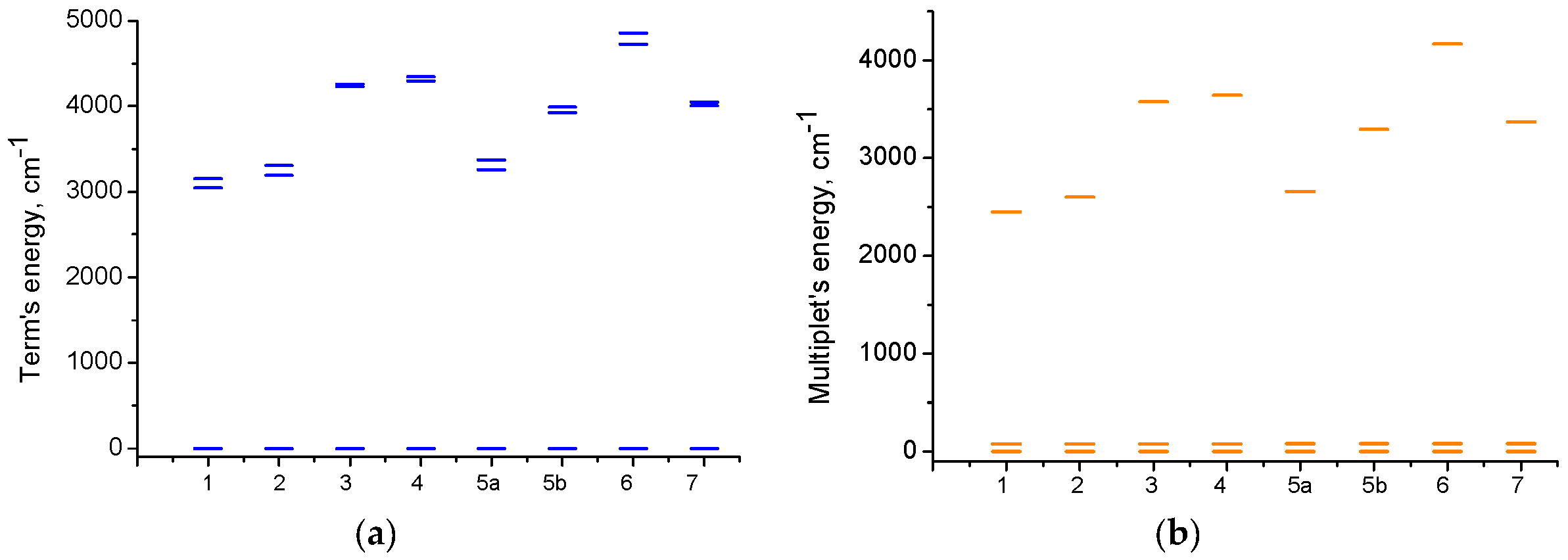
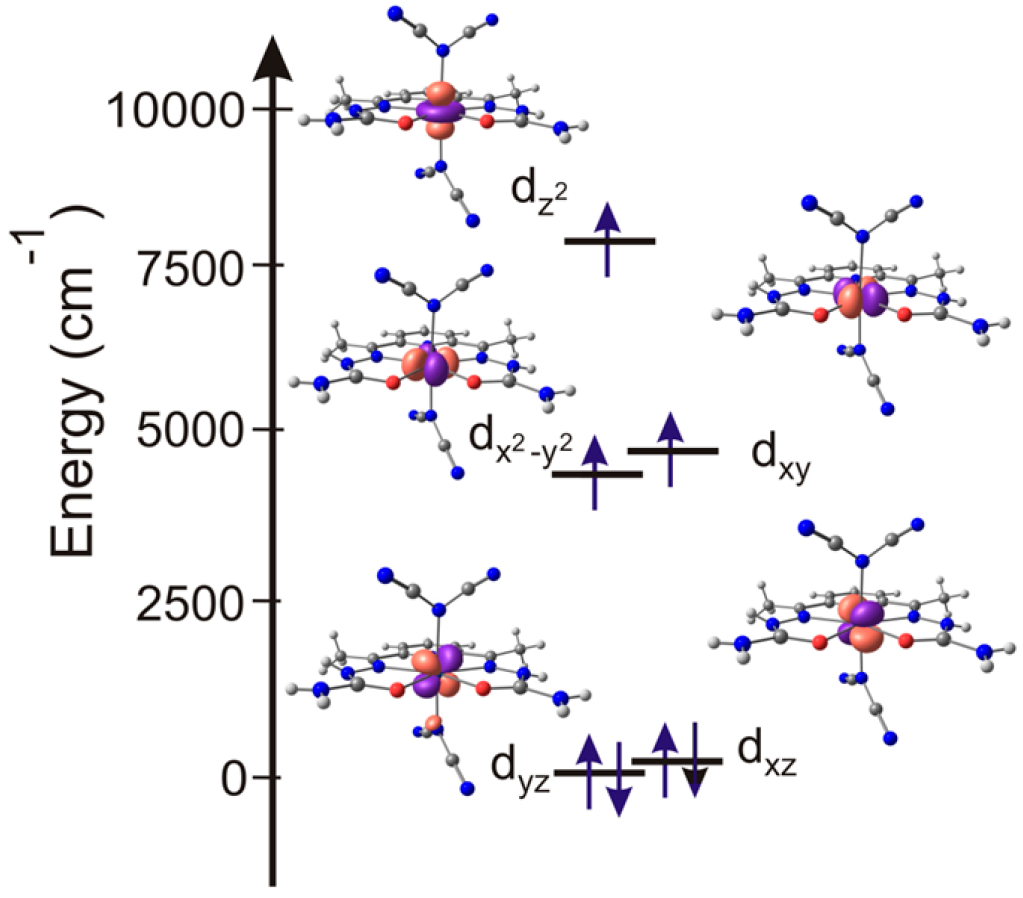
2.3.2. Quantum Chemical Calculations
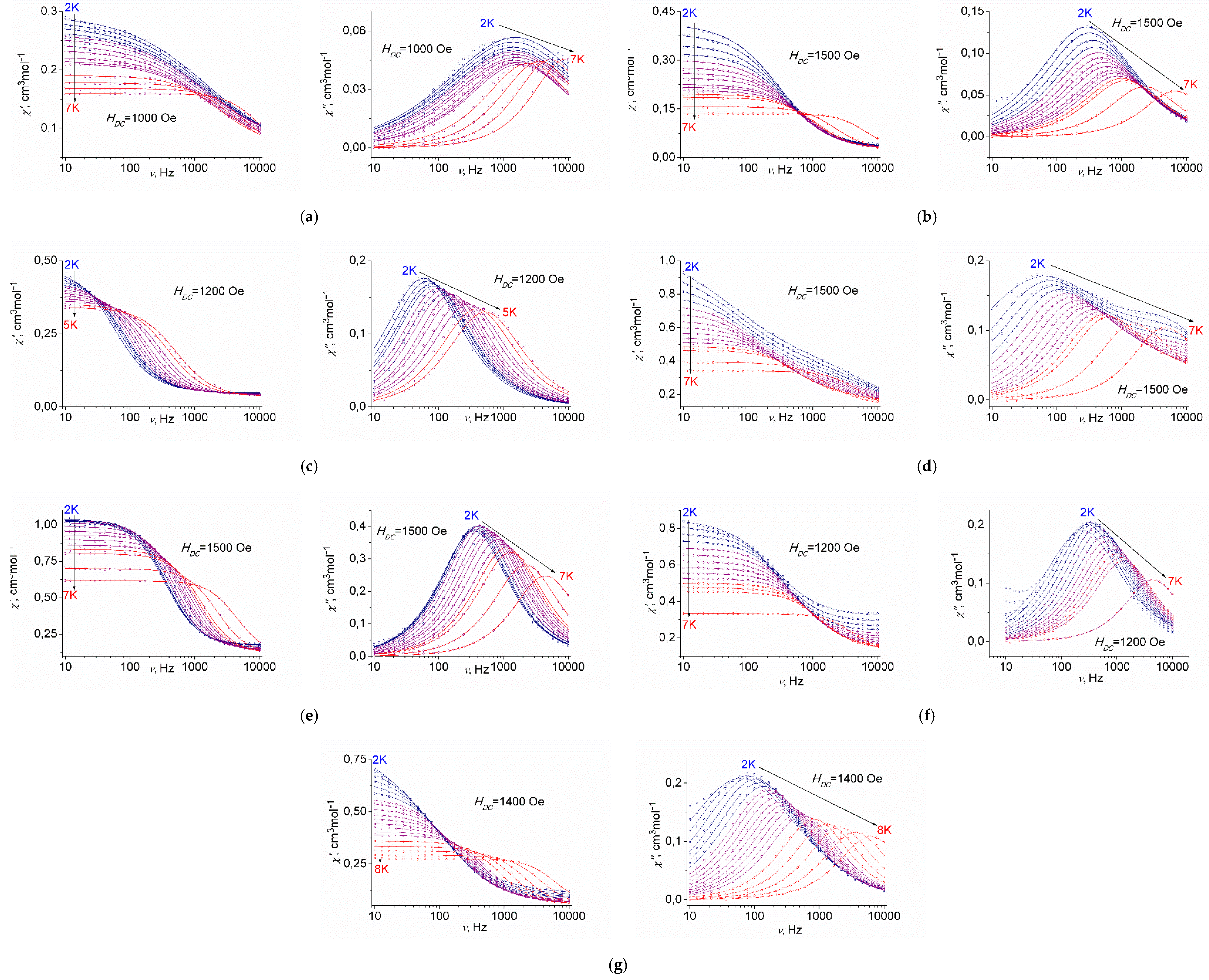
2.3.3. AC Susceptibility Data
3. Materials and Methods
3.1. Synthesis
3.2. X-ray Crystal Structure
3.3. Physical Measurements
3.4. Computational Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Milios, C.; Winpenny, R. Cluster-Based Single-Molecule Magnet. In Molecular Nanomagnets and Related Phenomena, 1st ed.; Gao, S., Ed.; Springer: Berlin/Heidelberg, Germany, 2015; Volume 164, pp. 1–109. [Google Scholar]
- Neese, F.; Pantazis, D. What is not required to make a single molecule magnet. Faraday Discuss. 2011, 148, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Waldmann, O. A Criterion for the Anisotropy Barrier in Single-Molecule Magnets. Inorg. Chem. 2007, 46, 10035–10037. [Google Scholar] [CrossRef] [PubMed]
- Aldoshin, S.M.; Korchagin, D.V.; Palii, A.V.; Tsukerblat, B.S. Some new trends in the design of single molecule magnets. Pure Appl. Chem. 2017, 89, 1119–1144. [Google Scholar] [CrossRef]
- Bar, A.K.; Pichon, C.; Sutter, J.P. Magnetic anisotropy in two- to eight-coordinated transition-metal complexes: Recent developments in molecular magnetism. Coord. Chem. Rev. 2016, 308, 346–380. [Google Scholar] [CrossRef]
- Craig, G.; Murrie, M. 3d single-ion magnets. Chem. Soc. Rev. 2015, 44, 2135–2147. [Google Scholar] [CrossRef]
- Zadrozny, J.M.; Xiao, D.J.; Atanasov, M.; Long, G.J.; Grandjean, F.; Neese, F.; Long, J.R. Magnetic blocking in a linear iron (I) complex. Nat. Chem. 2013, 5, 577–581. [Google Scholar] [CrossRef]
- Gómes-Coca, S.; Cremades, E.; Aliaga-Alcalde, N.; Ruiz, E. Mononuclear Single-Molecule Magnets: Tailoring the Magnetic Anisotropy of First-Row Transition-Metal Complexes. J. Amer. Chem. Soc. 2013, 135, 7010–7018. [Google Scholar] [CrossRef]
- Poulten, R.C.; Page, M.J.; Algarra, A.G.; Le Roy, J.J.; Lopez, I.; Carter, E.; Llobet, A.; Macgregor, S.A.; Mahon, M.F.; Murphy, D.M.; et al. Synthesis, Electronic Structure, and Magnetism of [Ni(6-Mes)2]+: A Two-Coordinate Nickel(I) Complex Stabilized by Bulky N-Heterocyclic Carbenes. J. Am. Chem. Soc. 2013, 135, 13640–13643. [Google Scholar] [CrossRef]
- Ruamps, R.; Maurice, R.; Batchelor, L.; Boggio-Pasqua, M.; Guillot, R.; Barra, A.; Liu, J.; Bendeif, E.; Pillet, S.; Hill, S.; et al. Giant Ising-Type Magnetic Anisotropy in Trigonal Bipyramidal Ni(II) Complexes: Experiment and Theory. J. Amer. Chem. Soc. 2013, 135, 3017–3026. [Google Scholar] [CrossRef]
- Saber, M.R.; Dunbar, K.R. Ligands effects on the magnetic anisotropy of tetrahedral cobalt complexes. Chem. Commun. 2014, 50, 12266–12269. [Google Scholar] [CrossRef]
- Zhang, Y.-Z.; Gómez-Coca, S.; Brown, A.J.; Saber, M.R.; Zhang, X.; Dunbar, K.R. Trigonal antiprismatic Co(II) single molecule magnets with large uniaxial anisotropies: importance of Raman and tunneling mechanisms. Chem. Sci. 2016, 7, 6519–6527. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.-N.; Du, J.-Z.; Zhang, Y.-Q.; Leng, X.-B.; Yang, M.-W.; Jiang, S.-D.; Wang, Z.-X.; Ouyang, Z.-W.; Deng, L.; Wang, B.-W.; et al. Two-Coordinate Co(II) Imido Complexes as Outstanding Single-Molecule Magnets. J. Am. Chem. Soc. 2017, 139, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Palii, A.V.; Korchagin, D.V.; Yureva, E.A.; Akimov, A.V.; Misochko, E.Y.; Shilov, G.V.; Talantsev, A.D.; Morgunov, R.B.; Aldoshin, S.M.; Tsukerblat, B.S. Single-Ion Magnet Et4N[CoII(hfac)3] with Nonuniaxial Anisotropy: Synthesis, Experimental Characterization, and Theoretical Modeling. Inorg. Chem. 2016, 55, 9696–9706. [Google Scholar] [CrossRef] [PubMed]
- Tupolova, Y.P.; Shcherbakov, I.N.; Popov, L.D.; Lebedev, V.E.; Tkachev, V.V.; Zakharov, K.V.; Vasiliev, A.N.; Korchagin, D.V.; Palii, A.V.; Aldoshin, S.M. Field-induced single-ion magnet behavior of a hexacoordinated Co(II) complex with easy-axis-type magnetic anisotropy. Dalton Trans. 2019, 48, 6960–6970. [Google Scholar] [CrossRef] [PubMed]
- Platas-Iglesias, C.; Vaiana, L.; Esteban-Gómez, D.; Avecilla, F.; Real, J.; de Blas, A.; Rodríguez-Blas, T. Electronic Structure Study of Seven-Coordinate First-Row Transition Metal Complexes Derived from 1,10-Diaza-15-crown-5: A Successful Marriage of Theory with Experiment. Inorg. Chem. 2005, 44, 9704–9713. [Google Scholar] [CrossRef]
- Batchelor, L.; Sangalli, M.; Guillot, R.; Guihéry, N.; Maurice, R.; Tuna, F.; Mallah, T. Pentanuclear Cyanide-Bridged Complexes Based on Highly Anisotropic CoII Seven-Coordinate Building Blocks: Synthesis, Structure, and Magnetic Behavior. Inorg. Chem. 2011, 50, 12045–12052. [Google Scholar] [CrossRef]
- Venkatakrishnan, T.; Sahoo, S.; Bréfuel, N.; Duhayon, C.; Paulsen, C.; Barra, A.; Ramasesha, S.; Sutter, J. Enhanced Ion Anisotropy by Nonconventional Coordination Geometry: Single-Chain Magnet Behavior for a [{FeIIL}2{NbIV(CN)8}] Helical Chain Compound Designed with Heptacoordinate Fe(II). J. Am. Chem. Soc. 2010, 132, 6047–6056. [Google Scholar] [CrossRef]
- Huang, X.; Zhou, C.; Shao, D.; Wang, X. Field-Induced Slow Magnet Relaxation in Cobalt(II) Compounds with Pentagonal Bipyramid Geometry. Inorg. Chem. 2014, 53, 12671–12673. [Google Scholar] [CrossRef]
- Ruamps, R.; Batchelor, L.; Maurice, R.; Gogoi, N.; Jiménez-Lozano, P.; Guihéry, N.; de Graaf, C.; Barra, A.; Sutter, J.; Mallah, T. Origin of the Magnetic Anisotropy in Heptacoordinate NiII and CoII Complexes. Chem. Eur. J. 2013, 19, 950–956. [Google Scholar] [CrossRef]
- Gogoi, N.; Thlijeni, M.; Duhayon, C.; Sutter, J. Heptacoordinated Nickel(II) as an Ising-Type Anisotropic Building Unit: Illustration with a Pentanuclear [(NiL)3{W(CN)8}2] Complex. Inorg. Chem. 2013, 52, 2283–2285. [Google Scholar] [CrossRef]
- Regueiro-Figueroa, M.; Lima, L.; Blanco, V.; Esteban-Gomez, D.; de Blas, A.; Rodriguez-Blas, T.; Delgado, R.; Platas-Iglesias, C. Reasons behind the relative abundances of heptacoordinate complexes along the late first-row transition metal series. Inorg. Chem. 2014, 53, 12859–12869. [Google Scholar] [CrossRef]
- Gavey, E.L.; Pilkington, M. Coordination complexes of 15-membered pentadentate aza, oxoaza and thiaaza Schiff base macrocycles “Old Complexes Offer New Attractions”. Coord. Chem. Rev. 2015, 296, 125–152. [Google Scholar] [CrossRef]
- Shao, D.; Zhang, S.; Zhang, Y.; Wang, X. Probing the Effect of Axial Ligands on Easy-Plany Anisotropy of Pentagonal-Bipyramidal Cobalt(II) Single-Ion Magnets. Inorg. Chem. 2016, 55, 10859–10869. [Google Scholar] [CrossRef]
- Drahoš, B.; Herchel, R.; Trávníček, Z. Impact of Halogenido Coligands on Magnetic Anisotropy in Seven-Coordinate Co(II) Complexes. Inorg. Chem. 2017, 56, 5076–5088. [Google Scholar] [CrossRef]
- Giordano, T.J.; Palenik, G.J.; Palenic, R.C.; Sullivan, D.A. Pentagonal-Bipyramidal Complexes. Synthesis and Characterization of Aqua(nitrato)[2,6-diacetylpyridinebis-(benzonic acid hydrazone)]cobalt(II) Nitrate and Diaqua[2,6-diacetylpyridinebis(benzonic acid hydrazone)]nickel(II) Nitrate Dihydrate. Inorg. Chem. 1979, 18, 2445–2450. [Google Scholar] [CrossRef]
- Ivanovic-Burmazovic, I.; Andjelkovic, K. Transition metal complexes with bis(hydrazone) ligands of 2,6-diacetylpyridine hepta-coordination of 3d metals. In Book Advances Inorganic Chemistry, 1st ed.; van Eldik, R., Ed.; Elsevier: Amsterdam, The Netherlands, 2004; Volume 55, pp. 315–360. [Google Scholar]
- Carcelli, M.; Ianelli, S.; Pelagatti, P.; Pelizzi, G. Structural characterization of a new ligand mode of 2,6-diacetylpyridine bis(semicarbazone), H2daps. Inorg. Chim. Acta 1999, 292, 121–126. [Google Scholar] [CrossRef]
- Palenik, G.J.; Wester, D.W. Pentagonal-bipyramidal complexes. Crystal and molecular structures of chloroaqua(2,6-diacetylpyridine bis(semicarbazone))manganese(II), -iron(II), -cobalt(II), and -zinc(II) chloride dihydrates. Inorg. Chem. 1978, 17, 864–870. [Google Scholar] [CrossRef]
- Palenik, G.J.; Wester, D.W.; Rychlewska, U.; Palenik, R.C. Pentagonal-bipyramidal complexes. Synthesis and crystal structures of diaqua[2,6-diacetylpyridine bis(semicarbazone)]chromium(III) hydroxide dinitrate hydrate and dichloro[2,6-diacetylpyridine bis(semicarbazone)]iron(III) chloride dihydrate. Inorg. Chem. 1976, 15, 1814–1819. [Google Scholar] [CrossRef]
- Bar, A.; Pichon, C.; Gogoi, N.; Duhayon, C.; Ramasesha, S.; Sutter, J. Single-ion magnet behaviour of heptacoordinated Fe(II) complexes: on the importance of supramolecular organization. Chem. Commun. 2015, 51, 3616–3619. [Google Scholar] [CrossRef]
- Habib, F.; Korobkov, I.; Murugesu, M. Exposing the intermolecular nature of the second relaxation pathway in a mononuclear cobalt(II) single-molecule magnet with positive anisotropy. Dalton Trans. 2015, 44, 6368–6373. [Google Scholar] [CrossRef] [PubMed]
- Dey, M.; Dutta, S.; Sarma, B.; Deka, R.C.; Gogoi, N. Modulation of the coordination environment: a convenient approach to tailor magnetic anisotropy in seven coordinate Co(II) complexes. Chem. Commun. 2016, 52, 753–756. [Google Scholar] [CrossRef] [PubMed]
- Mondal, A.K.; Mondal, A.; Dey, B.; Konar, S. Influence of the Coordination Environment on Easy-Plane Magnetic Anisotropy of Pentogonal Bipyramidal Cobalt(II) Complexes. Inorg. Chem. 2018, 57, 9999–10008. [Google Scholar] [CrossRef] [PubMed]
- Bar, A.; Gogoi, N.; Pichon, C.; Goli, V.; Thlijeni, M.; Duhayon, C.; Suaud, N.; Guihery, N.; Barra, A.; Ramasesha, S.; et al. Pentagonal Bipyramid FeII Complexes: Robust Ising-Spin Units towards Heteropolynuclear Nanomagnets. Chem. Eur. J. 2017, 23, 4380–4396. [Google Scholar] [CrossRef] [PubMed]
- Kopotkov, V.A.; Sasnovskaya, V.D.; Korchagin, D.V.; Morgunov, R.B.; Aldoshin, S.M.; Simonov, S.V.; Zorina, L.V.; Schaniel, D.; Woike, T.; Yagubskii, E.B. The first photochromic bimetallic assemblies based on Mn(III) and Mn(II) Schiff-base (salpn, dapsc) complexes and pentacyanonitrosylferrate(II). CrystEngComm. 2015, 17, 3866–3876. [Google Scholar] [CrossRef]
- Sasnovskaya, V.D.; Kopotkov, V.A.; Talantsev, A.D.; Morgunov, R.B.; Yagubskii, E.B.; Simonov, S.V.; Zorina, L.V.; Mironov, V.S. Synthesis, Structure, and Magnetic Properties of 1D {[MnIII(CN)6][MnII(dapsc)]}n Coordination Polymers: Origin of Unconventional Single-Chain Magnet Behavior. Inorg. Chem. 2017, 56, 8926–8943. [Google Scholar] [CrossRef]
- Pichon, C.; Suaud, N.; Duhayon, C.; Guihéry, N.; Sutter, J. Cyano-Bridged Fe(II)−Cr(III) Single-Chain Magnet Based on Pentagonal Bipyramid Units: On the Added Value of Aligned Axial Anisotropy. J. Amer. Chem. Soc. 2018, 140, 7698–7704. [Google Scholar] [CrossRef]
- Gomez-Coca, S.; Urtizberea, A.; Cremades, E.; Alonso, P.; Camon, A.; Ruiz, E.; Luis, F. Origin of slow magnetic relaxation in Kramers ions with non-uniaxial anisotropy. Nat. Commun. 2014, 5, 1–8. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Cryst. 2016, B72, 171–179. [Google Scholar] [CrossRef]
- Casanova, D.; Alemany, P.; Bofill, J.; Alvarez, S. Shape and symmetry of heptacoordinate transition-metal complexes: structural trends. Chem. Eur. J. 2003, 9, 1281–1295. [Google Scholar] [CrossRef]
- Alvarez, S.; Alemany, P.; Casanova, D.; Cirera, J.; Llunell, M.; Avnir, D. Shape maps and polyhedral interconversion paths in transition metal chemistry. Coord. Chem. Rev. 2005, 249, 1693–1708. [Google Scholar] [CrossRef]
- Mabbs, F.; Machin, D. Magnetism and Transition Metal Complexes, 2nd ed.; Dover Publications Inc.: New York, NY, USA, 2008; pp. 1–206. [Google Scholar]
- Guo, Y.; Xu, G.; Gamez, P.; Zhao, L.; Lin, S.; Deng, R.; Tang, J.; Zhang, H.J. Two-step relaxation in a linear tetranuclear dysprosium(III) aggregate showing single-molecule magnet behavior. J. Am. Chem. Soc. 2010, 132, 8538–8539. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Xu, G.F.; Guo, Y.; Tang, J. Relaxation dynamics of dysprosium(III) single molecule magnets. Dalton Trans. 2011, 40, 9953–9963. [Google Scholar] [CrossRef] [PubMed]
- Varga, F.; Rajnák, C.; Titiš, J.; Moncoľ, J.; Boča, R. Slow magnetic relaxation in a Co(II) octahedral-tetrahedral system formed of a [CoL3]2+ core with L = bis(diphenylphosphanoxido) methane and tetrahedral [CoBr4]2− counter anions. Dalton Trans. 2017, 46, 4148–4151. [Google Scholar] [CrossRef]
- Rajnák, C.; Titiš, J.; Moncoľ, J.; Renz, F.; Boča, R. Field-Supported Slow Magnetic Relaxation in Hexacoordinate CoII Complexes with Easy Plane Anisotropy. Eur. J. Inorg. Chem. 2017, 2017, 1520–1525. [Google Scholar] [CrossRef]
- Titiš, J.; Rajnák, C.; Valigura, D.; Boča, R. Breaking the Magic Border of One Second for Slow Magnetic Relaxation of Cobalt-Based Single Ion Magnets. Inorg. Chem. 2018, 57, 14314–14321. [Google Scholar]
- Mandal, S.; Mondal, S.; Rajnák, C.; Titiš, J.; Boča, R.; Mohanta, S. Syntheses, crystal structures and magnetic properties of two mixed-valence Co(III)Co(II) compounds derived from Schiff base ligands: field-supported single-ion-magnet behavior with easy-plane anisotropy. Dalton Trans. 2017, 46, 13135–13144. [Google Scholar] [CrossRef] [PubMed]
- Chilton, N.F. CC-fit, The University of Manchester, UK. 2014. Available online: http://www.nfchilton.com/cc-fit.html.
- Cole, K.S.; Cole, R.H. Dispersion and Absorption in Dielectrics, I. Alternating Current Characteristics. J. Chem. Phys. 1941, 9, 341–351. [Google Scholar] [CrossRef]
- Ishikawa, R.; Horii, Y.; Nakanishi, R.; Ueno, S.; Breedlove, B.; Yamashita, M.; Kawata, S. Field-Induced Single-Ion Magnetism Based on Spin-Phonon Relaxation in a Distorted Octahedral High-Spin Cobalt(II) Complex. Eur. J. Inorg. Chem. 2016, 2016, 3233–3239. [Google Scholar] [CrossRef]
- Korchagin, D.V.; Palii, A.A.V.; Yureva, E.A.; Akimov, A.V.; Misochko, E.Y.; Shilov, G.V.; Talantsev, A.D.; Morgunov, R.B.; Shakin, A.A.; Aldoshin, S.M.; et al. Evidence of field induced slow magnetic relaxation in: cis-[Co(hfac)2(H2O)2] exhibiting tri-axial anisotropy with a negative axial component. Dalton Trans. 2017, 46, 7540–7548. [Google Scholar] [CrossRef]
- Gatteschi, D.; Sessoli, R. Quantum tunneling of magnetization and related phenomena in molecular materials. Angew. Chem. Int. Ed. 2003, 42, 268–297. [Google Scholar] [CrossRef]
- Agilent. CrysAlis PRO Version 171.35.19; Agilent Technologies UK Ltd.: Yarnton, Oxfordshire, UK, 2011. [Google Scholar]
- Sheldrick, G.M. SHELXTL v. 6.14, Structure Determination Software Suite; Bruker AXS: Madison, WI, USA, 2000. [Google Scholar]
- Roos, B.O.; Taylor, P.R.; Siegbahn, P.E. A complete active space SCF method (CASSCF) using a density matrix formulated super-CI approach. Chem. Phys. 1980, 48, 157–173. [Google Scholar] [CrossRef]
- Per, S.; Heiberg, A.; Roos, B.; Levy, B. A Comparison of the Super-CI and the Newton-Raphson Scheme in the Complete Active Space SCF Method. Phys. Scr. 1980, 21, 323–327. [Google Scholar]
- Siegbahn, P.; Almlöf, J.; Heiberg, A.; Roos, B. The complete active space SCF (CASSCF) method in a Newton–Raphson formulation with application to the HNO molecule. J. Chem. Phys. 1981, 74, 2384–2396. [Google Scholar] [CrossRef]
- Angeli, C.; Cimiraglia, R.; Evangelisti, S.; Leininger, T.; Malrieu, J. Introduction of n-electron valence states for multireference perturbation theory. J. Chem. Phys. 2001, 114, 10252–10264. [Google Scholar] [CrossRef]
- Angeli, C.; Cimiraglia, R.; Malrieu, J. N-electron valence state perturbation theory: a fast implementation of the strongly contracted variant. Chem. Phys. Lett. 2001, 350, 297–305. [Google Scholar] [CrossRef]
- Angeli, C.; Cimiraglia, R. Multireference perturbation configuration interaction V. Third-order energy contributions in the Møller–Plesset and Epstein–Nesbet partitions. Theor. Chem. Acc. 2002, 107, 313–317. [Google Scholar] [CrossRef]
- Angeli, C.; Cimiraglia, R.; Malrieu, J. n-electron valence state perturbation theory: A spinless formulation and an efficient implementation of the strongly contracted and of the partially contracted variants. J. Chem. Phys. 2002, 17, 9138–9153. [Google Scholar] [CrossRef]
- Hess, B.A. Relativistic electronic-structure calculations employing a two-component no-pair formalism with external-field projection operators. Phys. Rev. A 1986, 33, 3742–3748. [Google Scholar] [CrossRef]
- Pantazis, D.A.; Chen, X.-Y.; Landis, C.R.; Neese, F. All-Electron Scalar Relativistic Basis Sets for Third-Row Transition Metal Atoms. J. Chem. Theory Comput. 2008, 4, 908–919. [Google Scholar] [CrossRef]
- Schafer, A.; Huber, C.; Ahlrichs, R. Fully optimized contracted Gaussian basis sets of triple zeta valence quality for atoms Li to Kr. J. Chem. Phys. 1994, 100, 5829–5835. [Google Scholar] [CrossRef]
- Schafer, A.; Horn, H.; Ahlrichs, R. Fully optimized contracted Gaussian basis sets for atoms Li to Kr. J. Chem. Phys. 1992, 97, 2571–2577. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Ganyushin, D.; Neese, F. First-principles calculations of zero-field splitting parameters. J. Chem. Phys. 2006, 125, 024103-11. [Google Scholar] [CrossRef] [PubMed]
- Neese, F. Efficient and accurate approximations to the molecular spin-orbit coupling operator and their use in molecular g-tensor calculations. J. Chem. Phys. 2005, 122, 034107-13. [Google Scholar] [CrossRef] [PubMed]
- Maurice, R.; Bastardis, R.; Graaf, C.; Suaud, N.; Mallah, T.; Guihéry, N. Universal Theoretical Approach to Extract Anisotropic Spin Hamiltonians. J. Chem. Theory Comput. 2009, 5, 2977–2984. [Google Scholar] [CrossRef]
- Atanasov, M.; Ganyushin, D.; Sivalingam, K.; Neese, F. A Modern First-Principles View on Ligand Field Theory Through the Eyes of Correlated Multireference Wavefunctionsin. In Molecular Electronic Structures of Transition Metal Complexes II, 1st ed.; Mingos, D., Day, P., Dahl, J., Eds.; Springer-Verlag: Berlin/Heidelberg, Germany, 2012; Volume 143, pp. 149–220. [Google Scholar]
- Singh, S.; Eng, J.; Atanasov, M.; Neese, F. Covalency and chemical bonding in transition metal complexes: An ab initio based ligand field perspective. Coord. Chem. Rev. 2017, 344, 2–25. [Google Scholar] [CrossRef]
- Neese, F. The ORCA program system. Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Neese, F. Software update: the ORCA program system, version 4.0. Comput. Mol. Sci. 2018, 8, e1327-6. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 | 2 | 3 | 4 | 5a | 5b | 6 | 7 | |
|---|---|---|---|---|---|---|---|---|
| ZFS and g values based on CASSCF/NEVPT2 calculations with CAS(7,5) | ||||||||
| D (cm−1) | 38.02 | 37.73 | 37.51 | 37.49 | 39.93 | 39.58 | 38.99 | 38.94 |
| E/D | 0.0227 | 0.0213 | 0.0065 | 0.0093 | 0.0162 | 0.0143 | 0.0143 | 0.0133 |
| gx | 2.3442 | 2.3366 | 2.3220 | 2.3206 | 2.3590 | 2.3490 | 2.3284 | 2.3511 |
| gy | 2.3641 | 2.3554 | 2.3269 | 2.3286 | 2.3736 | 2.3617 | 2.3559 | 2.3625 |
| gz | 2.0049 | 2.0023 | 1.9936 | 1.9943 | 1.9971 | 1.9949 | 1.9925 | 2.0009 |
| giso | 2.2377 | 2.2314 | 2.2148 | 2.2145 | 2.2432 | 2.2352 | 2.2256 | 2.2382 |
| Analysis of the experimental magnetic data | ||||||||
| D (cm−1) | 35.6 | 38.20 | 35.3 | 33.60 | 40.4 | 38.02 | 35.61 | |
| E/D | 0.17 | 0.00 | 0.101 | 0.149 | − | 0.018 | 0.16 | |
| gx,y/gz | 2.29/2.14 | 2.36/1.90 | 2.28/2.13 | 2.26/2.00 | 2.48/2.00 | 2.28/2.16 | 2.45/2.11 | |
| giso | 2.24 | 2.26 | 2.23 | 2.18 | 2.33 | 2.24 | 2.34 | |
| χTIP | − | 1.0 × 10−4 | 1.0 × 10−4 | 5.0 × 10−4 | − | 5.0 × 10−4 | − | |
| d-AO | 1 | 2 | 3 | 4 | 5a | 5b | 6 | 7 |
|---|---|---|---|---|---|---|---|---|
| dyz | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| dxz | 253.2 | 274.5 | 119.3 | 326.3 | 513.4 | 254.8 | 330.2 | 339.0 |
| dx2-y2 | 2888.0 | 3026.5 | 4437.7 | 4652.2 | 3779.5 | 4244.3 | 5142.2 | 4371.6 |
| dxy | 3499.5 | 3688.2 | 4741.3 | 4770.7 | 4019.6 | 4445.7 | 5241.4 | 4531.5 |
| dz2 | 8044.8 | 8141.7 | 7885.8 | 7817.5 | 7742.2 | 7005.5 | 6019.4 | 6732.7 |
| Excited State | Contributions to Total D, cm−1 | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| No. | Mult | 1 | 2 | 3 | 4 | 5a | 5b | 6 | 7 |
| 3 | 4 | 17.26 | 16.79 | 15.03 | 15.65 | 17.45 | 17.09 | 17.03 | 16.62 |
| 4 | 4 | 15.27 | 14.79 | 15.14 | 14.90 | 16.26 | 16.30 | 14.20 | 15.74 |
| 5 | 2 | - | - | 13.28 | 13.51 | 11.65 | 13.15 | 13.63 | 12.82 |
| 6 | 2 | - | 4.56 | - | |||||
| 7 | 2 | −1.55 | −1.51 | −1.33 | −1.61 | −1.58 | −1.53 | ||
| 8 | 2 | 6.48 | 4.71 | −1.59 | −1.55 | −1.59 | −1.57 | −1.51 | −1.63 |
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | |
|---|---|---|---|---|---|---|---|
| n | 9 (Fixed*) | 7.4(3) | 7.3(2) | 5.6(3) | 5.8(4) | 4.2(2) | 9 (Fixed*) |
| A, Oe−2s−1K−1 | 1.03(2) × 10−3 | 4.10(9) × 10−4 | 1.29(2) × 10−4 | 7.7(6) × 10−5 | 2.9(1) × 10−4 | 2.9(1) × 10−4 | 1.11(3) × 10−4 |
| C, s−1K−n | 1.06(6) × 10−3 | 0.02(1) | 0.017(6) | 0.4(2) | 0.26(6) | 0.26(6) | 2.43(5) × 10−4 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kopotkov, V.A.; Korchagin, D.V.; Sasnovskaya, V.D.; Gilmutdinov, I.F.; Yagubskii, E.B. A Series of Field-Induced Single-Ion Magnets Based on the Seven-Coordinate Co(II) Complexes with the Pentadentate (N3O2) H2dapsc Ligand. Magnetochemistry 2019, 5, 58. https://doi.org/10.3390/magnetochemistry5040058
Kopotkov VA, Korchagin DV, Sasnovskaya VD, Gilmutdinov IF, Yagubskii EB. A Series of Field-Induced Single-Ion Magnets Based on the Seven-Coordinate Co(II) Complexes with the Pentadentate (N3O2) H2dapsc Ligand. Magnetochemistry. 2019; 5(4):58. https://doi.org/10.3390/magnetochemistry5040058
Chicago/Turabian StyleKopotkov, Vyacheslav A., Denis V. Korchagin, Valentina D. Sasnovskaya, Ildar F. Gilmutdinov, and Eduard B. Yagubskii. 2019. "A Series of Field-Induced Single-Ion Magnets Based on the Seven-Coordinate Co(II) Complexes with the Pentadentate (N3O2) H2dapsc Ligand" Magnetochemistry 5, no. 4: 58. https://doi.org/10.3390/magnetochemistry5040058
APA StyleKopotkov, V. A., Korchagin, D. V., Sasnovskaya, V. D., Gilmutdinov, I. F., & Yagubskii, E. B. (2019). A Series of Field-Induced Single-Ion Magnets Based on the Seven-Coordinate Co(II) Complexes with the Pentadentate (N3O2) H2dapsc Ligand. Magnetochemistry, 5(4), 58. https://doi.org/10.3390/magnetochemistry5040058