Characterization of Halogen Bonded Adducts in Solution by Advanced NMR Techniques


Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. 1D Nuclear Magnetic Resonance (NMR) Techniques: Applications
3. Advanced NMR Techniques: Theory
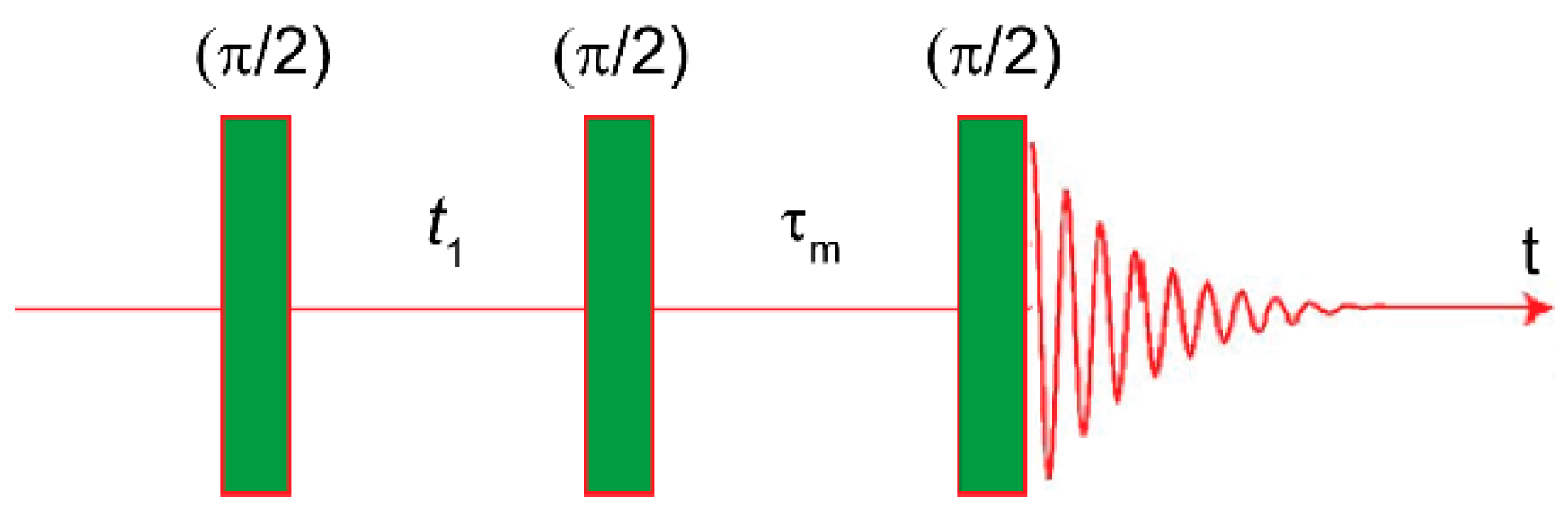
3.1. Nuclear Overhauser Effect
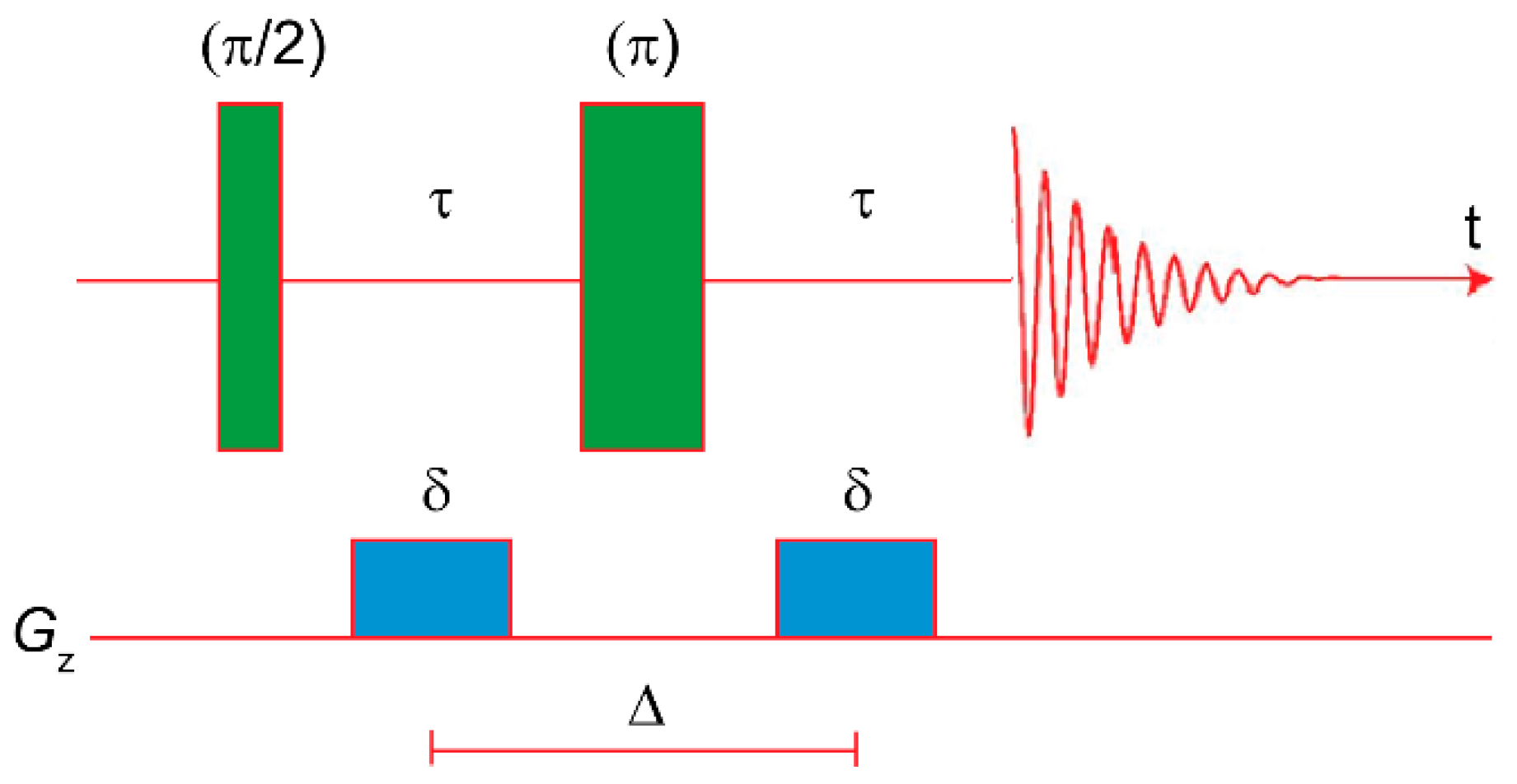
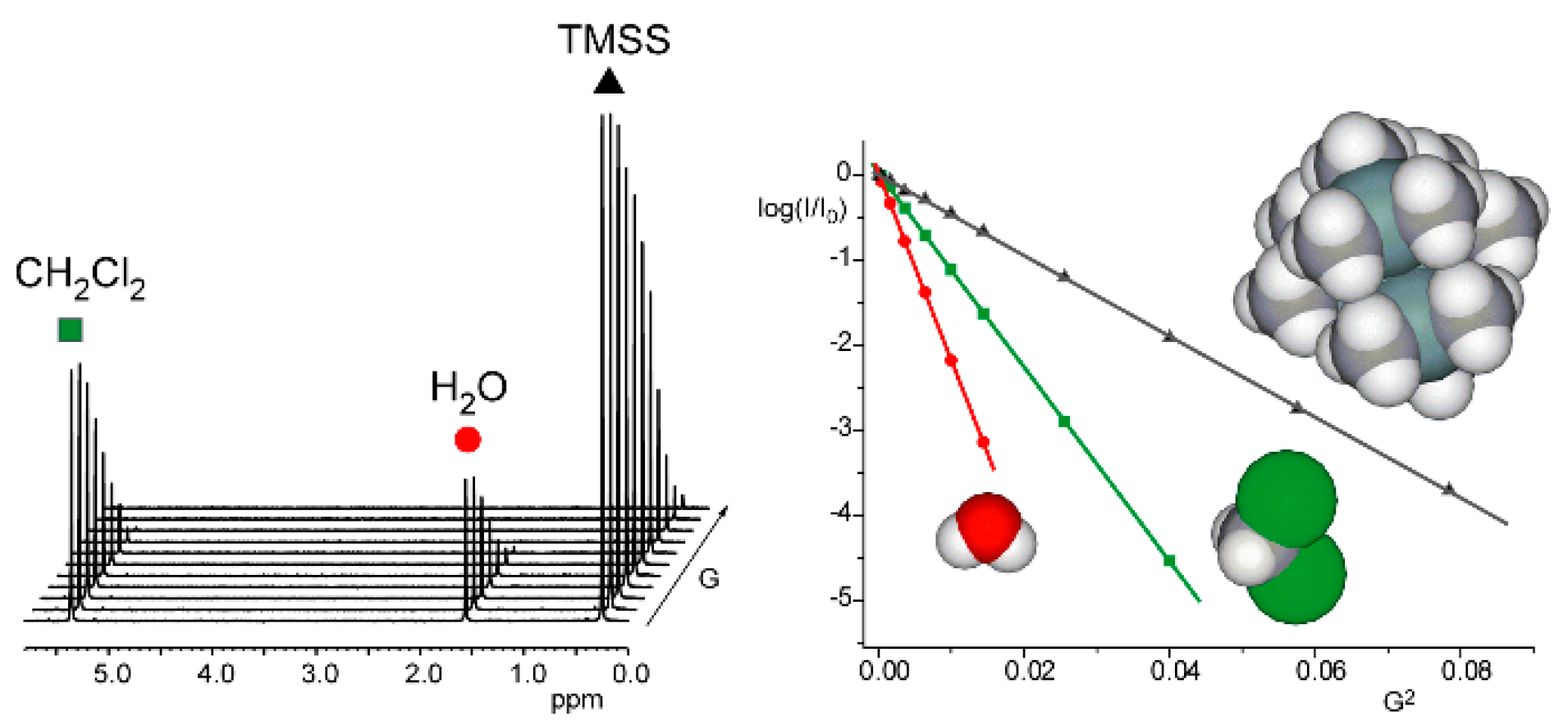
3.2. Diffusion NMR
4. Advanced NMR Techniques: Applications
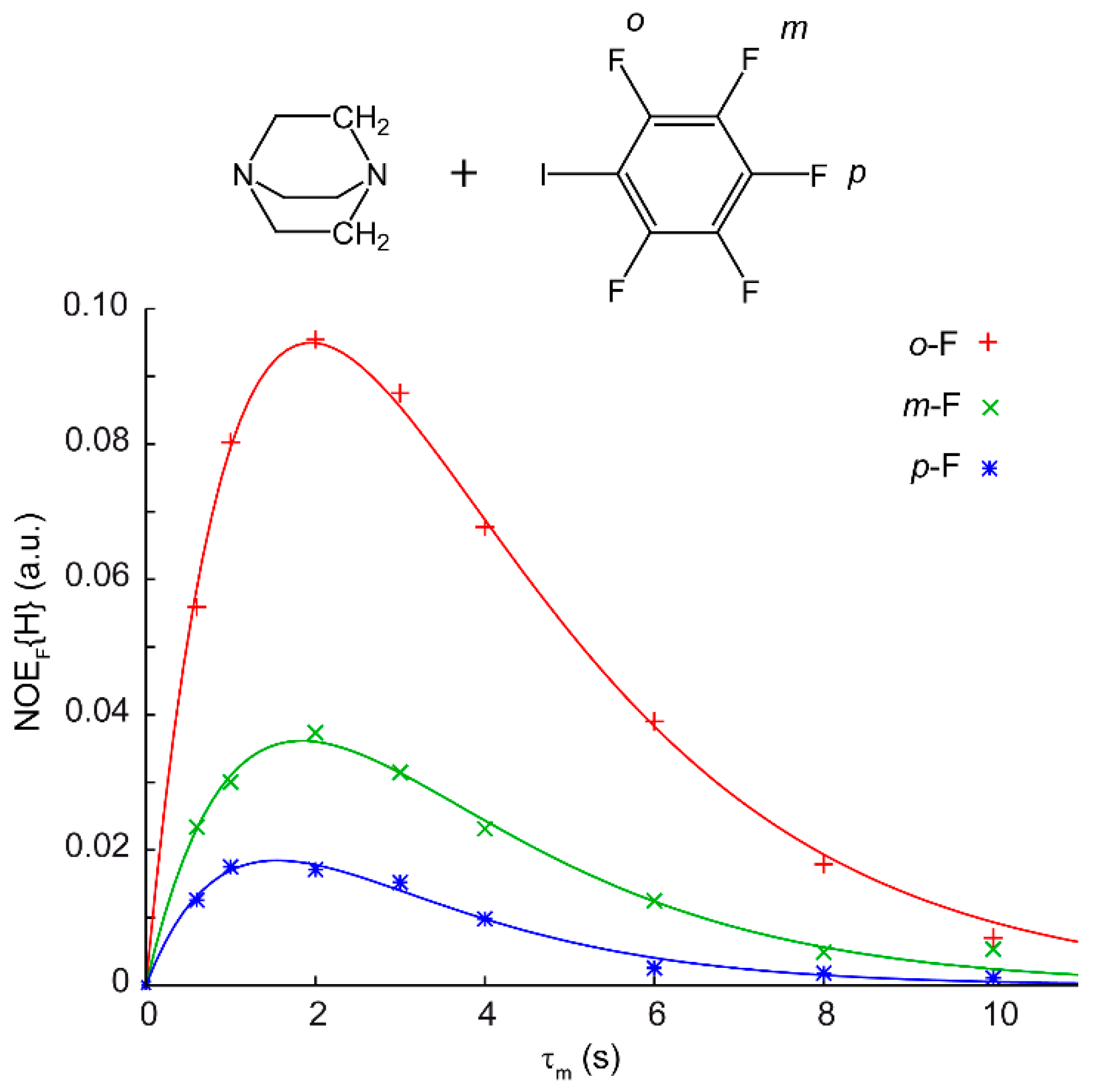
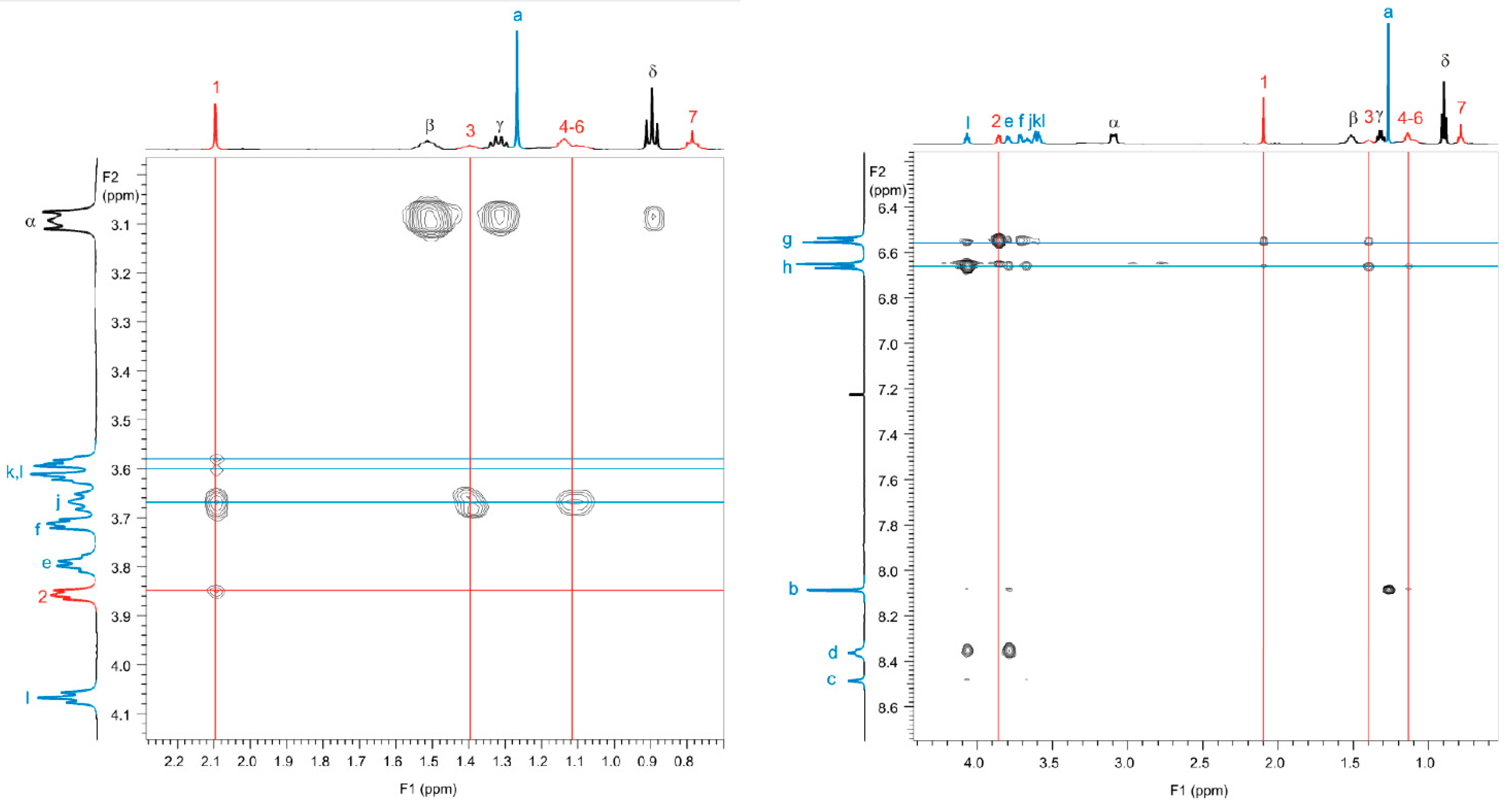
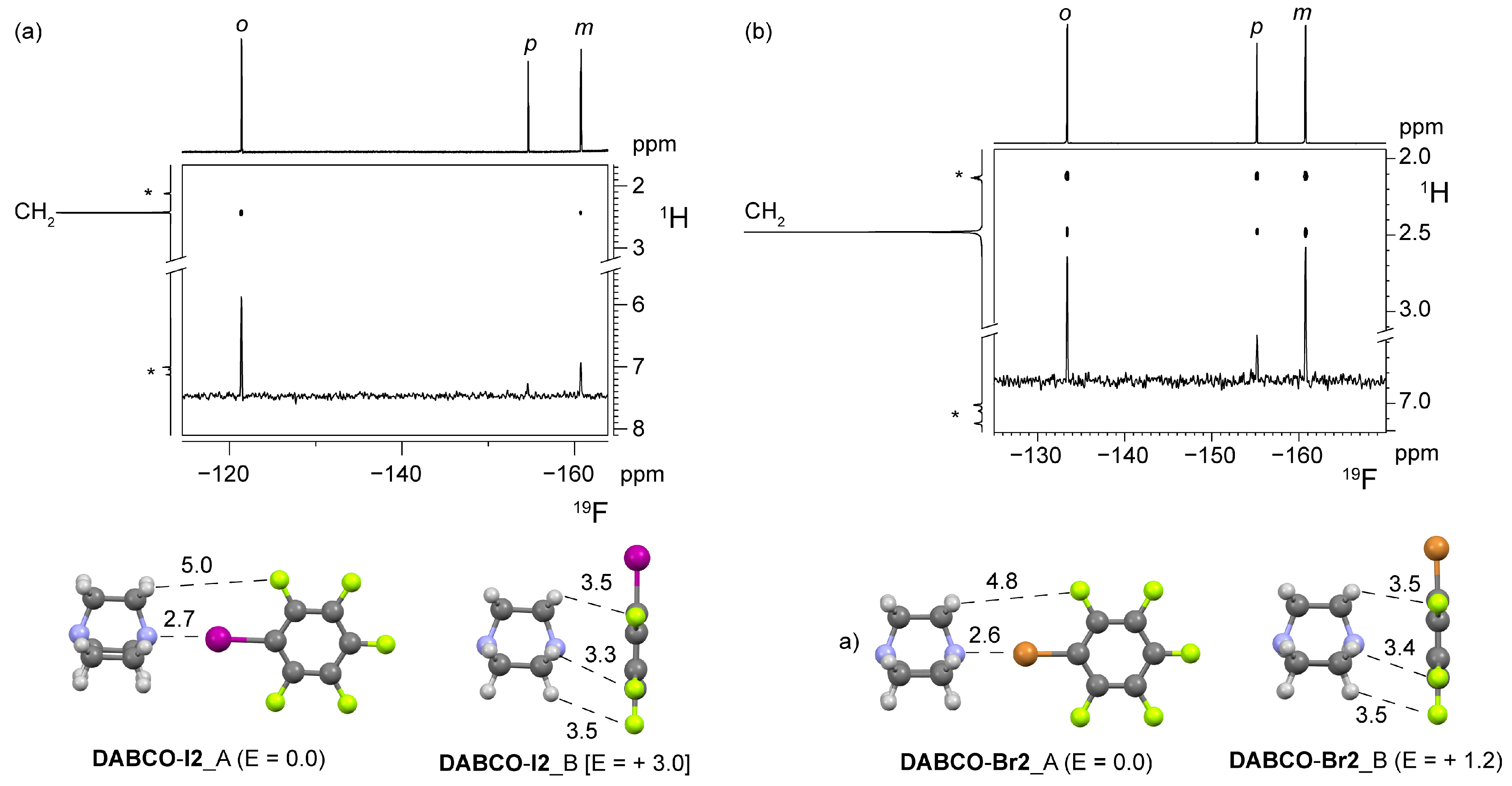
4.1. Nuclear Overhauser Effect
4.2. Diffusion NMR
5. Conclusions
Conflicts of Interest
References and Notes
- Hassel, O.; Hvoslef, J. The Structure of Bromine 1,4-Dioxanate. Acta Chem. Scand. 1954, 8, 873. [Google Scholar] [CrossRef]
- Hassel, O. Structural aspects of interatomic charge-transfer bonding. Science 1970, 170, 497–502. [Google Scholar] [CrossRef] [PubMed]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The Halogen Bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef] [PubMed]
- Beale, T.M.; Chudzinski, M.G.; Sarwar, M.G.; Taylor, M.S. Halogen bonding in solution: Thermodynamics and applications. Chem. Soc. Rev. 2013, 42, 1667–1680. [Google Scholar] [CrossRef] [PubMed]
- Bulfield, D.; Huber, S.M. Halogen Bonding in Organic Synthesis and Organocatalysis. Chem. Eur. J. 2016, 22, 14434–14450. [Google Scholar] [CrossRef] [PubMed]
- Kolár, M.H.; Hobza, P. Computer Modeling of Halogen Bonds and Other σ-Hole Interactions. Chem. Rev. 2016, 116, 5155–5187. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, W.; Jin, W.J. σ-Hole Bond vs π-Hole Bond: A Comparison Based on Halogen Bond. Chem. Rev. 2016, 116, 5072–5104. [Google Scholar] [CrossRef] [PubMed]
- Desiraju, G.R.; Ho, P.S.; Kloo, L.; Legon, A.C.; Marquardt, R.; Metrangolo, P.; Politzer, P.; Resnati, G.; Rissanen, K. Definition of the Halogen Bond (IUPAC Recommendations 2013). Pure Appl. Chem. 2013, 85, 1711–1713. [Google Scholar] [CrossRef]
- Zhang, Q.; Xu, Z.; Shi, J.; Zhu, W. The Underestimated Halogen Bonds Forming with Protein Backbone in Protein Data Bank. J. Chem. Inf. Model. 2017, 57, 1529–1534. [Google Scholar] [CrossRef] [PubMed]
- Aragoni, M.C.; Arca, M.; Devillanova, F.A.; Isaia, F.; Lippolis, V. Adducts of S/Se Donors with Dihalogens as a Source of Information for Categorizing the Halogen Bonding. Cryst. Growth Des. 2012, 12, 2769–2779. [Google Scholar] [CrossRef]
- Poznański, J.; Poznańska, A.; Shugar, D. A Protein Data Bank Survey Reveals Shortening of Intermolecular Hydrogen Bonds in Ligand-Protein Complexes When a Halogenated Ligand Is an H-Bond Donor. PLoS ONE 2014, 6, e99984. [Google Scholar] [CrossRef] [PubMed]
- Mooibroek, T.J.; Gamez, P. Halogen bonding versus hydrogen bonding: What does the Cambridge Database reveal? Cryst. Eng. Comm. 2013, 15, 4565–4570. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, W.; Liu, Y.; Lu, Y. Influence of transition metal coordination on halogen bonding: CSD survey and theoretical study. Chem. Phys. Lett. 2013, 578, 38–42. [Google Scholar] [CrossRef]
- Farina, A.; Meille, S.V.; Messina, M.T.; Metrangolo, P.; Resnati, G.; Vecchio, G. Resolution of Racemic 1,2-Dibromohexa-fluoropropane through Halogen-Bonded Supramolecular Helices. Angew. Chem. Int. Ed. Engl. 1999, 38, 2433–2436. [Google Scholar] [CrossRef]
- Takeuchi, T.; Minato, Y.; Takase, M.; Shinmori, H. Molecularly Imprinted Polymers with Halogen Bonding-Based Molecular Recognition Sites. Tetrahedron Lett. 2005, 46, 9025–9027. [Google Scholar] [CrossRef]
- Martí-Rujas, J.; Meazza, L.; Keat Lim, G.; Terraneo, G.; Pilati, T.; Harris, K.D.M.; Metrangolo, P.; Resnati, G. An Adaptable and Dynamically Porous Organic Salt Traps Unique Tetrahalide Dianions. Angew. Chem. Int. Ed. 2013, 52, 13444–13448. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.L.; Horton, P.N.; Hursthouse, M.B.; Legon, A.C.; Bruce, D.W. Halogen Bonding: A New Interaction for Liquid Crystal Formation. J. Am. Chem. Soc. 2004, 126, 16–17. [Google Scholar] [CrossRef] [PubMed]
- Bolton, O.; Lee, K.; Kim, H.-J.; Lin, K.Y.; Kim, J. Activating Efficient Phosphorescence from Purely Organic Materials by Crystal Design. Nat. Chem. 2011, 3, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Yan, D.; Delori, A.; Lloyd, G.O.; Friščić, T.; Day, G.M.; Jones, W.; Lu, J.; Wei, M.; Evans, D.G.; Duan, X. A Cocrystal Strategy to Tune the Luminescent Properties of Stilbene-Type Organic Solid-State Materials. Angew. Chem. Int. Ed. 2011, 50, 12483–12486. [Google Scholar] [CrossRef] [PubMed]
- Atzori, M.; Serpe, A.; Deplano, P.; Schlueter, J.A.; Laura Mercuri, M. Tailoring Magnetic Properties of Molecular Materials through Non-Covalent Interactions. Inorg. Chem. Front. 2015, 2, 108–115. [Google Scholar] [CrossRef]
- Fourmigué, M.; Batail, P. Activation of Hydrogen- and Halogen-Bonding Interactions in Tetrathiafulvalene-Based Crystalline Molecular Conductors. Chem. Rev. 2004, 104, 5379–5418. [Google Scholar] [CrossRef] [PubMed]
- Sarwar, M.G.; Dragisić, B.; Dimitrijević, E.; Taylor, M.S. Halogen Bonding between Anions and Iodoperfluoroorganics: Solution-Phase Thermodynamics and Multidentate-Receptor Design. Chem. Eur. J. 2013, 19, 2050–2058. [Google Scholar] [CrossRef] [PubMed]
- Sarwar, M.G.; Dragisic, B.; Sagoo, S.; Taylor, M.S. A Tridentate Halogen-Bonding Receptor for Tight Binding of HalideAnions. Angew. Chem. Int. Ed. 2010, 49, 1674–1677. [Google Scholar] [CrossRef] [PubMed]
- Gilday, L.C.; White, N.G.; Beer, P.D. Halogen- and hydrogen-bonding triazole-functionalised porphyrin-based receptors for anion recognition. Dalton Trans. 2013, 42, 15766–15773. [Google Scholar] [CrossRef] [PubMed]
- Walter, S.M.; Kniep, F.; Herdtweck, E.; Huber, S.M. Halogen-Bond-Induced Activation of a Carbon-Heteroatom Bond. Angew. Chem. Int. Ed. 2011, 50, 7187–7191. [Google Scholar] [CrossRef] [PubMed]
- Coulembier, O.; Meyer, F.; Dubois, P. Controlled Room Temperature ROP of L-Lactide by ICl3: A Simple Halogen-Bonding Catalyst. Polym. Chem. 2010, 1, 434–437. [Google Scholar] [CrossRef]
- He, W.; Ge, Y.C.; Tan, C.H. Halogen-Bonding-Induced Hydrogen Transfer to C=N Bond with Hantzsch Ester. Org. Lett. 2014, 16, 3244–3247. [Google Scholar] [CrossRef] [PubMed]
- Thordarson, P. Determining association constants from titration experiments in supramolecular chemistry. Chem. Soc. Rev. 2011, 40, 1305–1323. [Google Scholar] [CrossRef] [PubMed]
- Laurence, C.; Queignec-Cabanetos, M.; Dziembowska, T.; Queignec, R.; Wojtkowiak, B. 1-Iodoacetylenes. 1. Spectroscopic evidence of their complexes with Lewis bases. A spectroscopic scale of soft basicity. J. Am. Chem. Soc. 1981, 103, 2567–2573. [Google Scholar] [CrossRef]
- Walker, O.J. Absorption spectra of iodine solutions and the influence of the solvent. Trans. Faraday Soc. 1935, 31, 1432–1438. [Google Scholar] [CrossRef]
- Erdélyi, M. Halogen bonding in solution. Chem. Soc. Rev. 2012, 41, 3547–3557. [Google Scholar] [CrossRef] [PubMed]
- Webb, J.E.A.; Crossley, M.J.; Turner, P.; Thordarson, P. Pyromellitamide Aggregates and Their Response to Anion Stimuli. J. Am. Chem. Soc. 2007, 129, 7155–7162. [Google Scholar] [CrossRef] [PubMed]
- Pastor, A.; Martınez-Viviente, E. NMR spectroscopy in coordination supramolecular chemistry: A unique and powerful methodology. Coord. Chem. Rev. 2008, 252, 2314–2345. [Google Scholar] [CrossRef]
- Ciancaleoni, G.; Zuccaccia, C.; Zuccaccia, D.; Macchioni, A. Techniques in Inorganic Chemistry; Fackler, J.P., Jr., Falvello, L., Eds.; CRC: Boca Raton, FL, USA, 2011; pp. 129–180. ISBN 978-1-4398-1514-4. [Google Scholar]
- Macchioni, A.; Ciancaleoni, G.; Zuccaccia, C.; Zuccaccia, D. Diffusion Ordered NMR Spectroscopy (DOSY). In Supramolecular Chemistry: From Molecules to Nanomaterial; Gale, P.A., Steed, J.W., Eds.; John Wiley & Sons, Ltd.: New York, NY, USA, 2012; Volume 2, Chapter 4; ISBN 978-0-470-74640-0. [Google Scholar]
- Bellachioma, G.; Ciancaleoni, G.; Zuccaccia, C.; Zuccaccia, D.; Macchioni, A. NMR investigation of non-covalent aggregation of coordination compounds ranging from dimers and ion pairs up to nano-aggregates. Coord. Chem. Rev. 2008, 252, 2224–2238. [Google Scholar] [CrossRef]
- Rocchigiani, L.; Macchioni, A. Disclosing the multi-faceted world of weakly interacting inorganic systems by means of NMR spectroscopy. Dalton Trans. 2016, 45, 2785–2790. [Google Scholar] [CrossRef] [PubMed]
- Neuhaus, D.; Williamson, M. The Nuclear Overhauser Effect in Structural and Conformational Analysis, 2nd ed.; WILEY-VCH: New York, NY, USA, 2000; ISBN 978-0-471-24675-6. [Google Scholar]
- Stilbs, P. Fourier transform pulsed-gradient spin-echo studies of molecular diffusion. Prog. Nucl. Magn. Reson. Spectrosc. 1987, 19, 1–45. [Google Scholar] [CrossRef]
- Price, W.S. Pulsed-field gradient nuclear magnetic resonance as a tool for studying translational diffusion. Part I. Basic theory. Concepts Magn. Res. 1997, 9, 299–336. [Google Scholar] [CrossRef]
- Price, W.S. Pulsed-field gradient nuclear magnetic resonance as a tool for studying translational diffusion. Part II. Experimental aspects. Concepts Magn. Res. 1998, 10, 197–237. [Google Scholar] [CrossRef]
- Bertrán, J.F.; Rodríguez, M. Detection of halogen bond formation by correlation of proton solvent shifts. 1. Haloforms in n-electron donor solventes. Org. Magn. Reson. 1979, 12, 92–94. [Google Scholar] [CrossRef]
- Metrangolo, P.; Resnati, G. Halogen Bonding: A Paradigm in Supramolecular Chemistry. Chem. Eur. J. 2001, 7, 2511–2519. [Google Scholar] [CrossRef]
- Carlsson, A.-C.C.; Veiga, A.X.; Erdelyi, M. Halogen Bonding in Solution. Top. Curr. Chem. 2015, 359, 49–76. [Google Scholar] [CrossRef] [PubMed]
- Thorson, R.A.; Woller, G.R.; Driscoll, Z.L.; Geiger, B.E.; Moss, C.A.; Schlapper, A.L.; Speetzen, E.D.; Bosch, E.; Erdélyi, M.; Bowling, N.P. Intramolecular Halogen Bonding in Solution: 15N, 13C, and19F NMR Studies of Temperature and Solvent Effects. Eur. J. Org. Chem. 2015, 2015, 1685–1695. [Google Scholar] [CrossRef]
- Hakkert, S.B.; Gräfenstein, J.; Erdelyi, M. The 15N NMR chemical shift in the characterization of weak halogen bonding in solution. Faraday Discuss. 2017. [Google Scholar] [CrossRef] [PubMed]
- Maugeri, L.; Asencio-Hernández, J.; Lébl, T.; Cordes, D.B.; Slawin, A.M.Z.; Delsuc, M.-A.; Philp, D. Neutral iodotriazoles as scaffolds for stable halogen-bonded assemblies in solution. Chem. Sci. 2016, 7, 6422–6428. [Google Scholar] [CrossRef] [PubMed]
- Webb, J.A.; Klijn, J.E.; Hill, P.A.; Bennett, J.L.; Goroff, N.S. Experimental Studies of the 13C NMR of Iodoalkynes in Lewis-Basic Solvents. J. Org. Chem. 2004, 69, 660–664. [Google Scholar] [CrossRef] [PubMed]
- Ma, N.; Zhang, Y.; Ji, B.; Tian, A.; Wang, W. Structural Competition between Halogen Bonds and Lone-Pair···π Interactions in Solution. Chem. Phys. Chem. 2012, 13, 1411–1414. [Google Scholar] [CrossRef] [PubMed]
- Rocchigiani, L.; Ciancaleoni, G.; Zuccaccia, C.; Macchioni, A. Probing the Association of Frustrated Phosphine—Borane Lewis Pairs in Solution by NMR Spectroscopy. J. Am. Chem. Soc. 2014, 136, 112–115. [Google Scholar] [CrossRef] [PubMed]
- Korenaga, T.; Shoji, T.; Onoue, K.; Sakai, T. Demonstration of the existence of intermolecular lone pair⋯π interaction between alcoholic oxygen and the C6F5 group in organic solvent. Chem. Commun. 2009, 4678–4680. [Google Scholar] [CrossRef] [PubMed]
- Viger-Gravel, J.; Leclerc, S.; Korobkov, I.; Bryce, D.L. Correlation between 13C chemical shifts and the halogen bonding environment in a series of solid para-diiodotetrafluorobenzene complexes. CrystEngComm 2013, 15, 3168–3177. [Google Scholar] [CrossRef]
- Vioglio, P.C.; Chierotti, M.R.; Gobetto, R. Solid-state nuclear magnetic resonance as a tool for investigating the halogen bond. Cryst. Eng. Comm. 2016, 18, 9173–9184. [Google Scholar] [CrossRef]
- Vioglio, P.C.; Catalano, L.; Vasylyeva, V.; Nervi, C.; Chierotti, M.R.; Resnati, G.; Gobetto, R.; Metrangolo, P. Natural Abundance 15N and 13C Solid-State NMR Chemical Shifts: High Sensitivity Probes of the Halogen Bond Geometry. Chem. Eur. J. 2016, 22, 16819–16828. [Google Scholar] [CrossRef] [PubMed]
- Cabot, R.; Hunter, C.A. Non-covalent interactions between iodo-perfluorocarbons and hydrogen bond acceptors. Chem. Commun. 2009, 2005–2007. [Google Scholar] [CrossRef] [PubMed]
- Shen, Q.J.; Jin, W.J. Strong halogen bonding of 1,2-diiodoperfluoroethane and 1,6-diiodoperfluorohexane with halide anions revealed by UV-Vis, FT-IR, NMR spectroscopes and crystallography. Phys. Chem. Chem. Phys. 2011, 13, 13721–13729. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.-X.; Sun, Y.; Feng, Y.; Chen, H.; He, Y.-M.; Fan, Q.-H. Halogen-Bonding for Visual Chloride Ion Sensing: A Case Study Using Supramolecular Poly(aryl ether) Dendritic Organogel System. Chem. Commun. 2016, 52, 2269–2272. [Google Scholar] [CrossRef] [PubMed]
- Mungalpara, D.; Stegmüller, S.; Kubik, S. A neutral halogen bonding macrocyclic anion receptor based on a pseudocyclopentapeptide with three 5-iodo-1,2,3-triazole subunits. Chem. Commun. 2017, 53, 5095–5098. [Google Scholar] [CrossRef] [PubMed]
- Noggle, J.H.; Schirmer, R.E. The Nuclear Overhauser Effect; Academic Press: New York, NY, USA, 1971; ISBN 9780323141390. [Google Scholar]
- Keeler, J. Understanding NMR Spectroscopy. 2002. Available online: http://www-keeler.ch.cam.ac.uk/lectures/Irvine/ (accessed on 28 August 2017).
- Ciancaleoni, G.; Bertani, R.; Rocchigiani, L.; Sgarbossa, P.; Zuccaccia, C.; Macchioni, A. Discriminating Halogen-Bonding from Other Noncovalent Interactions by a Combined NOE NMR/DFT Approach. Chem. Eur. J. 2015, 21, 440–447. [Google Scholar] [CrossRef] [PubMed]
- Zuccaccia, C.; Bellachioma, G.; Cardaci, G.; Macchioni, A. Solution structure investigation of Ru(II) complex ion pairs: Quantitative NOE measurements and determination of average interionic distances. J. Am. Chem. Soc. 2001, 123, 11020–11028. [Google Scholar] [CrossRef] [PubMed]
- Zuccaccia, C.; Bellachioma, G.; Cardaci, G.; Macchioni, A. Specificity of interionic contacts and estimation of average interionic distances by NOE NMR measurements in solution of cationic Ru(II) organometallic complexes bearing unsymmetrical counterions. Organometallics 1999, 18, 1–3. [Google Scholar] [CrossRef]
- Ciancaleoni, G.; Zuccaccia, C.; Zuccaccia, D.; Macchioni, A. Diffusion and NOE NMR studies on the interactions of neutral amino-acidate arene ruthenium(II) supramolecular aggregates with ions and ion pairs. Magn. Reson. Chem. 2008, 46, S72–S79. [Google Scholar] [CrossRef] [PubMed]
- Geldbach, T. J.; Ruegger, H.; Pregosin, P. S. NOESY, HOESY, T1 and solid-state NMR studies on [RuH(h6-toluene)(Binap)](CF3SO3): A molecule with a strongly distorted piano-stool structure. Magn. Reson. Chem. 2003, 41, 703–708. [Google Scholar] [CrossRef]
- Lingscheid, Y.; Arenz, S.; Giernoth, R. Heteronuclear NOE Spectroscopy of Ionic Liquids. Chem. Phys. Chem. 2012, 13, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Braun, D.; Steinhauser, O. The intermolecular NOE is strongly influenced by dynamics. Phys. Chem. Chem. Phys. 2015, 17, 8509–8517. [Google Scholar] [CrossRef] [PubMed]
- Gabl, S.; Schröder, C.; Braun, D.; Weingärtner, H.; Steinhauser, O. Pair dynamics and the intermolecular nuclear Overhauser effect (NOE) in liquids analysed by simulation and model theories: Application to an ionic liquid. J. Chem. Phys. 2012, 140, 184503. [Google Scholar] [CrossRef] [PubMed]
- Crank, J. The Mathematics of Diffusion, 2nd ed.; Clarendon Press: Oxford, UK, 1975; ISBN 978-0198534112. [Google Scholar]
- Einstein, A. Investigations in the Theory of Brownian Movements; Dover: New York, NY, USA, 1956; ISBN 9781607962854. [Google Scholar]
- Hahn, E.L. Spin echoes. Phys. Rev. 1950, 80, 580–594. [Google Scholar] [CrossRef]
- Stejskal, E.O.; Tanner, J.E. Spin diffusion measurements: Spin echoes in the presence of a time-dependent field gradient. J. Chem. Phys. 1965, 42, 288–292. [Google Scholar] [CrossRef]
- Macchioni, A.; Ciancaleoni, G.; Zuccaccia, C.; Zuccaccia, D. Determining accurate molecular sizes in solution through NMR diffusion spectroscopy. Chem. Soc. Rev. 2008, 37, 479–489. [Google Scholar] [CrossRef] [PubMed]
- By using this formulation, the effect of T2 can be neglected, since it is the same with and without G.
- Edward, J.T. Molecular volumes and the Stokes-Einstein equation. J. Chem. Educ. 1970, 47, 261–270. [Google Scholar] [CrossRef]
- Chen, H.-C.; Chen, S.-H. Diffusion of crown ethers in alcohols. J. Phys. Chem. 1984, 88, 5118–5121. [Google Scholar] [CrossRef]
- Sinnaeve, D. Simultaneous solvent and J-modulation suppression in PGSTE-based diffusion experiments. J. Magn. Res. 2014, 245, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.; Wu, D.; Johnson, C.S., Jr. Determination of Molecular Weight Distributions for Polymers by Diffusion-Ordered NMR. J. Am. Chem. Soc. 1995, 117, 7965–7970. [Google Scholar] [CrossRef]
- Marega, R.; Aroulmoji, V.; Bergamin, M.; Feruglio, L.; Dinon, F.; Bianco, A.; Murano, E.; Prato, M. Two-Dimensional Diffusion-Ordered NMR Spectroscopy as a Tool for Monitoring Functionalized Carbon Nanotube Purification and Composition. ACS Nano 2010, 4, 2051–2058. [Google Scholar] [CrossRef] [PubMed]
- Rocchigiani, L.; Bellachioma, G.; Ciancaleoni, G.; Crocchianti, S.; Lagana, A.; Zuccaccia, C.; Zuccaccia, D.; Macchioni, A. Anion-Dependent Tendency of Di-Long-Chain Quaternary Ammonium Salts to Form Ion Quadruples and Higher Aggregates in Benzene. Chem. Phys. Chem. 2010, 11, 3243–3254. [Google Scholar] [CrossRef] [PubMed]
- Pettirossi, S.; Bellachioma, G.; Ciancaleoni, G.; Zuccaccia, C.; Zuccaccia, D.; Macchioni, A. Diffusion and NOE NMR Studies on Multicationic DAB-Organoruthenium Dendrimers: Size-Dependent Noncovalent Self-Assembly to Megamers and Ion Pairing. Chem. Eur. J. 2009, 15, 5337–5347. [Google Scholar] [CrossRef] [PubMed]
- Pregosin, P.S. NMR diffusion methods in inorganic and organometallic chemistry. Spectrosc. Prop. Inorg. Organomet. Compd. 2011, 42, 248–268. [Google Scholar] [CrossRef]
- Rocchigiani, L.; Busico, V.; Pastore, A.; Macchioni, A. Probing the interactions between all components of the catalytic pool for homogeneous olefin polymerisation by diffusion NMR spectroscopy. Dalton Trans. 2013, 42, 9104–9111. [Google Scholar] [CrossRef] [PubMed]
- Allouche, L.; Marquis, A.; Lehn, J.M. Discrimination of Metallosupramolecular Architectures in Solution by Using Diffusion Ordered Spectroscopy (DOSY) Experiments: Double-Stranded Helicates of Different Lengths. Chem. Eur. J. 2006, 12, 7520–7525. [Google Scholar] [CrossRef] [PubMed]
- Ciancaleoni, G.; Zuccaccia, C.; Zuccaccia, D.; Clot, E.; Macchioni, A. Self-Aggregation Tendency of All Species Involved in the Catalytic Cycle of Bifunctional Transfer Hydrogenation. Organometallics 2009, 28, 960–967. [Google Scholar] [CrossRef]
- Tatko, C.D.; Waters, M.L. Effect of Halogenation on Edge—Face Aromatic Interactions in a β-Hairpin Peptide: Enhanced Affinity with Iodo-Substituents. Org. Lett. 2006, 6, 3969–3972. [Google Scholar] [CrossRef] [PubMed]
- Serpell, C.J.; Kila, N.L.; Costa, P.J.; Félix, V.; Beer, P.D. Halogen Bond Anion Templated Assembly of an Imidazolium Pseudorotaxane. Angew. Chem. Int. Ed. 2010, 49, 5322–5326. [Google Scholar] [CrossRef] [PubMed]
- Gilday, L.C.; Beer, P.D. Halogen- and Hydrogen-Bonding catenanes for Halide-Anion Recognition. Chem. Eur. J. 2014, 20, 8379–8385. [Google Scholar] [CrossRef] [PubMed]
- Huber, S.M.; Scanlon, J.D.; Jimenez-Izal, E.; Ugalde, J.M.; Infante, I. On the directionality of halogen bonding. Phys. Chem. Chem. Phys. 2013, 15, 10350–10357. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen Bonding: An Electrostatically-Driven Highly Directional Noncovalent Interaction. Phys. Chem. Chem. Phys. 2010, 12, 7748–7757. [Google Scholar] [CrossRef] [PubMed]
- Kozuch, S.; Martin, J.M.L. Halogen Bonds: Benchmarks and Theoretical Analysis. J. Chem. Theory Comput. 2013, 9, 1918–1931. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.Q.; Zhao, X.R.; Wang, H.; Jin, W.J. The Competition of σ-Hole···Cl− and π-Hole···Cl− Bonds between C6F5X (X = F, Cl, Br, I) and the Chloride Anion and Its Potential Application in Separation Science. J. Phys. Chem. B 2014, 118, 1080–1087. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Yan, X.; He, W.; Li, X.; Li, Z.; Mo, Y.; Liu, M.; Jiang, Y.-B. C−I···π Halogen Bonding Driven Supramolecular Helix of Bilateral N-Amidothioureas Bearing β-Turns. J. Am. Chem. Soc. 2017, 139, 6605–6610. [Google Scholar] [CrossRef] [PubMed]
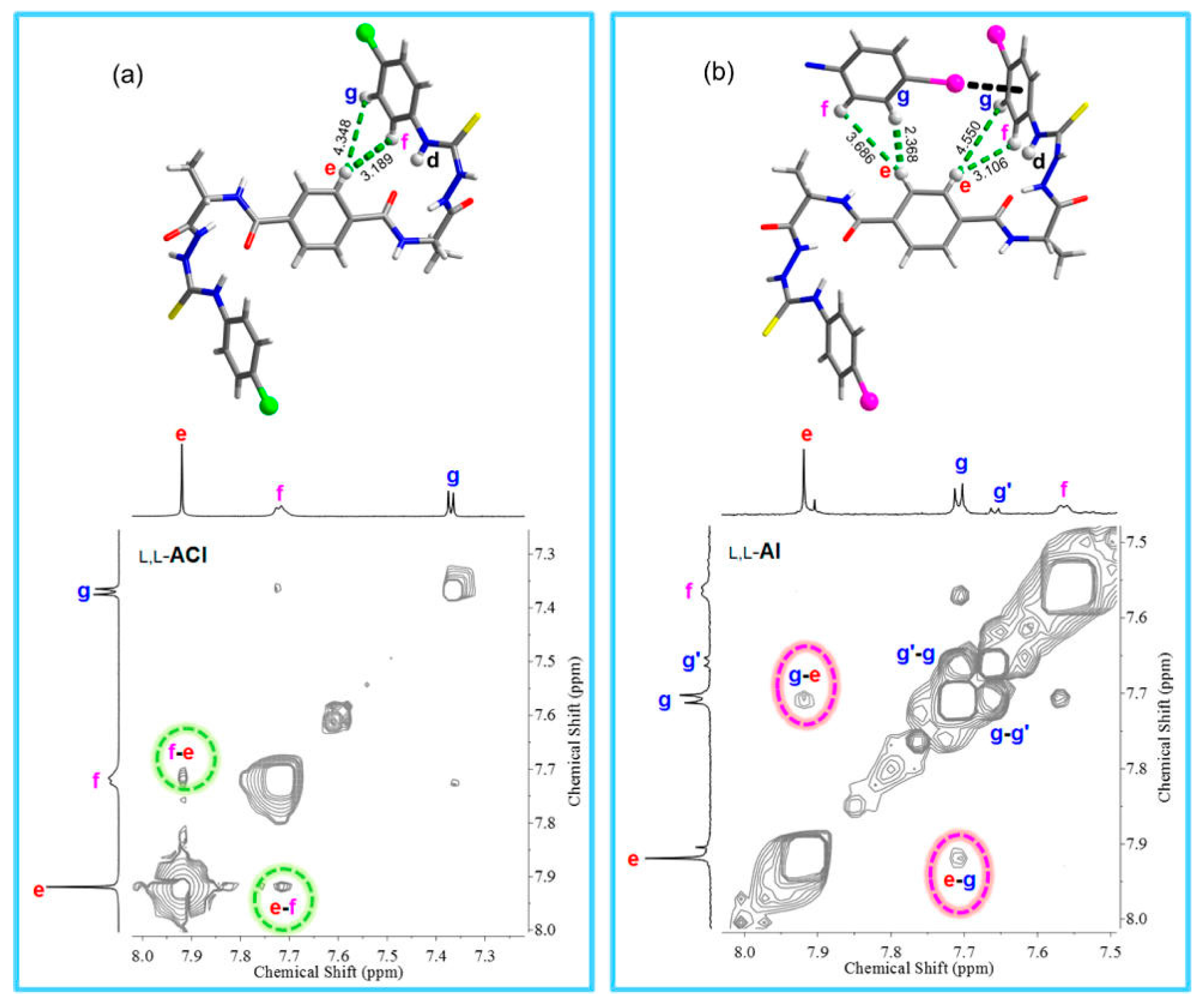
- In the author’s opinion, NOESY and peak intensities should have been coupled with a simple DOSY experiment (see below) that would have proved the existence of supramolecular oligomers more effectively.
- Casnati, A.; Liantonio, R.; Metrangolo, P.; Resnati, G.; Ungaro, R.; Ugozzoli, F. Molecular and Supramolecular Homochirality: Enantiopure Perfluorocarbon Rotamers and Halogen-Bonded Fluorous Double Helices. Angew. Chem. Int. Ed. 2006, 45, 1915–1918. [Google Scholar] [CrossRef] [PubMed]
- Danelius, E.; Andersson, H.; Jarvoll, P.; Lood, K.; Gräfenstein, J.; Erdélyi, M. Halogen Bonding: A Powerful Tool for Modulation of Peptide Conformation. Biochemistry 2017, 56, 3265–3272. [Google Scholar] [CrossRef] [PubMed]
- Carlsson, A.-C.C.; Gräfenstein, J.; Budnjo, A.; Laurila, J.L.; Bergquist, J.; Karim, A.; Kleinmaier, R.; Brath, U.; Erdélyi, M. Symmetric Halogen Bonding Is Preferred in Solution. J. Am. Chem. Soc. 2012, 134, 5706–5715. [Google Scholar] [CrossRef] [PubMed]
- Perrin, C.L. Are Short, Low-Barrier Hydrogen Bonds Unusually Strong? Acc. Chem. Res. 2010, 43, 1550–1557. [Google Scholar] [CrossRef] [PubMed]
- Macchioni, A. Ion Pairing in Transition-Metal Organometallic Chemistry. Chem. Rev. 2005, 105, 2039–2074. [Google Scholar] [CrossRef] [PubMed]
- For example, in [L1AuL2]X ion pairs, the positive charge is formally located on the gold, but the anion position depends on the nature of the ligands. See references 101–103.
- Zuccaccia, D.; Belpassi, L.; Tarantelli, F.; Macchioni, A. Ion Pairing in Cationic Olefin–Gold(I) Complexes. J. Am. Chem. Soc. 2009, 131, 3170–3171. [Google Scholar] [CrossRef] [PubMed]
- Ciancaleoni, G.; Belpassi, L.; Tarantelli, F.; Zuccaccia, D.; Macchioni, A. A combined NMR/DFT study on the ion pair structure of [(PR12R2)Au(η2–3-hexyne)]BF4 complexes. Dalton Trans. 2013, 42, 4122–4131. [Google Scholar] [CrossRef] [PubMed]
- Biasiolo, L.; Ciancaleoni, G.; Belpassi, L.; Bistoni, G.; Macchioni, A.; Tarantelli, F.; Zuccaccia, D. Relationship between the anion/cation relative orientation and the catalytic activity of nitrogen acyclic carbene–gold catalysts. Catal. Sci. Technol. 2015, 5, 1558–1567. [Google Scholar] [CrossRef]
- Bedin, M.; Karim, A.; Reitti, M.; Carlsson, A.-C.C.; Topić, F.; Cetina, M.; Pan, F.; Havel, V.; Al-Ameri, F.; Sindelar, V.; et al. Counterion influence on the N–I–N halogen bond. Chem. Sci. 2015, 6, 3746–3756. [Google Scholar] [CrossRef]
- Karim, A.; Reitti, M.; Carlsson, A.-C.C.; Gräfenstein, J.; Erdélyi, M. The nature of [N–Cl–N]+ and [N–F–N]+ halogen bonds in solution. Chem. Sci. 2014, 5, 3226–3233. [Google Scholar] [CrossRef]
- Zapata, F.; Caballero, A.; White, N.G.; Claridge, T.D.W.; Costa, P.J.; Félix, V.; Beer, P.D. Fluorescent Charge-Assisted Halogen-Bonding Macrocyclic Halo-Imidazolium Receptors for Anion Recognition and Sensing in Aqueous Media. J. Am. Chem. Soc. 2012, 134, 11533–11541. [Google Scholar] [CrossRef] [PubMed]
- Ciancaleoni, G.; Macchioni, A.; Rocchigiani, L.; Zuccaccia, C. A PGSE NMR approach to the characterization of single and multi-site halogen-bonded adducts in solution. RSC Adv. 2016, 6, 80604–80612. [Google Scholar] [CrossRef]
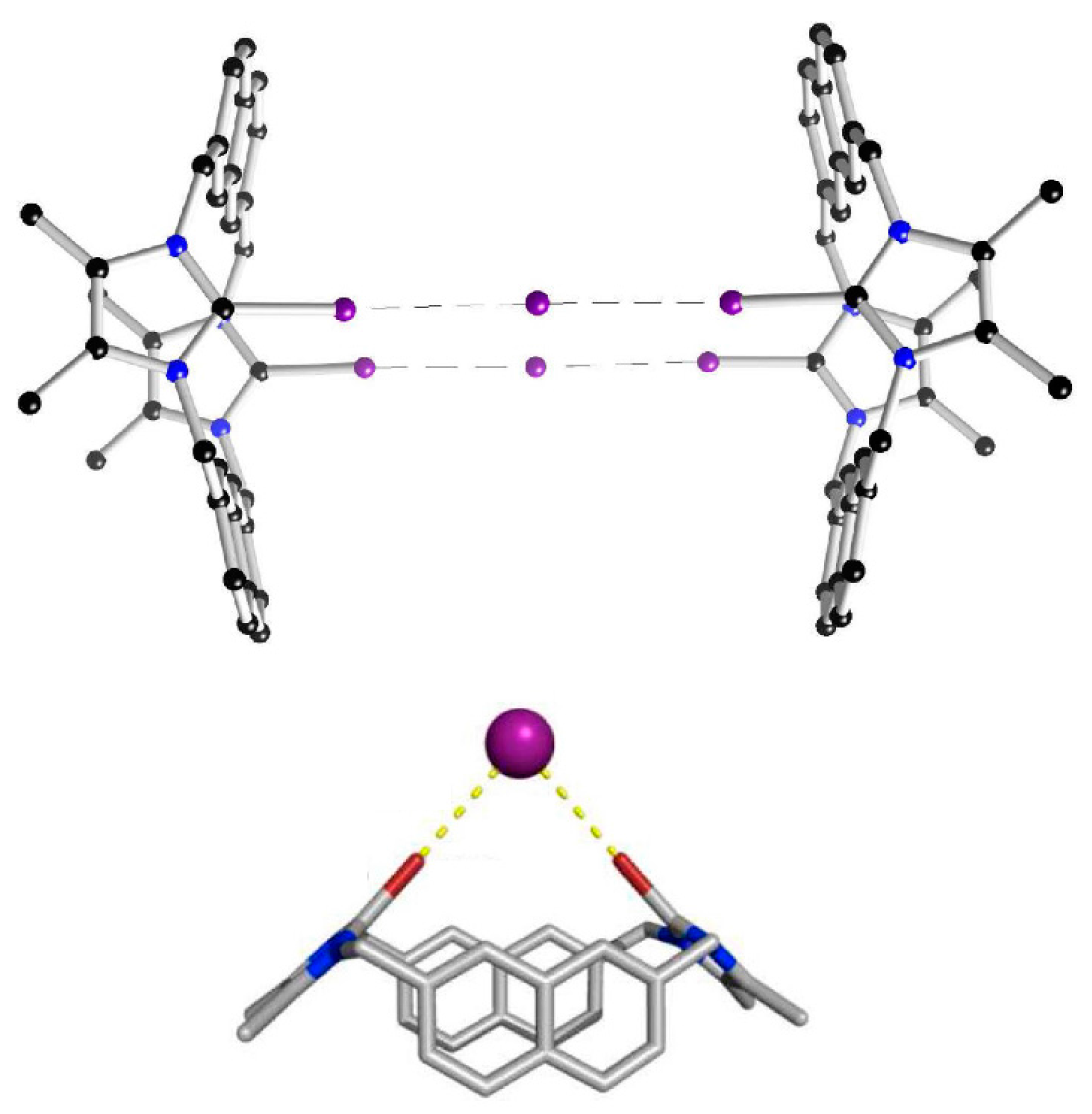
- If inter-penetrating adducts are involved, as in the case of helices or cages, this could be not true.
- If the adduct and the components are not in rapid exchange, the Equation (11) is not valid anymore. But the kinetics of XB formation is generally very fast.
- Belpassi, L.; Infante, I.; Tarantelli, F.; Visscher, L. The Chemical Bond between Au(I) and the Noble Gases. Comparative Study of NgAuF and NgAu+ (Ng = Ar, Kr, Xe) by Density Functional and Coupled Cluster Methods. J. Am. Chem. Soc. 2008, 130, 1048–1060. [Google Scholar] [CrossRef] [PubMed]
- Ciancaleoni, G.; Santi, C.; Ragni, M.; Braga, A.M. Charge–displacement analysis as a tool to study chalcogen bonded adducts and predict their association constants in solution. Dalton Trans. 2015, 44, 20168–20175. [Google Scholar] [CrossRef] [PubMed]
- Ciancaleoni, G.; Belpassi, L.; Marchetti, F. Back-Donation in High-Valent d0 Metal Complexes: Does It Exist? The Case of NbV. Inorg. Chem. 2017, 56, 11266–11274. [Google Scholar] [CrossRef] [PubMed]
- Bistoni, G.; Rampino, S.; Scafuri, N.; Ciancaleoni, G.; Zuccaccia, D.; Belpassi, L.; Tarantelli, F. How π back-donation quantitatively controls the CO stretching response in classical and non-classical metal carbonyl complexes. Chem. Sci. 2016, 7, 1174–1184. [Google Scholar] [CrossRef]
- Raatikainen, K.; Rissanen, K. Interaction between amines and N-haloimides: A new motif for unprecedentedly short Br⋯N and I⋯N halogen bonds. CrystEngComm 2011, 13, 6972–6977. [Google Scholar] [CrossRef]










© 2017 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ciancaleoni, G. Characterization of Halogen Bonded Adducts in Solution by Advanced NMR Techniques. Magnetochemistry 2017, 3, 30. https://doi.org/10.3390/magnetochemistry3040030
Ciancaleoni G. Characterization of Halogen Bonded Adducts in Solution by Advanced NMR Techniques. Magnetochemistry. 2017; 3(4):30. https://doi.org/10.3390/magnetochemistry3040030
Chicago/Turabian StyleCiancaleoni, Gianluca. 2017. "Characterization of Halogen Bonded Adducts in Solution by Advanced NMR Techniques" Magnetochemistry 3, no. 4: 30. https://doi.org/10.3390/magnetochemistry3040030
APA StyleCiancaleoni, G. (2017). Characterization of Halogen Bonded Adducts in Solution by Advanced NMR Techniques. Magnetochemistry, 3(4), 30. https://doi.org/10.3390/magnetochemistry3040030