
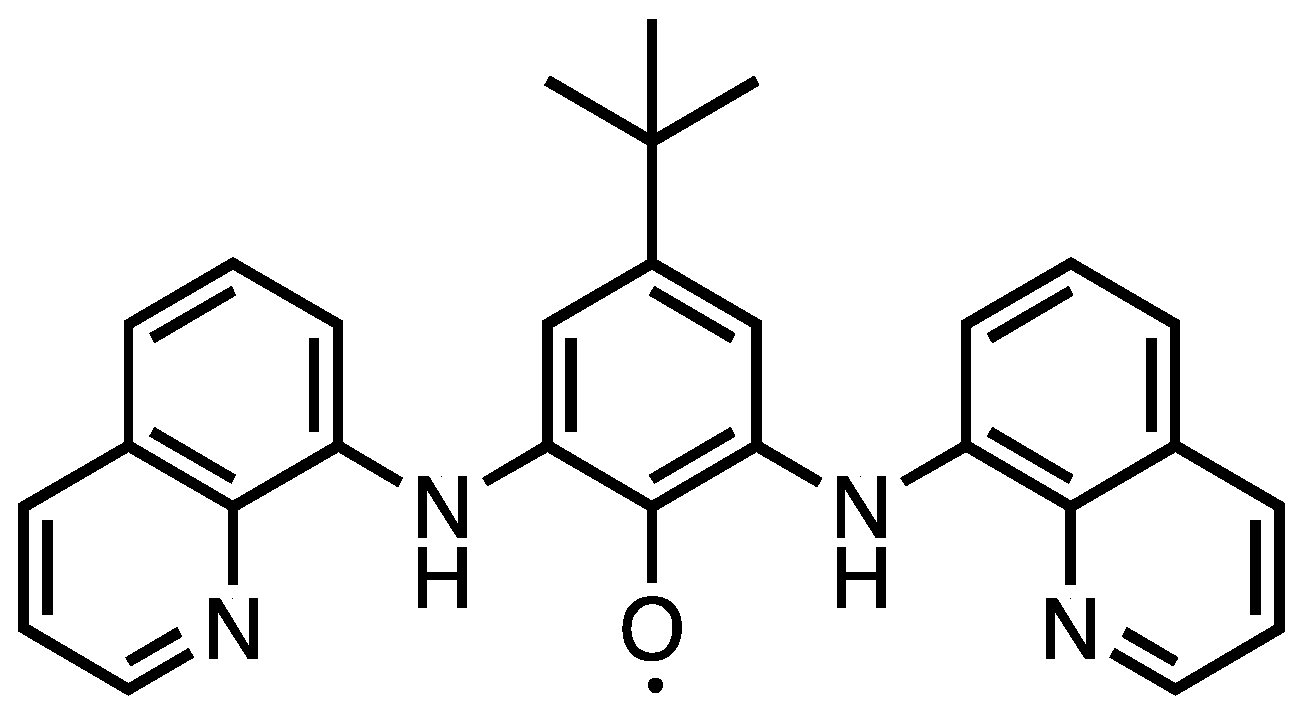
Synthesis, Characterization and Copper(2+) Coordination Chemistry of a Polytopic Paramagnetic Ligand


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
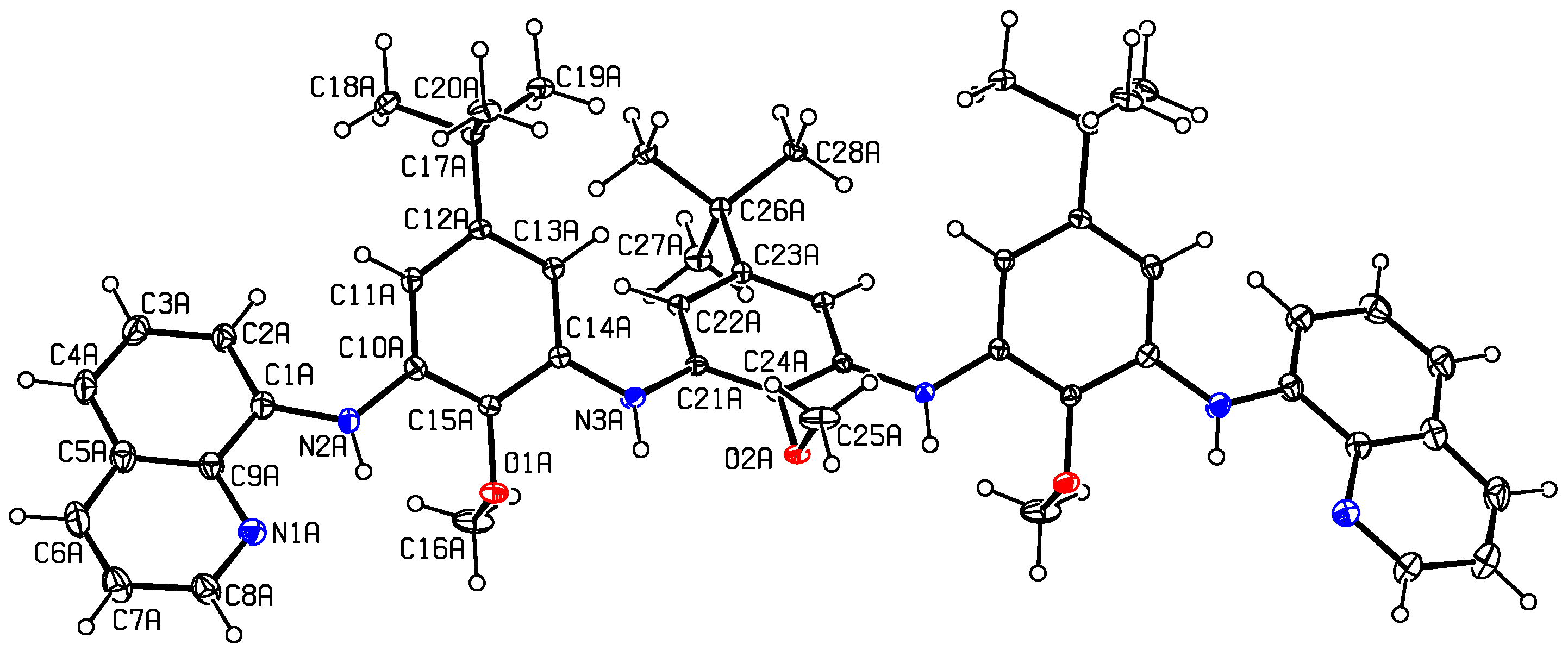
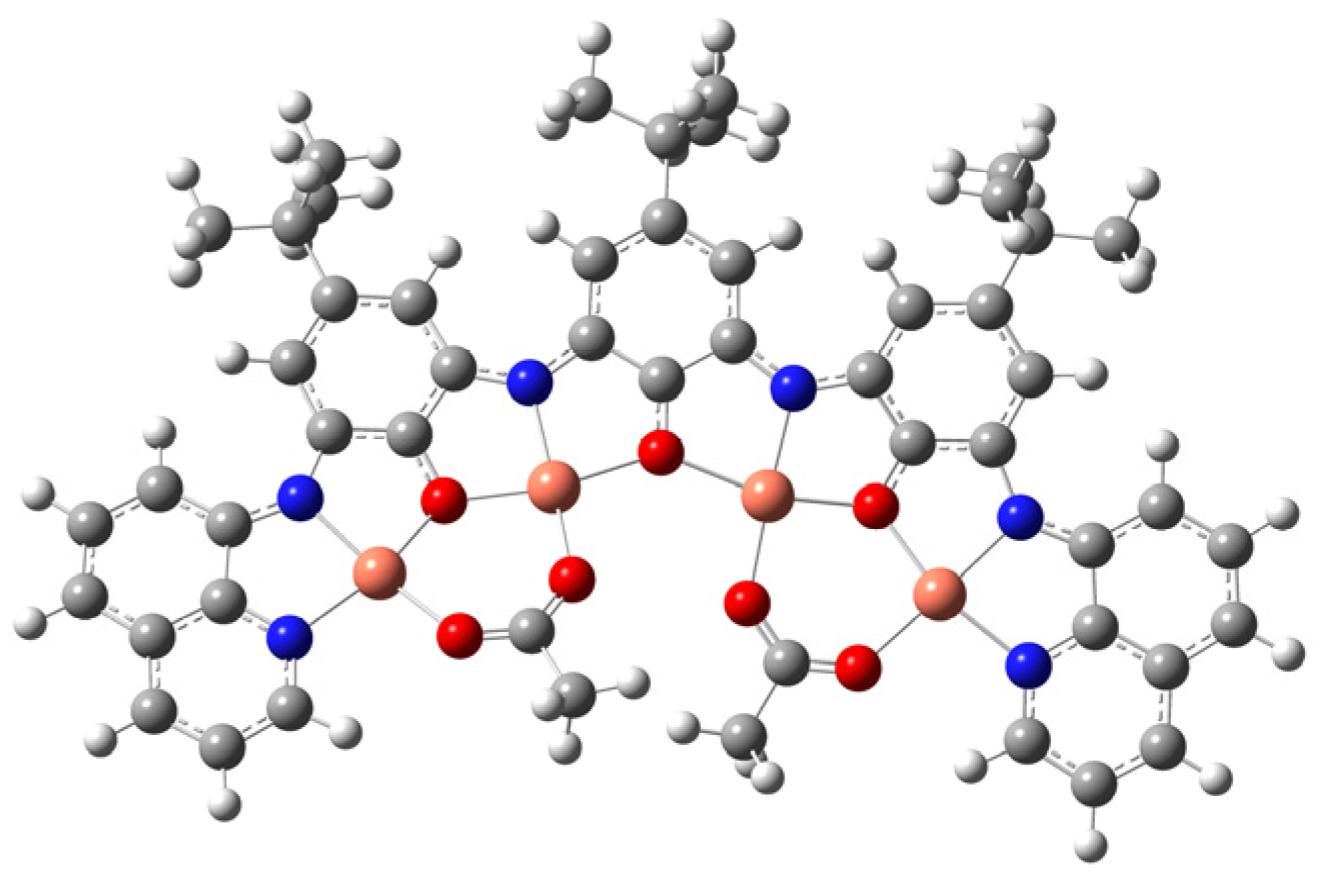
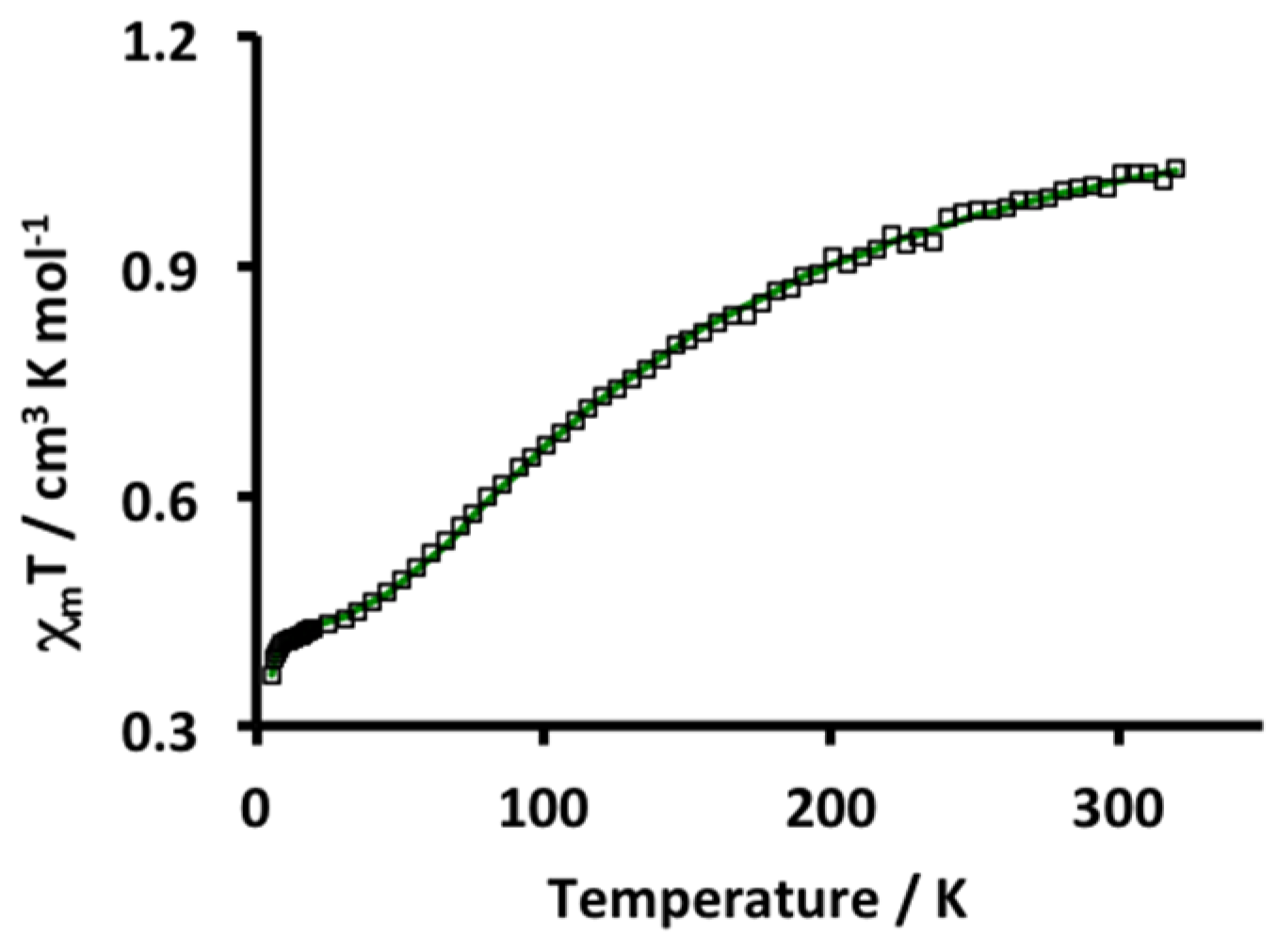
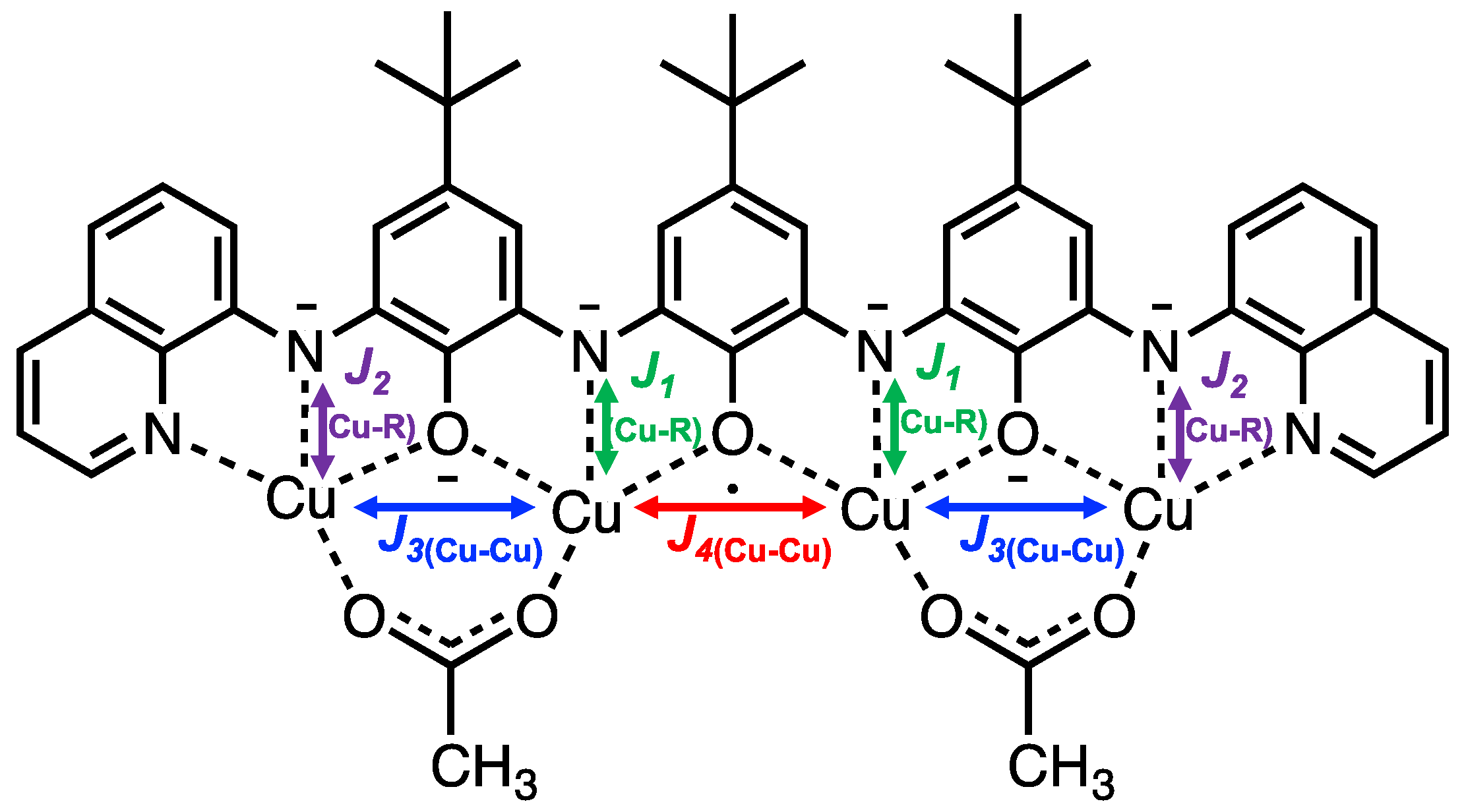
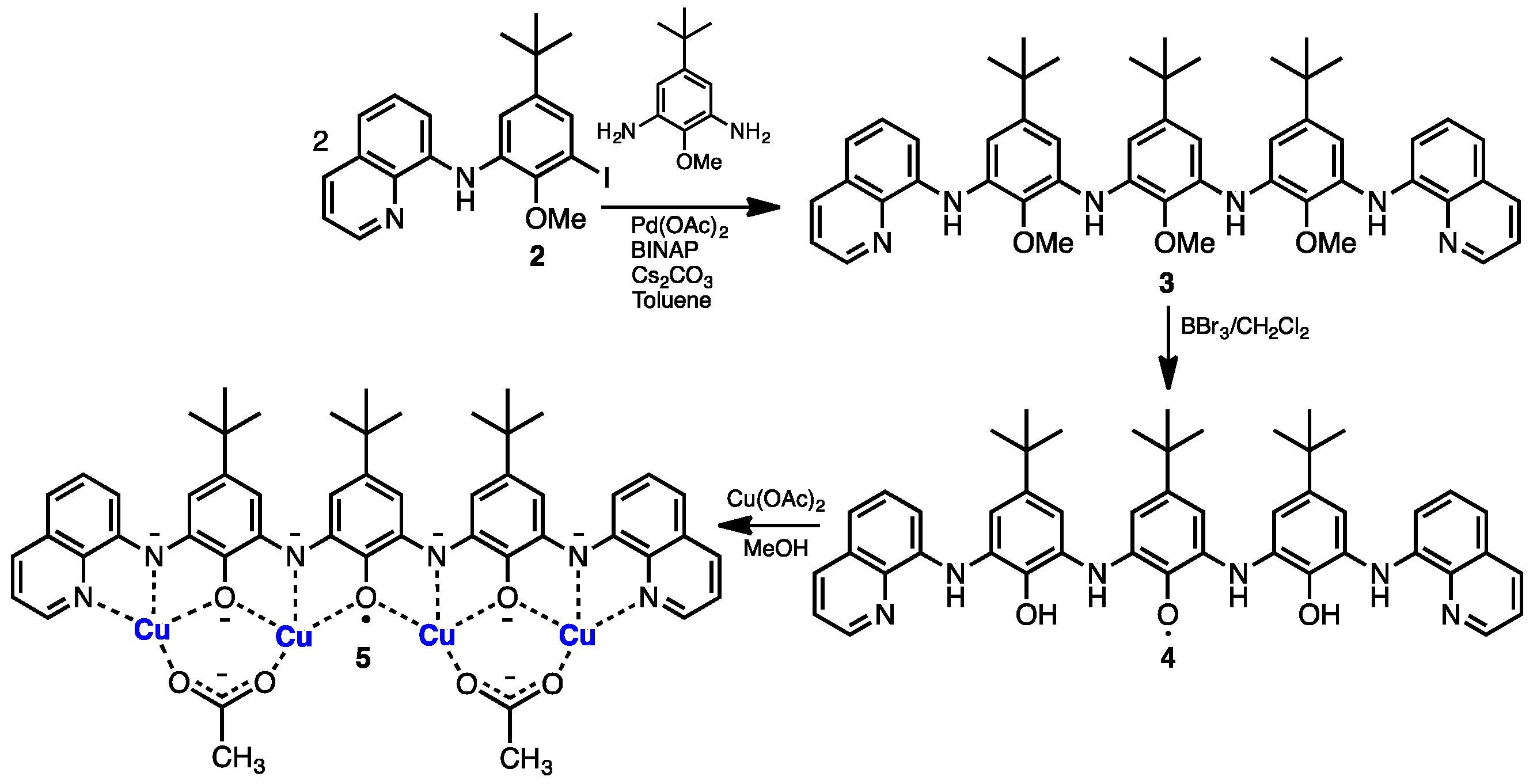
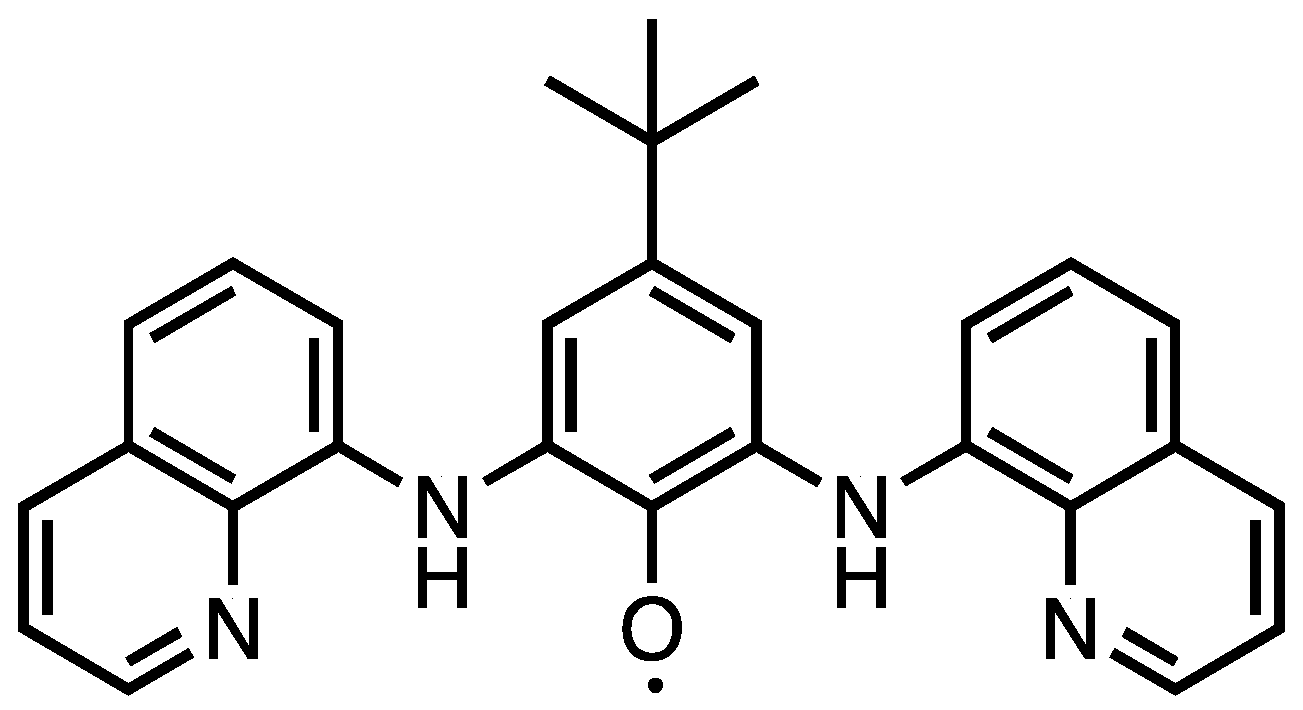
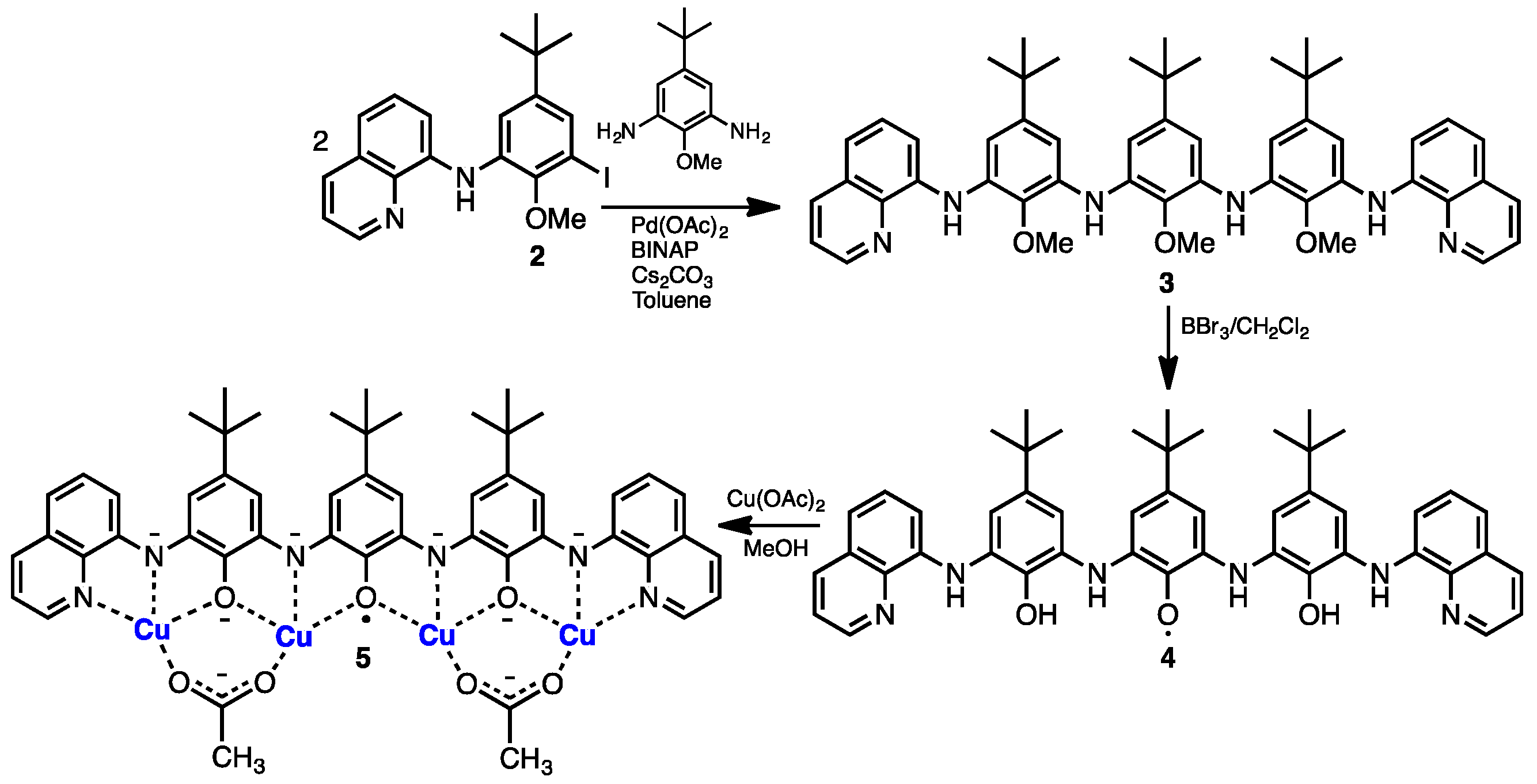
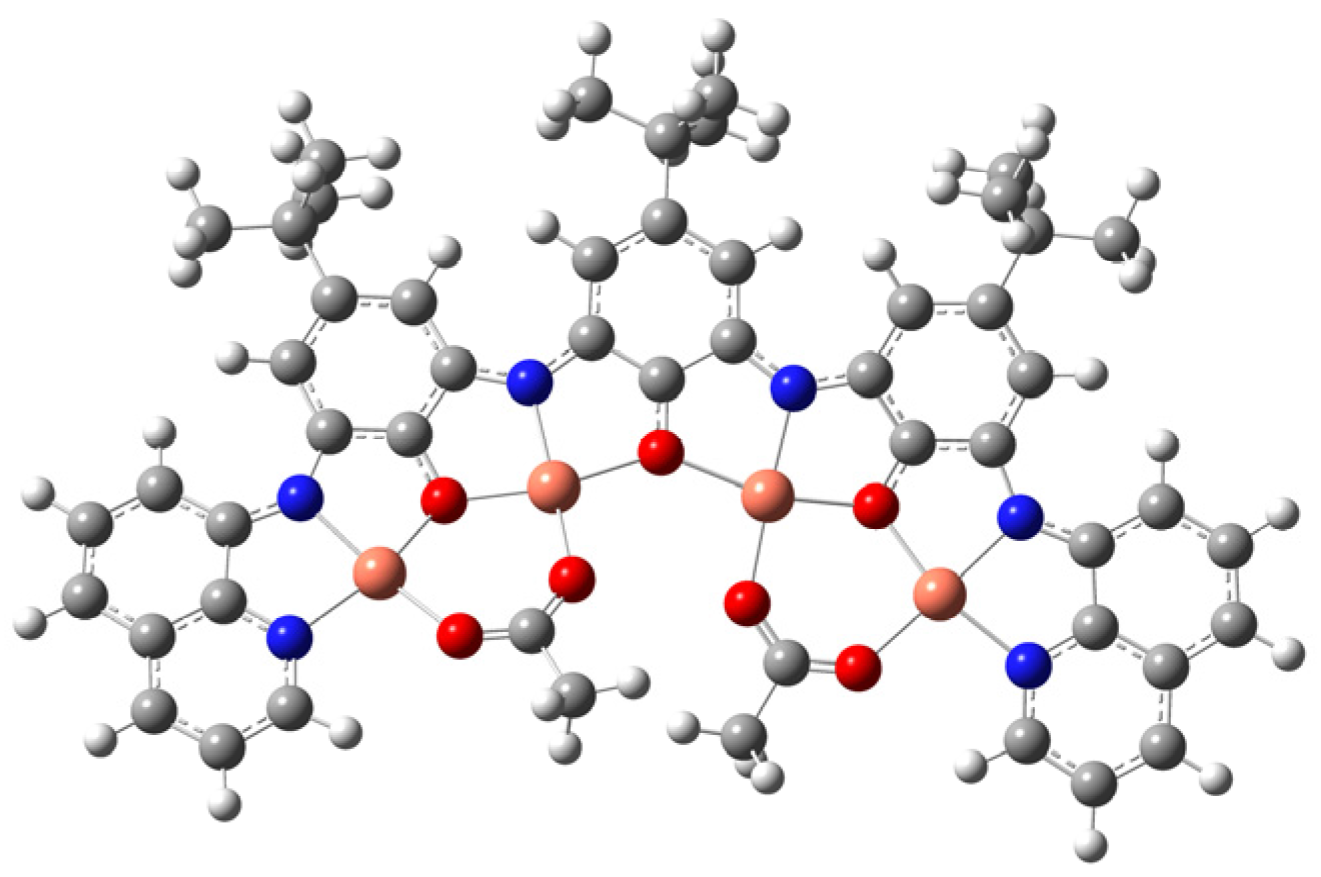
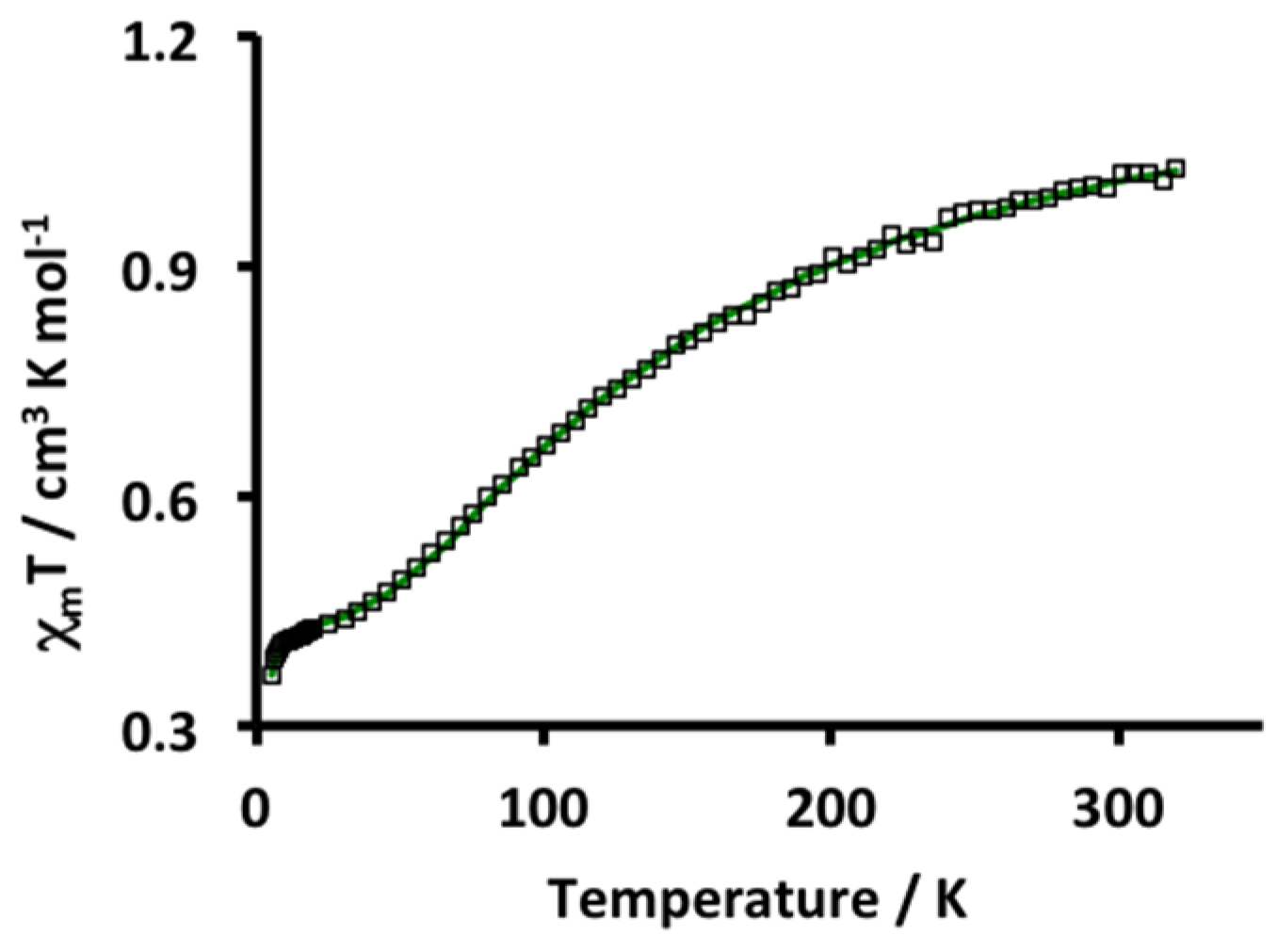
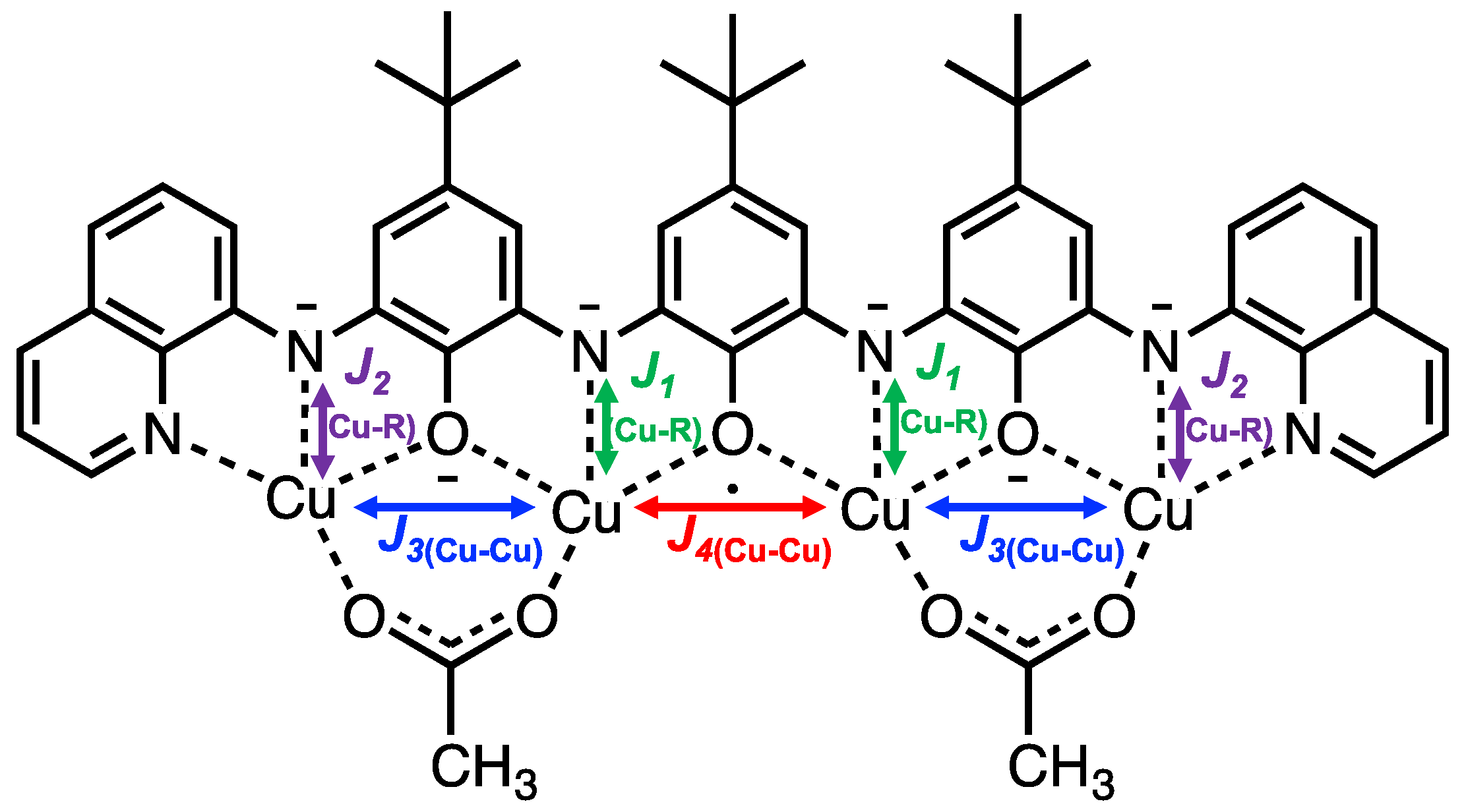
2. Results and Discussion
3. Materials and Methods
3.1. General Considerations
3.2. Computational Details
3.3. Synthesis of 2–5
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Luca, O.R.; Crabtree, R.H. Redox-active ligands in catalysis. Chem. Soc. Rev. 2013, 42, 1440–1459. [Google Scholar] [CrossRef] [PubMed]
- Lyaskovskyy, V.; De Bruin, B. Redox non-innocent ligands: Versatile new tools to control catalytic reactions. ACS Catal. 2012, 2, 270–279. [Google Scholar] [CrossRef]
- Praneeth, V.K.K.; Ringenberg, M.R.; Ward, T.R. Redox-active ligands in catalysis. Angew. Chem. Int. Ed. 2012, 51, 10228–10234. [Google Scholar] [CrossRef] [PubMed]
- Kaim, W.; Schwederski, B. Non-innocent ligands in bioinorganic chemistry—An overview. Coord. Chem. Rev. 2010, 254, 1580–1588. [Google Scholar] [CrossRef]
- Lyons, C.T.; Stack, T.D.P. Recent advances in phenoxyl radical complexes of salen-type ligands as mixed-valent galactose oxidase models. Coord. Chem. Rev. 2013, 257, 528–540. [Google Scholar] [CrossRef] [PubMed]
- Paul, N.D.; Rana, U.; Goswami, S.; Mondal, T.K.; Goswami, S. Azo anion radical complex of rhodium as a molecular memory switching device: isolation, characterization, and evaluation of current-voltage characteristics. J. Am. Chem. Soc. 2012, 134, 6520–6523. [Google Scholar] [CrossRef] [PubMed]
- D’Alessandro, D.M. Exploiting redox activity in metal–organic frameworks: Concepts, trends and perspectives. Chem. Commun. 2016, 52, 8957–8971. [Google Scholar] [CrossRef] [PubMed]
- Tezgerevska, T.; Alley, K.G.; Boskovic, C. Valence tautomerism in metal complexes: Stimulated and reversible intramolecular electron transfer between metal centers and organic ligands. Coord. Chem. Rev. 2014, 268, 23–40. [Google Scholar] [CrossRef]
- DeGayner, J.A.; Jeon, I.-R.; Harris, T.D. A series of tetraazalene radical-bridged M2 (M = Cr III, Mn II, Fe II, Co II ) complexes with strong magnetic exchange coupling. Chem. Sci. 2015, 6639–6648. [Google Scholar] [CrossRef]
- Jeon, I.-R.; Park, J.G.; Xiao, D.J.; Harris, T.D. An Azophenine Radical-Bridged Fe2 Single-Molecule Magnet with Record Magnetic Exchange Coupling. J. Am. Chem. Soc. 2013, 135, 16845–16848. [Google Scholar] [CrossRef] [PubMed]
- Demir, S.; Jeon, I.; Long, J.R.; Harris, T.D. Radical ligand-containing single-molecule magnets. Coord. Chem. Rev. 2015, 289–290, 149–176. [Google Scholar] [CrossRef]
- Bonanno, N.M.; Lough, A.J.; Prosser, K.E.; Walsby, C.J.; Poddutoori, P.K.; Lemaire, M.T. A stable open-shell redox active ditopic ligand. Dalton Trans. 2016, 3, 5460–5463. [Google Scholar] [CrossRef] [PubMed]
- Surry, D.S.; Buchwald, S.L. Dialkylbiaryl phosphines in Pd-catalyzed amination: A user's guide. Chem. Sci. 2011, 2, 27–50. [Google Scholar] [CrossRef] [PubMed]
- Broere, D.L.J.; Plessius, R.; van der Vlugt, J.I. New avenues for ligand-mediated processes—Expanding metal reactivity by the use of redox-active catechol, o-aminophenol and o-phenylenediamine ligands. Chem. Soc. Rev. 2015, 44, 6886–6915. [Google Scholar] [CrossRef] [PubMed]
- Min, K.S.; Weyhermüller, T.; Wieghardt, K. O,N-Coordinated o-iminobenzoquinone and o-iminobenzosemiquinonato(1−) ligands in complexes of Ni(II), Co(III) and Fe(III). Dalton Trans. 2003, 1126–1132. [Google Scholar]
- Min, K.S.; Weyhermüller, T.; Wieghardt, K. Coordination chemistry of 2-(8-aminoquinolino)-4,6-di-tert-butylphenol with manganese(IV), iron(III), and cobalt(II/III):N,O-coordinated o-iminobenzosemiquinonate(1−) pi radical monoanions vs.o-iminophenolate(2−) dianions. Dalton Trans. 2004, 178–186. [Google Scholar]
- Chilton, N.F.; Anderson, R.P.; Turner, L.D.; Soncini, A.; Murray, K.S. PHI: a powerful new program for the analysis of anisotropic monomeric and exchange-coupled polynuclear d- and f-block complexes. J. Comput. Chem. 2013, 34, 1164–1175. [Google Scholar] [CrossRef] [PubMed]
- Ahlrichs, R.; Bär, M.; Häser, M.; Horn, H.; Kölmel, C. Electronic structure calculations on workstation computers: The program system turbomole. Chem. Phys. Lett. 1989, 162, 165–169. [Google Scholar] [CrossRef]
- Treutler, O.; Ahlrichs, R. Efficient molecular numerical integration schemes. J. Chem. Phys. 1995, 102, 346–354. [Google Scholar] [CrossRef]
- Ahlrichs, R.; Bäcker, S.; Ehrig, M.; Eichkorn, K.; Elliott, S.; Haase, F.; Häser, M.; Horn, H.; Huber, C.; Huniar, U.; et al. TURBOMOLE V6.5 2013, a Development of University of Karlsruhe, and Forschungszentrum Karlsruhe GmbH, 1987–2007, TURBMOLE GmbH, since 2007. Available online: http://www.turbomole.com (accessed on 13 March 2017).
- Schafer, A.; Horn, H.; Ahlrichs, R. Fully Optimized Contracted Gaussian-Basis Sets for Atoms Li to Kr. J. Chem. Phys. 1992, 97, 2571–2577. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: a critical analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef]
- Perdew, J.P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B 1986, 33, 8822–8824. [Google Scholar] [CrossRef]
- Eichkorn, K.; Htiser, M.; Ahlrichs, R.; Eichkorn, K.; Treutler, O.; Marco, H.; Ahlrichs, R. Auxiliary basis sets to approximate Coulomb potentials. Chem. Phys. Lett. 1995, 240, 283–290. [Google Scholar] [CrossRef]
- Eichkorn, K.; Weigend, F.; Treutler, O.; Ahlrichs, R. Auxiliary basis sets for main row atoms and transition metals and their use to approximate Coulomb potentials. Theor. Chem. Acc. 1997, 97, 119–124. [Google Scholar] [CrossRef]
- Sierka, M.; Hogekamp, A.; Ahlrichs, R. Fast evaluation of the Coulomb potential for electron densities using multipole accelerated resolution of identity approximation. J. Chem. Phys. 2003, 118, 9136–9148. [Google Scholar] [CrossRef]
- Bauernschmitt, R.; Häser, M.; Treutler, O.; Ahlrichs, R. Calculation of excitation energies within time-dependent density functional theory using auxiliary basis set expansions. Chem. Phys. Lett. 1997, 264, 573–578. [Google Scholar] [CrossRef]
- Deglmann, P.; May, K.; Furche, F.; Ahlrichs, R. Nuclear second analytical derivative calculations using auxiliary basis set expansions. Chem. Phys. Lett. 2004, 384, 103–107. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D. 01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Skripnikov, L.V.; Chemissian, V. 4.01, Visualization Computer Program. 2014. Available online: www.chemissian.com (accessed on 13 March 2017).

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bonanno, N.M.; Lough, A.J.; Poddutoori, P.K.; Lemaire, M.T. Synthesis, Characterization and Copper(2+) Coordination Chemistry of a Polytopic Paramagnetic Ligand. Magnetochemistry 2017, 3, 15. https://doi.org/10.3390/magnetochemistry3010015
Bonanno NM, Lough AJ, Poddutoori PK, Lemaire MT. Synthesis, Characterization and Copper(2+) Coordination Chemistry of a Polytopic Paramagnetic Ligand. Magnetochemistry. 2017; 3(1):15. https://doi.org/10.3390/magnetochemistry3010015
Chicago/Turabian StyleBonanno, Nico M., Alan J. Lough, Prashanth K. Poddutoori, and Martin T. Lemaire. 2017. "Synthesis, Characterization and Copper(2+) Coordination Chemistry of a Polytopic Paramagnetic Ligand" Magnetochemistry 3, no. 1: 15. https://doi.org/10.3390/magnetochemistry3010015
APA StyleBonanno, N. M., Lough, A. J., Poddutoori, P. K., & Lemaire, M. T. (2017). Synthesis, Characterization and Copper(2+) Coordination Chemistry of a Polytopic Paramagnetic Ligand. Magnetochemistry, 3(1), 15. https://doi.org/10.3390/magnetochemistry3010015