

Complete Chloroplast Genome Sequence of Rosa lucieae and Its Characteristics
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Taxon Sampling
2.2. DNA Extraction and Sequencing
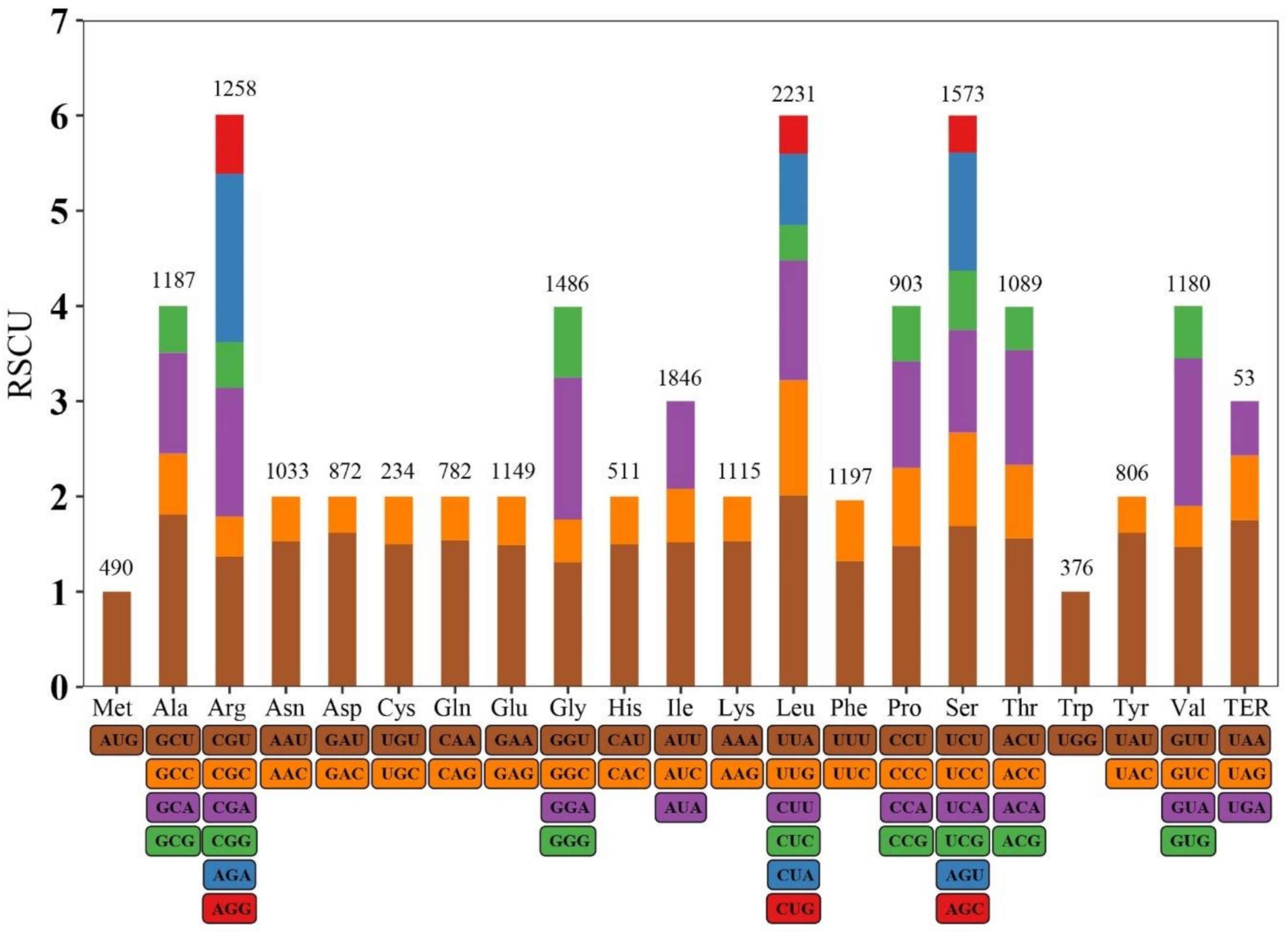
2.3. Chloroplast Genome Assembly, Gene Annotation, and Relative Synonymous Codon Usage
2.4. Repeat Sequence and SSR Analysis
2.5. Contraction and Expansion of IRs
2.6. Sliding Window Analysis
2.7. Positive Selection Analysis
2.8. Phylogenetic Analyses
3. Results and Discussion
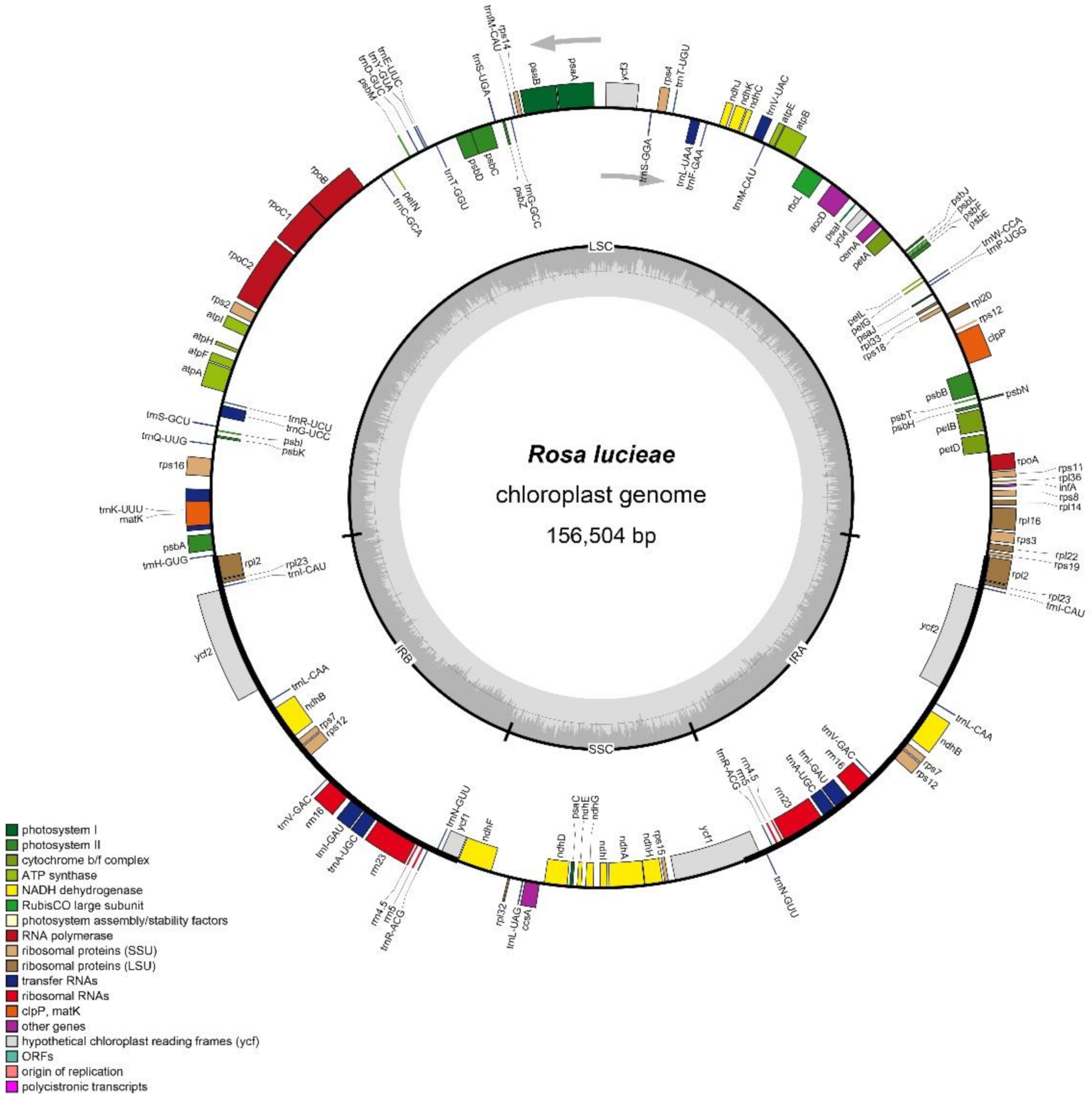
3.1. Chloroplast Genome Characteristics of R. lucieae
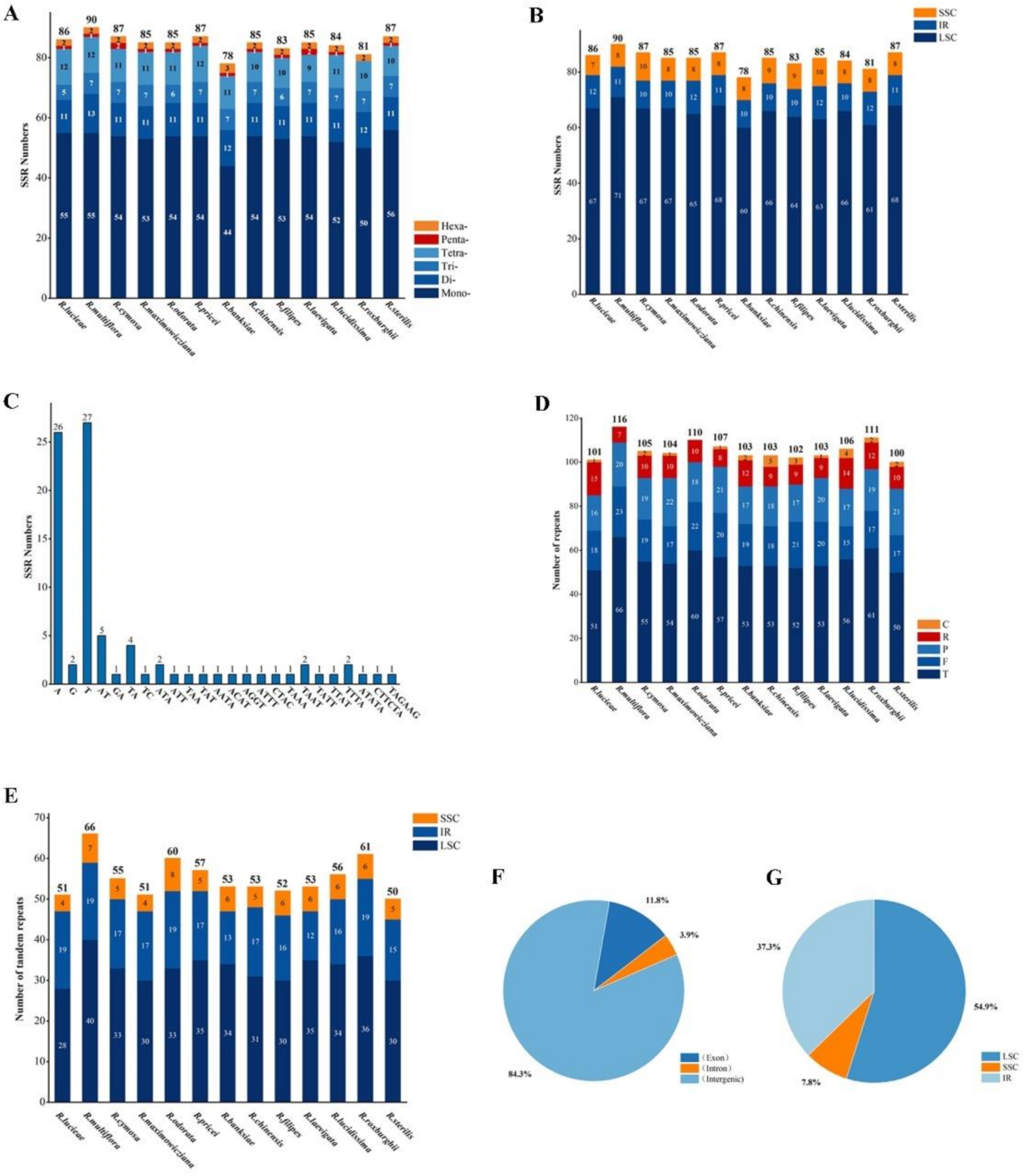
3.2. Repeat Sequence and SSR Analysis
3.3. Inverted Repeat Contraction and Expansion Analysis
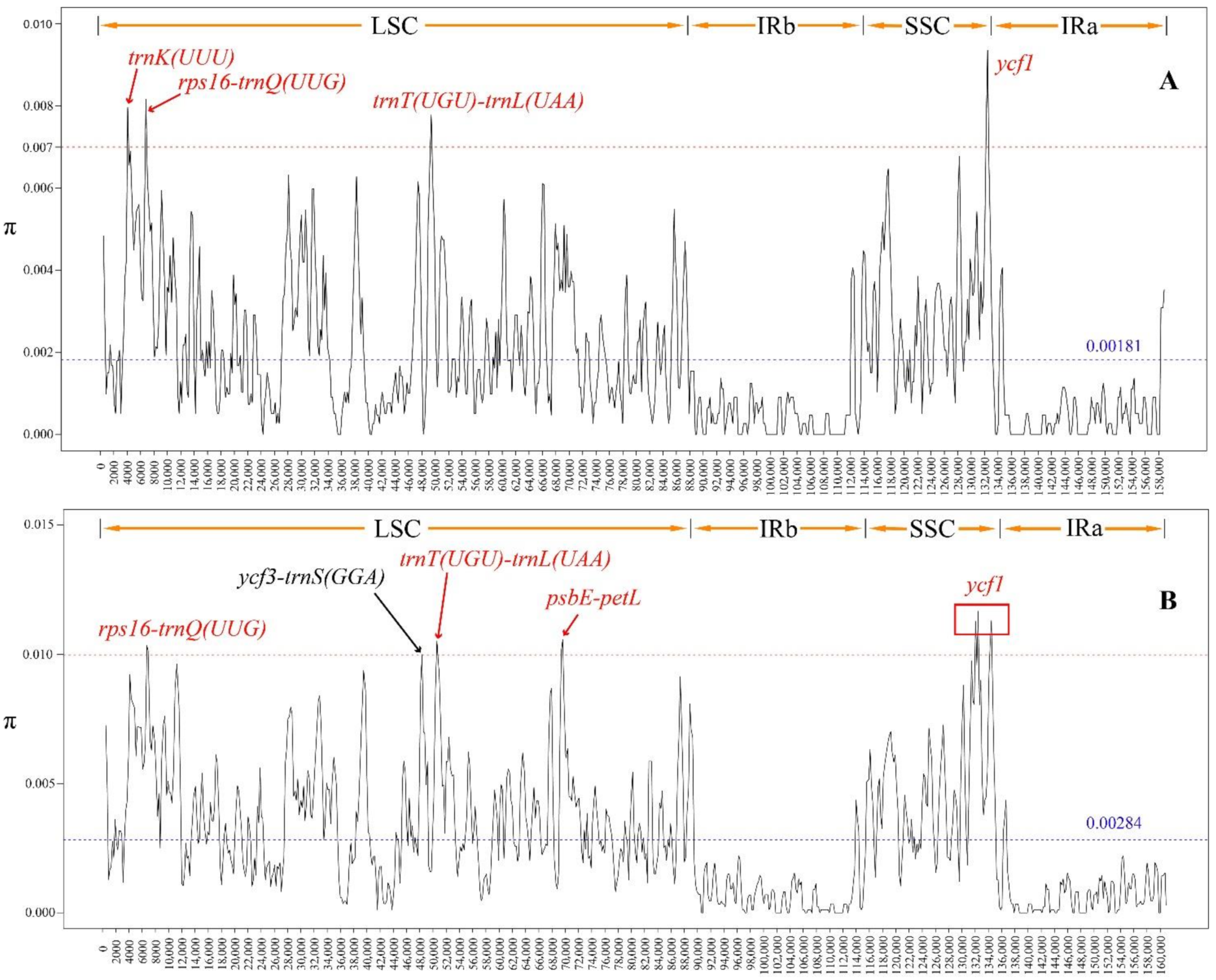
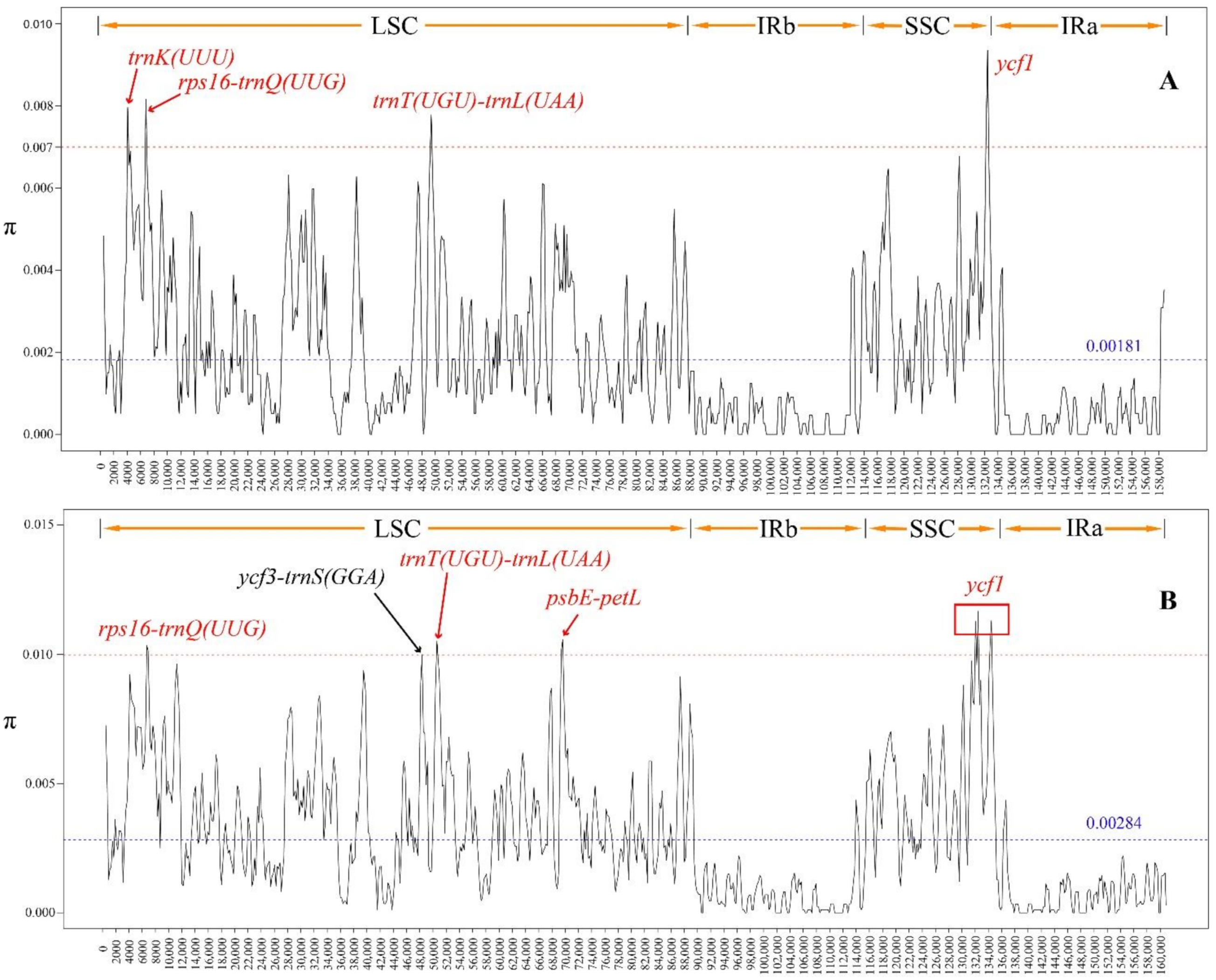
3.4. Sliding Window Analysis
3.5. Positive Selection Analysis
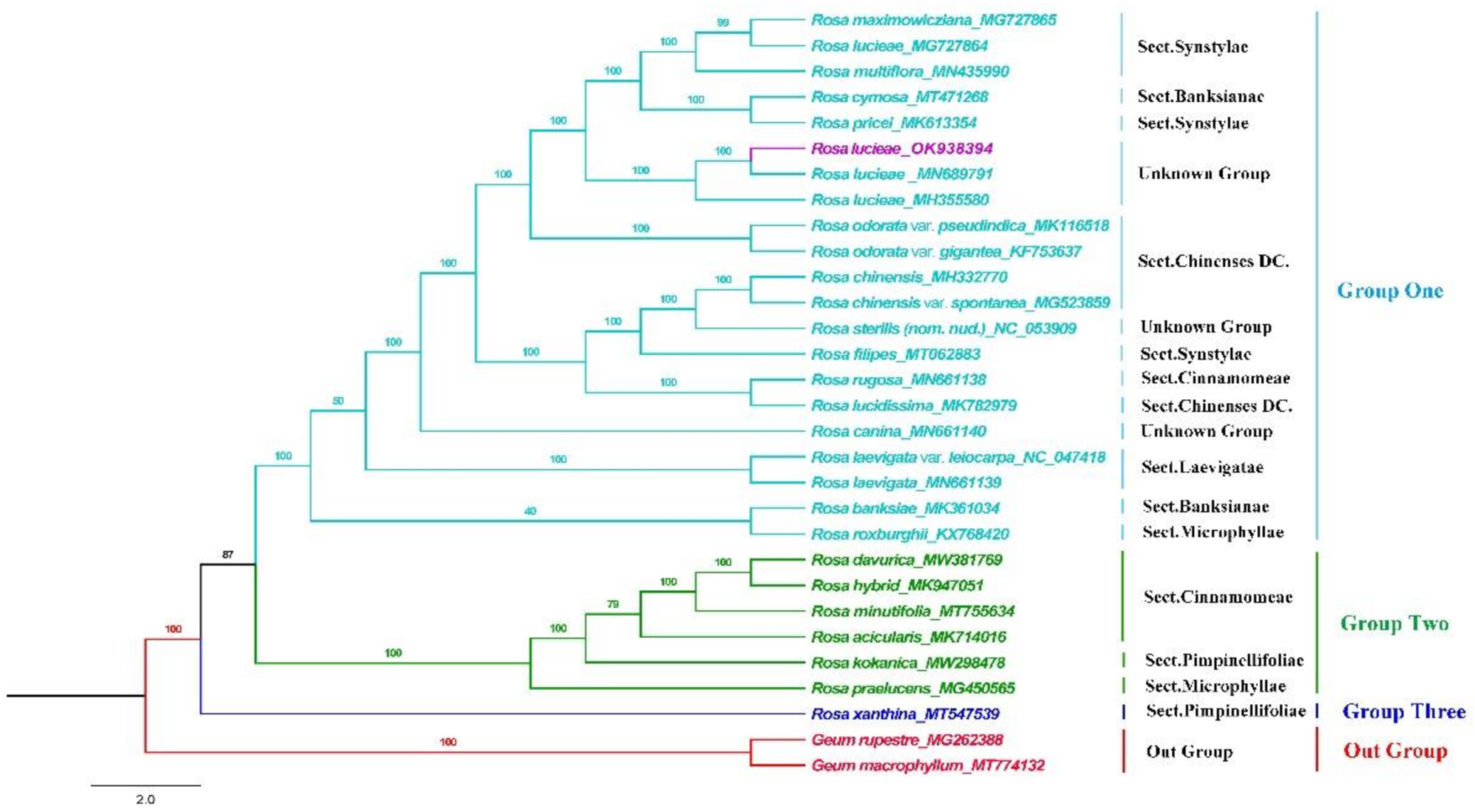
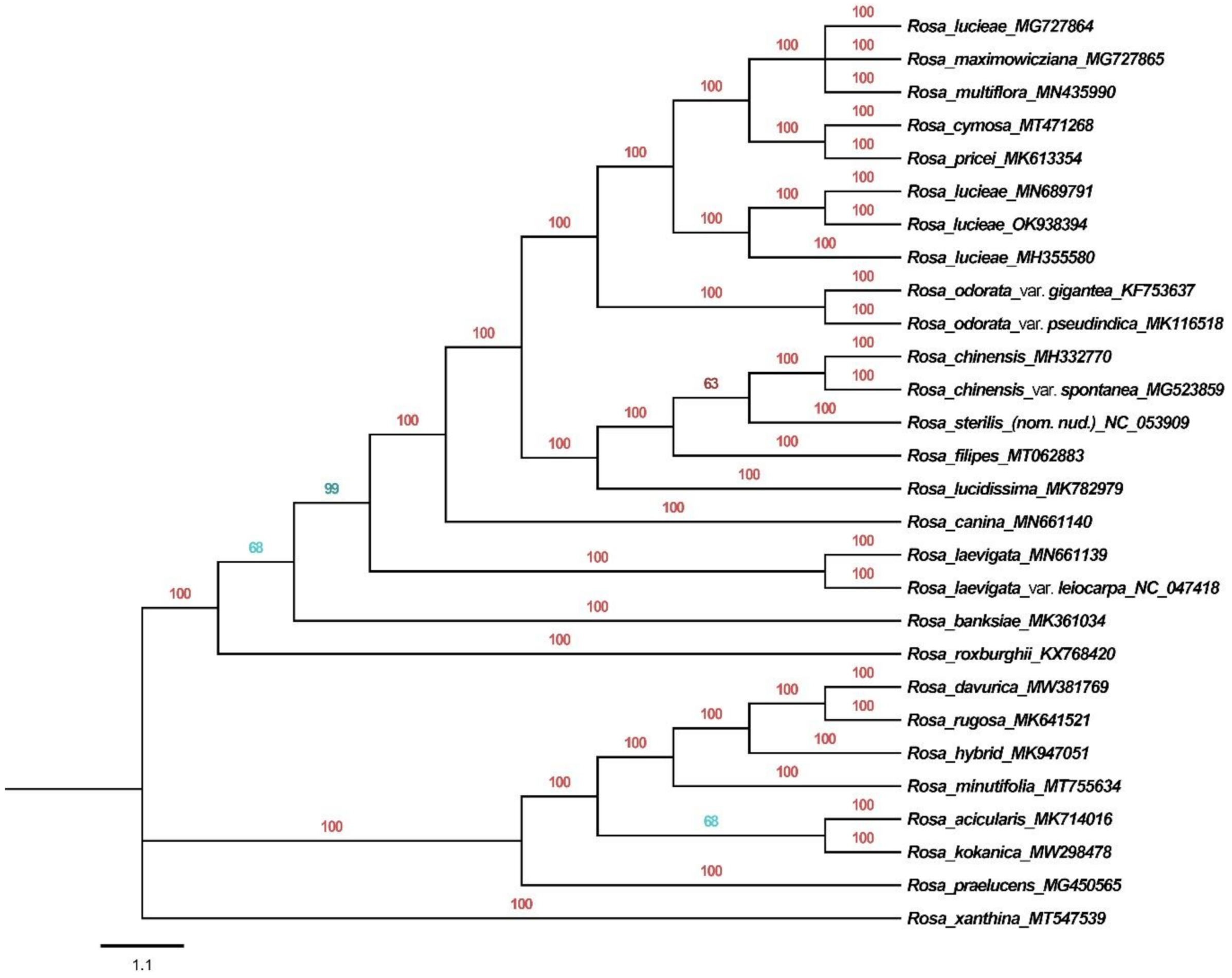
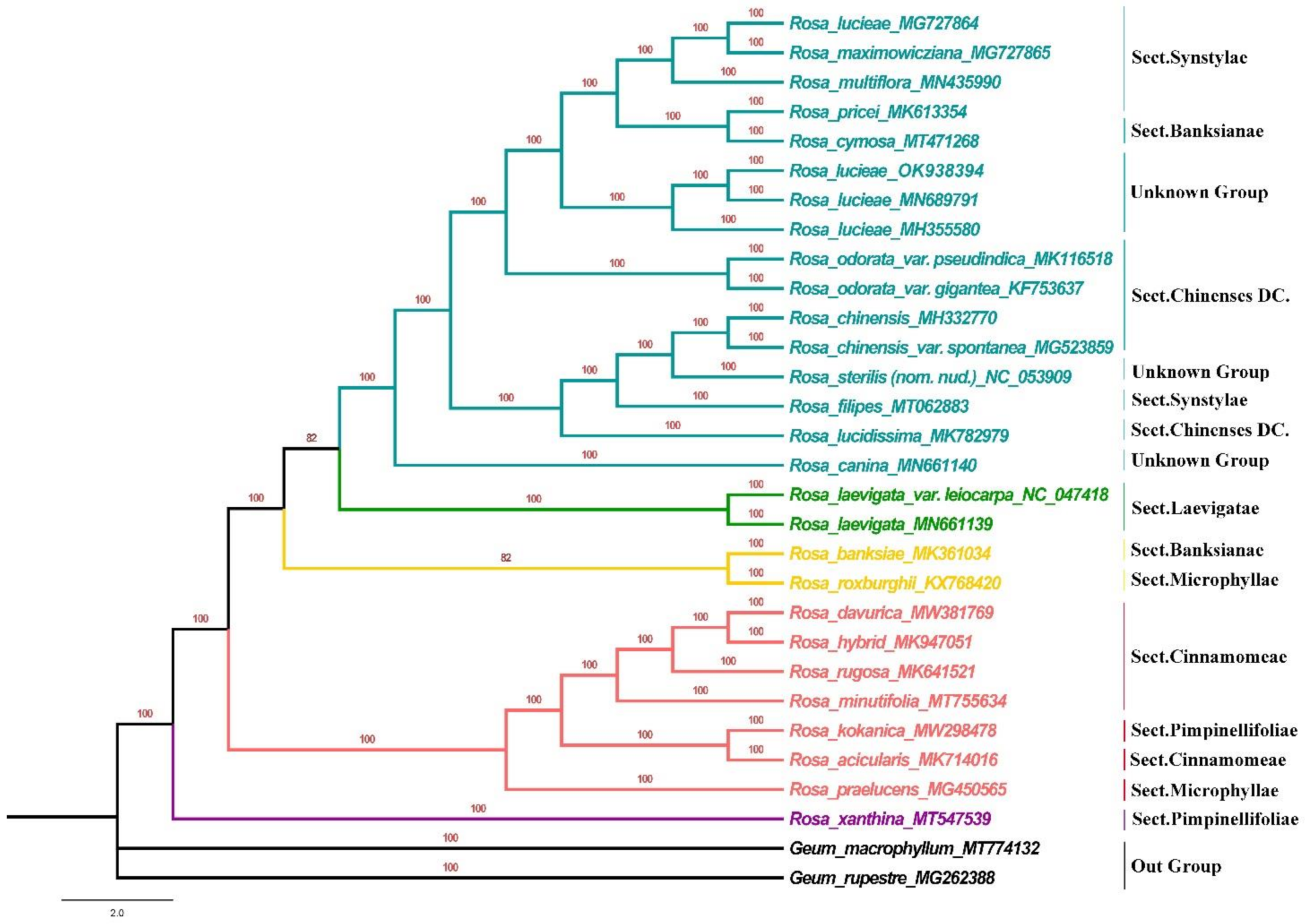
3.6. Phylogenetic Analysis
4. Discussion
4.1. Comparison of cp Genomes in the Rosa Species
4.2. Sliding Window Analysis
4.3. Positive Selection Analysis
4.4. Phylogenetic Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A
References
- Jeon, J.H.; Kim, S.C. Comparative analysis of the complete chloroplast genome sequences of three closely related East-Asian wild roses (Rosa sect. Synstylae; Rosaceae). Genes 2019, 10, 23. [Google Scholar] [CrossRef] [PubMed]
- Debener, T.; Linde, M. Exploring complex ornamental genomes: The Rose as a model plant. CRC Crit. Rev. Plant Sci. 2009, 28, 267–280. [Google Scholar] [CrossRef]
- Lv, J.J. Establishment of plant regeneration system and preliminary studies on genetic transformation system of Rosa wichuriana ‘Basye’s thornless. Master’s Thesis, Huazhong Agricultural University, Wuhan, China, 2013. [Google Scholar]
- Jin, J.; Jin, P.; Wu, H.E.; Dong, W.P.; Yang, C.H.; Zhou, H.Y. Investigation and Application of Rosa plant Resources in Guizhou. Seed 2020, 39, 61–65,69. [Google Scholar] [CrossRef]
- Wang, S.Q.; Zhu, Z.M. Relationship between species richness patterns of Rosa L. and environmental factors in China. Acta Ecol. Sin. 2022, 42, 209–219. [Google Scholar] [CrossRef]
- Xing, S.C.; Clarke, J.L. Progress in Chloroplast Genome Analysis. Prog. Biochem. Biophys. 2008, 4, 21–28. [Google Scholar]
- Raubeson, L.A.; Jansen, R.K. Chloroplast Genomes of Plants, Plant Diversity and Evolution: Genotypic and Phenotypic Variation in Higher Plants; CABI Publishing: Cambridge, UK, 2005. [Google Scholar] [CrossRef]
- Jansen, R.K.; Ruhlman, T.A. Plastid Genomes of Seed Plants. In Genomics of Chloroplasts and Mitochondria; Springer: Dordrecht, The Netherlands, 2012. [Google Scholar] [CrossRef]
- Daniell, H.; Lin, C.S.; Yu, M.; Chang, W.J. Chloroplast genomes: Diversity, evolution, and applications in genetic engineering. Genome Biol. 2016, 17, 134. [Google Scholar] [CrossRef]
- Liang, X.; Li, P.; Xu, D.M.; Jia, X.Y.; Wang, W.B. Characteristics Analysis of Whole Chloroplast Genome in Perilla frustescens. J. Shanxi Agric. Sci. 2021, 49, 265–272. [Google Scholar] [CrossRef]
- Day, P.D.; Madeleine, B.; Laurence, H.; Fay, M.F.; Leitch, A.R.; Leitch, I.J.; Kelly, L.J. Evolutionary relationships in the medicinally important genus Fritillaria L. (Liliaceae). Mol. Phylogenetics Evol. 2014, 80, 11–19. [Google Scholar] [CrossRef]
- Li, M.; Ye, Q.; Song, Y.F.; Wu, S.H.; Yi, X.G.; Wang, X.R. The Analysis of the Codon Usage Bias in the Chloroplast Genome of Prunus sargentii. Mol. Plant Breed. 2021. Available online: https://kns.cnki.net/kcms/detail/46.1068.S.20210728.0854.002.html (accessed on 8 May 2022).
- Dong, Z.H.; Qu, S.H.; Liu, C.; Ye, P.; Xin, P.Y. The complete chloroplast genome sequence of Eriobotrya fragrans. Mitochondrial DNA Part B 2019, 4, 3549–3550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, Y.Y.; Xin, J.; Feng, F.Y.; Dong, Z.H.; Qu, S.H.; Wang, H.Y. Codon Usage Bais in Chloroplast Genome of Eriobotrya fragrans Champ. Ex Benth. J. Northwest For. Univ. 2021, 36, 138–144,158. [Google Scholar]
- Su, Y.; Liu, J.J.; Wan, B.; Zhang, P.J.; Cheng, Z.G.; Suzuki, J.J.; Wang, C. Chloroplast Genome Structure Characteristic and Phylogenetic Analysis of Mulgedium tataricum. J. Agric. Sci. Technol. 2021, 23, 33–42. [Google Scholar] [CrossRef]
- Wang, Q.R.; Huang, Z.R.; Gao, C.S.; Ge, Y.Q.; Cheng, R.B. The complete chloroplast genome sequence of Rubus hirsutus Thunb. and a comparative analysis within Rubus species. Genetica 2021, 149, 299–311. [Google Scholar] [CrossRef]
- Yu, J.J.; Fu, J.; Fang, Y.P.; Xiang, J.; Dong, H.J. Complete chloroplast genomes of Rubus species (Rosaceae) and comparative analysis within the genus. BMC Genom. 2022, 23, 32. [Google Scholar] [CrossRef]
- Li, Q.Q.; Wen, J. The complete chloroplast genome of Geum macrophyllum (Rosaceae: Colurieae). Mitochondrial DNA Part B 2021, 6, 297–298. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.P.; Wang, L.; Lu, X. Complete chloroplast genome of Geum aleppicum (Rosaceae). Mitochondrial DNA B 2022, 7, 234–235. [Google Scholar] [CrossRef] [PubMed]
- Xin, Y.X.; Li, R.Z.; Li, X.; Chen, L.Q.; Tang, J.R.; Qu, Y.Y.; Xin, P.; Li, Y. Analysis on codon usage bias of chloroplast genome in Mangifera indica. J. Cent. South Univ. For. Technol. 2021, 41, 148–156+165. [Google Scholar] [CrossRef]
- Moore, M.J.; Dhingra, A.; Soltis, P.S.; Shaw, R.; Farmerie, W.G.; Folta, K.M.; Soltis, D.E. Rapid and accurate pyrosequencing of angiosperm plastid genomes. BMC Plant Biol. 2006, 6, 17. [Google Scholar] [CrossRef]
- Bayly, M.J.; Rigault, P.; Spokevicius, A.; Ladiges, P.Y.; Ades, P.K.; Anderson, C.; Bossinger, G.; Merchant, A.; Udovicic, F.; Woodrow, I.E.; et al. Chloroplast genome analysis of Australian eucalypts—Eucalyptus, Corymbia, Angophora, Allosyncarpia and Stockwellia (Myrtaceae). Mol. Phylogenetics Evol. 2013, 69, 704–716. [Google Scholar] [CrossRef]
- Matsumotoa, S.; Kouchib, M.; Yabukib, J.; Kusunokia, M.; Uedac, Y.; Fukuib, H. Phylogenetic analyses of the genus Rosa using the matK sequence: Molecular evidence for the narrow genetic background of modern roses. Sci. Hortic. 1998, 77, 73–82. [Google Scholar] [CrossRef]
- Cui, W.H.; Zhong, M.C.; Du, X.Y.; Qu, X.J.; Jiang, X.D.; Sun, Y.B.; Wang, D.; Chen, S.Y.; Hu, J.Y. The complete chloroplast genome sequence of a rambler rose, Rosa wichuraiana (Rosaceae). Mitochondrial DNA Part B 2020, 5, 252–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, C.W.; Wu, C.H.; Zhang, Q.; Wu, M.X.; Chen, R.R.; Zhao, Y.L.; Guo, A.; Li, Z. Sequence and phylogenetic analysis of the chloroplast genome for Rosa xanthina. Mitochondrial DNA Part B 2020, 5, 2940–2941. [Google Scholar] [CrossRef]
- Zhao, X.; Gao, C.W. The complete chloroplast genome sequence of Rosa minutifolia. Mitochondrial DNA Part B 2020, 5, 3338–3339. [Google Scholar] [CrossRef] [PubMed]
- Doyle, J.J.; Doyle, J.L. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar] [CrossRef]
- Jin, J.J.; Yu, W.B.; Yang, J.B.; Song, Y.; dePamphilis, C.W.; Yi, T.S.; Li, D.Z. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020, 21, 241. [Google Scholar] [CrossRef] [PubMed]
- Wick, R.R.; Schultz, M.B.; Zobel, J.; Holt, K.E. Bandage: Interactive visualization of de novo genome assemblies. Bioinformatics 2015, 31, 3350–3352. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.D.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Greiner, S.; Lehwark, P.; Bock, R. OrganellarGenomeDRAW (OGDRAW) version 1.3.1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 2019, 47, 59–64. [Google Scholar] [CrossRef]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef] [Green Version]
- Amiryousefi, A.; Hyvönen, J.; Poczai, P. IRscope: An online program to visualize the junction sites of chloroplast genomes. Bioinformatics 2018, 34, 3030–3031. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D. MAFFT multiple sequence alignment software version improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large datasets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Gao, F.L.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2019, 20, 348–355. [Google Scholar] [CrossRef]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. Creating the CIPRES science gateway for inference of large phylogenetic trees. In Proceedings of the Gateway Computing Environments Workshop (GCE), New Orleans, LA, USA, 14 November 2010; pp. 1–8. [Google Scholar] [CrossRef]
- Gao, F.; Chen, C.; Arab, D.A.; Du, Z.; He, Y.; Ho, S.Y.W. EasyCodeML: A visual tool for analysis of selection using CodeML. Ecol. Evol. 2019, 9, 3891–3898. [Google Scholar] [CrossRef]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.Q.; Chen, J.F.; Zhu, Z.M. The complete chloroplast genome sequence of Rosa filipes (Rosaceae). Mitochondrial DNA Part B 2020, 5, 1376–1377. [Google Scholar] [CrossRef]
- Ding, M.Y.; Liao, M.; Liu, P.Q.; Tan, G.Q.; Chen, Y.Q.; Shi, S. The complete chloroplast genome of Rosa cymosa (Rosaceae), a traditional medicinal plant in South China. Mitochondrial DNA B 2020, 5, 2571–2572. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.S.; Li, P.; Qiu, Y.X. The complete chloroplast genomes of three Cardiocrinum (Liliaceae) species: Comparative genomic and phylogenetic analyses. Front. Plant Sci. 2017, 10, 2054. [Google Scholar] [CrossRef] [PubMed]
- Raubeson, L.A.; Peery, R.; Chumley, T.W.; Dziubek, C.; Fourcade, H.M.; Boore, J.L.; Jansen, R.K. Comparative chloroplast genomics: Analyses including new sequences from the angiosperms Nuphar advena and Ranunculus macranthus. BMC Genom. 2007, 8, 174. [Google Scholar] [CrossRef]
- Li, R.; Ma, P.F.; Wen, J.; Yi, T.S. Complete sequencing of five Araliaceae chloroplast genomes and the phylogenetic implications. PLoS ONE 2013, 8, e78568. [Google Scholar] [CrossRef]
- Song, Y.; Dong, W.; Liu, B.; Xu, C.; Yao, X.; Gao, J.; Corlett, R.T. Comparative analysis of complete chloroplast genome sequences of two tropical trees Machilus yunnanensis and Machilus balansae in the family Lauraceae. Front. Plant Sci. 2015, 6, 662. [Google Scholar] [CrossRef]
- Kim, K.J.; Lee, H.L. Complete chloroplast genome sequences from Korean ginseng (Panax schinseng Nees) and comparative analysis of sequence evolution among 17 vascular plants. DNA Res. Int. J. Rapid Publ. Rep. Genes Genomes 2004, 11, 247–261. [Google Scholar] [CrossRef]
- Yang, G.F.; Su, K.L.; Zhao, Y.R.; Song, Z.B.; Sun, J. Analysis of codon usage in the chloroplast genome of Medicago truncatula. Acta Prataculturae Sin. 2015, 24, 171–178. [Google Scholar] [CrossRef]
- Ye, Y.J.; Ni, Z.X.; Bai, T.D.; Xu, L. The analysis of chloroplast genome codon usage bais in Pinus massoniana. Genom. Appl. Biol. 2018, 37, 4464–4471. [Google Scholar] [CrossRef]
- Yuan, X.L.; Li, Y.Q.; Zhang, J.F.; Wang, Y. Analysis of codon usage bias in the chloroplast genome of Dalbergia odorifera. Guihaia 2021, 41, 622–630. [Google Scholar] [CrossRef]
- Shaw, J.; Lickey, E.B.; Schilling, E.E.; Small, R.L. Comparison of whole chloroplast genome sequences to choose noncoding regions for phylogenetic studies in angiosperms: The tortoise and the hare III. Am. J. Bot. 2007, 94, 275–288. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.H.; Yang, L.; Zhang, C.J.; Xu, H.J.; Chen, Z.L. Characteristics of the complete chloroplast genome of Dendrobium ochreatum and its comparative analysis. Chin. J. Trop. Crops 2021, 42, 3111–3119. [Google Scholar] [CrossRef]
- Sun, L.Y.; Jiang, Z.; Wan, X.X.; Zou, X.; Yao, X.Y.; Wang, Y.L.; Yin, Z.F. The complete chloroplast genome of Magnolia polytepala: Comparative analyses offer implication for genetics and phylogeny of Yulania. Gene 2020, 736, 100010. [Google Scholar] [CrossRef] [PubMed]
- Ninio, J. The neutral theory of molecular evolution: Edited by Mooto Kimura Cambridge University Press. Cambridge, 1983, 366 pages. FEBS Lett. 1984, 170, 210–211. [Google Scholar] [CrossRef]
- Yang, Z.; Nielsen, R. Estimating synonymous and nonsynonymous substitution rates under realistic evolutionary models. Mol. Biol. Evol. 2000, 17, 32–43. [Google Scholar] [CrossRef]
- Nawae, W.; Yundaeng, C.; Naktang, C.; Kongkachana, W.; Yoocha, T.; Sonthirod, C.; Narong, N.; Somta, P.; Laosatit, K.; Tangphatsornruang, S.; et al. The Genome and Transcriptome Analysis of the Vigna mungo Chloroplast. Plants 2020, 9, 1247. [Google Scholar] [CrossRef]
- Sen, L.; Fares, M.A.; Liang, B.; Gao, L.; Wang, B.; Wang, T.; Su, Y.J. Molecular evolution of rbcL in three gymnosperm families: Identifying adaptive and coevolutionary patterns. Biol. Direct 2011, 6, 29. [Google Scholar] [CrossRef]
- Rono, P.C.; Dong, X.; Yang, J.X.; Mutie, F.M.; Oulo, M.A.; Malombe, I. Initial complete chloroplast genomes of Alchemilla (Rosaceae): Comparative analysis and phylogenetic relationships. Front. Genet. 2020, 11. [Google Scholar] [CrossRef]
- Sheng, J.; Yan, M.; Wang, J.; Zhao, L.; Zhou, F.; Hu, Z.; Jin, S.; Diao, Y. The complete chloroplast genome sequences of five Miscanthus species, and comparative analyses with other grass plastomes. Ind. Crops Prod. 2021, 162, 113248. [Google Scholar] [CrossRef]
- Huang, S.N.; Ge, X.J.; Cano, A.; Salazar, B.G.M.; Deng, Y.F. Comparative analysis of chloroplast genomes for five Dicliptera species (Acanthaceae): Molecular structure, phylogenetic relationships, and adaptive evolution. PeerJ 2020, 8, e8450. [Google Scholar] [CrossRef] [Green Version]
- Xie, D.F.; Yu, Y.; Deng, Y.Q.; Li, J.; Liu, H.Y.; Zhou, S.D.; He, X.J. Comparative analysis of the chloroplast genomes of the chinese endemic genus Urophysa and their contribution to chloroplast phylogeny and adaptive evolution. Int. J. Mol. Sci. 2018, 19, 1847. [Google Scholar] [CrossRef] [PubMed]
- Bock, D.G.; Andrew, R.L.; Rieseberg, L.H. On the adaptive value of cytoplasmic genomes in plants. Mol. Ecol. 2014, 23, 4899–4911. [Google Scholar] [CrossRef] [PubMed]
- Galmes, J.; Andralojc, P.J.; Kapralov, M.V.; Flexas, J.; Keys, A.J.; Molins, A.; Conesa, M.À. Environmentally driven evolution of Rubisco and improved photosynthesis and growth within the C3 genus Limonium (Plumbaginaceae). New Phytol. 2014, 203, 989–999. [Google Scholar] [CrossRef] [PubMed]
- Kapralov, M.V.; Smith, J.A.C.; Filatov, D.A. Rubisco evolution in C4 eudicots: An analysis of Amaranthaceae sensu lato. PLoS ONE 2012, 7, e52974. [Google Scholar] [CrossRef]
- Drescher, A.; Ruf, S.; Calsa, T.; Carrer, H., Jr.; Bock, R. The two largest chloroplast genome-encoded open reading frames of higher plants are essential genes. Plant J. 2000, 22, 97–104. [Google Scholar] [CrossRef]
- Kikuchi, S.; Bedard, J.; Hirano, M.; Hirabayashi, Y.; Oishi, M.; Imai, M.; Takasetoru, M.; Ide, T.; Nakai, M. Uncovering the protein translocon at the chloroplast inner envelope membrane. Science 2013, 339, 571–574. [Google Scholar] [CrossRef]
- Gao, L.Z.; Liu, Y.L.; Zhang, D.; Li, W.; Gao, J.; Liu, Y.; Li, K.; Shi, C.; Zhao, Y.; Zhao, Y.-J.; et al. Evolution of Oryza chloroplast genomes promotedad aptation to diverse ecological habitats. Commun. Biol. 2019, 2, 278. [Google Scholar] [CrossRef] [Green Version]
- Zhu, S.X. Systematics of Chaetoseris and Stenoseris (Compositae-Lactuceae). Master’s Thesis, Graduate School of Chinese Academy of Sciences (Institute of Botany), Beijing, China, 2014. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Taxon | Accession Number | Gene Number | Length (bp) | GC (%) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| CDS | tRNA | rRNA | Genome | Genome | LSC | SSC | IR | |||
| R. acicularis | MK714016 | 84 | 37 | 8 | 130 | 156,527 | 85,673 | 18,748 | 26,053 | 37.2% |
| R. banksiae | MK361034 | 84 | 37 | 8 | 130 | 156,575 | 85,792 | 18,767 | 26,008 | 37.2% |
| R. canina | MN661140 | 85 | 37 | 8 | 130 | 156,501 | 85,653 | 18,742 | 26,053 | 37.3% |
| R. chinensis | MH332770 | 85 | 37 | 8 | 130 | 156,591 | 85,737 | 18,766 | 26,044 | 37.2% |
| R. chinensis var. spontanea | MG523859 | 84 | 37 | 8 | 130 | 156,590 | 85,825 | 18,677 | 26,044 | 37.2% |
| R. cymosa | MT471268 | 92 | 39 | 8 | 140 | 156,607 | 85,722 | 18,763 | 26,061 | 37.2% |
| R. davurica | MW381769 | 85 | 37 | 8 | 131 | 156,971 | 86,032 | 18,837 | 26,051 | 37.2% |
| R. filipes | MT062883 | 90 | 37 | 8 | 137 | 156,624 | 85,754 | 18,784 | 26,043 | 37.2% |
| R. hybrid | MK947051 | 84 | 37 | 8 | 130 | 156,989 | 86,227 | 18,816 | 25,973 | 37.2% |
| R. kokanica | MW298478 | 85 | 37 | 8 | 131 | 156,793 | 85,890 | 18,773 | 26,065 | 37.2% |
| R. laevigata | MN661139 | 85 | 37 | 8 | 130 | 156,333 | 85,452 | 18,785 | 26,048 | 37.3% |
| R. laevigata var. leiocarpa | NC_047418 | 92 | 39 | 8 | 140 | 156,373 | 85,494 | 18,785 | 26,047 | 37.3% |
| R. lucidissima | MK782979 | 83 | 37 | 8 | 129 | 156,588 | 85,713 | 18,779 | 26,048 | 37.2% |
| R. lucieae | OK938394 | 85 | 37 | 8 | 130 | 156,504 | 85,660 | 18,744 | 26,050 | 37.2% |
| R. lucieae | MN689791 | 85 | 37 | 8 | 130 | 156,504 | 85,661 | 18,743 | 26,050 | 37.2% |
| R. lucieae | MH355580 | 85 | 37 | 8 | 130 | 156,500 | 85,651 | 18,751 | 26,049 | 37.2% |
| R. lucieae | MG727864 | 88 | 37 | 8 | 134 | 156,506 | 85,631 | 18,759 | 26,058 | 37.2% |
| R. maximowicziana | MG727865 | 88 | 37 | 8 | 134 | 156,405 | 85,529 | 18,760 | 26,058 | 37.2% |
| R. minutifolia | MT755634 | 86 | 39 | 8 | 135 | 157,396 | 86,547 | 18,903 | 25,973 | 37.2% |
| R. multiflora | MN435990 | 88 | 37 | 8 | 96 | 157,385 | 86,255 | 19,014 | 26,058 | 37.2% |
| R. odorata var. gigantea | KF753637 | 88 | 40 | 8 | 139 | 156,634 | 85,767 | 18,761 | 26,053 | 37.2% |
| R. odorata var. pseudindica | MK116518 | 85 | 37 | 8 | 133 | 156,652 | 85,785 | 18,761 | 26,053 | 37.2% |
| R. praelucens | MG450565 | 84 | 37 | 8 | 130 | 157,186 | 86,313 | 18,743 | 26,065 | 37.2% |
| R. pricei | MK613354 | 86 | 39 | 8 | 137 | 156,599 | 85,731 | 18,750 | 26,059 | 37.2% |
| R. roxburghii | KX768420 | 88 | 39 | 8 | 139 | 156,749 | 85,852 | 18,791 | 26,053 | 37.2% |
| R. rugosa | MK641521 | 85 | 37 | 8 | 135 | 157,110 | 86,215 | 18,819 | 26,038 | 37.2% |
| R. sterilis (nom. nud.) | NC_053909 | 84 | 37 | 8 | 130 | 156,561 | 85,701 | 18,746 | 26,057 | 37.2% |
| R. xanthina | MT547539 | 86 | 39 | 8 | 137 | 157,214 | 86,302 | 18,800 | 26,056 | 37.2% |
| Geum macrophyllum | MT774132 | 85 | 37 | 8 | 130 | 155,940 | 85,307 | 18,329 | 26,152 | 36.6% |
| Geum rupestre | MG262388 | 87 | 39 | 8 | 138 | 155,479 | 85,771 | 18,550 | 25,579 | 36.8% |
| Category | Gene Group | Gene Name | Number |
|---|---|---|---|
| Photosynthesis gene | Photosystem I gene | psaA, psaB, psaC, psaI, psaJ | 5 |
| Photosystem II gene | psbA, psbB, psbC, psbD, psbE, psbF, psbH, psbI, psbJ, psbK, psbL, psbM, psbN, psbT, psbZ | 15 | |
| Cytochrome b/f complex gene | petA, petB, petD, petG, petL, petN | 6 | |
| ATP synthase gene | atpA, atpB, atpE, atpF, atpH, atpI | 6 | |
| NADH dehydrogenase gene | ndhA, ndhB C, ndhC, ndhD, ndhE, ndhF, ndhG, ndhH, ndhI, ndhJ, ndhK | 11 | |
| Rubis CO large subunit gene | rbcL | 1 | |
| Self-replication gene | RNA polymerase gene | rpoA, rpoB, rpoC1, rpoC2 | 4 |
| Ribosomal proteins (SSU) gene | rps2, rps3, rps4, rps7 C, rps8, rps11, rps12 A,C, rps14, rps15, rps16, rps18, rps19c | 12 | |
| Ribosomal proteins (LSU) gene | rpl2 C, rpl14, rpl16, rpl20, rpl22, rpl23 C, rpl32, rpl33, rpl36 | 9 | |
| Ribosomal RNAs gene | rrn4.5 C, rrn5 C, rrn16 C, rrn23 C | 4 | |
| Transfer RNAs gene | trnA-UGC A,C, trnC-GCA, trnD-GUC, trnE-UUC, trnF-GAA, trnfM-CAU, trnG-GCC, trnG-UCC A, trnH-GUG, trnI-CAU C, trnI-GAU A,C, trnK-UUU A, trnL-CAA C, trnL-UAA A, trnL-UAG, trnM-CAU, trnN-GUU C, trnP-UGG, trnQ-UUG, trnR-ACG C, trnS-GCU, trnS-GGA, trnS-UGA, trnT-GGU, trnT-UGU, trnV-GAC C, trnV-UAC A, trnW-CCA, trnY-GUA | 29 | |
| Other genes | Translational initiation factor gene | infA | 1 |
| Maturase K gene | matK | 1 | |
| Subunit of acetyl-Co A gene | accD | 1 | |
| Envelop membrane protein gene | cemA | 1 | |
| c-type cytochrome synthesis gene | ccsA | 1 | |
| Protease gene | clpP | 1 | |
| Hypothetical chloroplast reading frames (ycf) | ycf1 C, ycf2 C, ycf3, ycf4 | 4 |
| Gene Name | M8 | Gene Name | M8 | ||
|---|---|---|---|---|---|
| Selected Site | Score | Selected Site | Score | ||
| atpF | 108L | 0.989 * | rpl20 | 72N | 0.955 * |
| matK | 83F | 1.000 ** | rpl23 | 24S | 0.960 * |
| ndhD | 72R | 1.000 ** | rpoA | 271Y | 0.958 * |
| ndhH | 269M | 0.971 * | 326I | 0.993 ** | |
| ndhJ | 93G | 0.965 * | 328K | 0.964 * | |
| ndhK | 173N | 0.967 * | 329H | 0.951 * | |
| petB | 2S | 1.000 ** | ycf1 | 615K | 0.965 * |
| psaA | 148G | 0.988 * | 1460I | 0.997 ** | |
| 209G | 0.989 * | 1768I | 0.969 * | ||
| psbA | 155T | 0.998 ** | ycf2 | 933L | 0.983 * |
| psbB | 494T | 1.000 * | 1997A | 0.998 ** | |
| psbC | 280A | 0.985 | 1999V | 0.996 ** | |
| 427A | 0.999 ** | 2001S | 0.994 ** | ||
| rbcL | 91A | 0.956 * | 2006E | 0.982 * | |
| 225I | 1.000 ** | 2007M | 0.955 * | ||
| 249D | 0.974 * | 2009I | 0.981 * | ||
| 255V | 0.975 * | 2010G | 0.984 * | ||
| 279T | 0.989 * | 2011F | 0.971 * | ||
| 309M | 0.977 * | 2012M | 0.967 * | ||
| 340E | 0.973 * | ycf4 | 141I | 0.978 * | |
| 365T | 0.959 * | ||||
| 475L | 1.000 * | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shen, W.; Dong, Z.; Zhao, W.; Ma, L.; Wang, F.; Li, W.; Xin, P. Complete Chloroplast Genome Sequence of Rosa lucieae and Its Characteristics. Horticulturae 2022, 8, 788. https://doi.org/10.3390/horticulturae8090788
Shen W, Dong Z, Zhao W, Ma L, Wang F, Li W, Xin P. Complete Chloroplast Genome Sequence of Rosa lucieae and Its Characteristics. Horticulturae. 2022; 8(9):788. https://doi.org/10.3390/horticulturae8090788
Chicago/Turabian StyleShen, Weixiang, Zhanghong Dong, Wenzhi Zhao, Luyao Ma, Fei Wang, Weiying Li, and Peiyao Xin. 2022. "Complete Chloroplast Genome Sequence of Rosa lucieae and Its Characteristics" Horticulturae 8, no. 9: 788. https://doi.org/10.3390/horticulturae8090788
APA StyleShen, W., Dong, Z., Zhao, W., Ma, L., Wang, F., Li, W., & Xin, P. (2022). Complete Chloroplast Genome Sequence of Rosa lucieae and Its Characteristics. Horticulturae, 8(9), 788. https://doi.org/10.3390/horticulturae8090788