
Transcriptomic and Chemical Analyses Reveal the Hub Regulators of Flower Color Variation from Camellia japonica Bud Sport


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials and Chroma Measurements
2.2. Extraction and Quantitative and Qualitative Analyses of the Anthocyanins
2.3. Library Construction for Next-Generation Sequencing (NGS) and RNA Extraction
2.4. Functional Annotation and Identification of the Open Reading Frames
2.5. Identification of DEGs and the Weighted Gene Co-Expression Network Analysis (WGCNA)
2.6. Cloning and Cis-Element Analysis of the Promoter
2.7. qRT-PCR Analysis
2.8. Statistical Analysis
3. Results
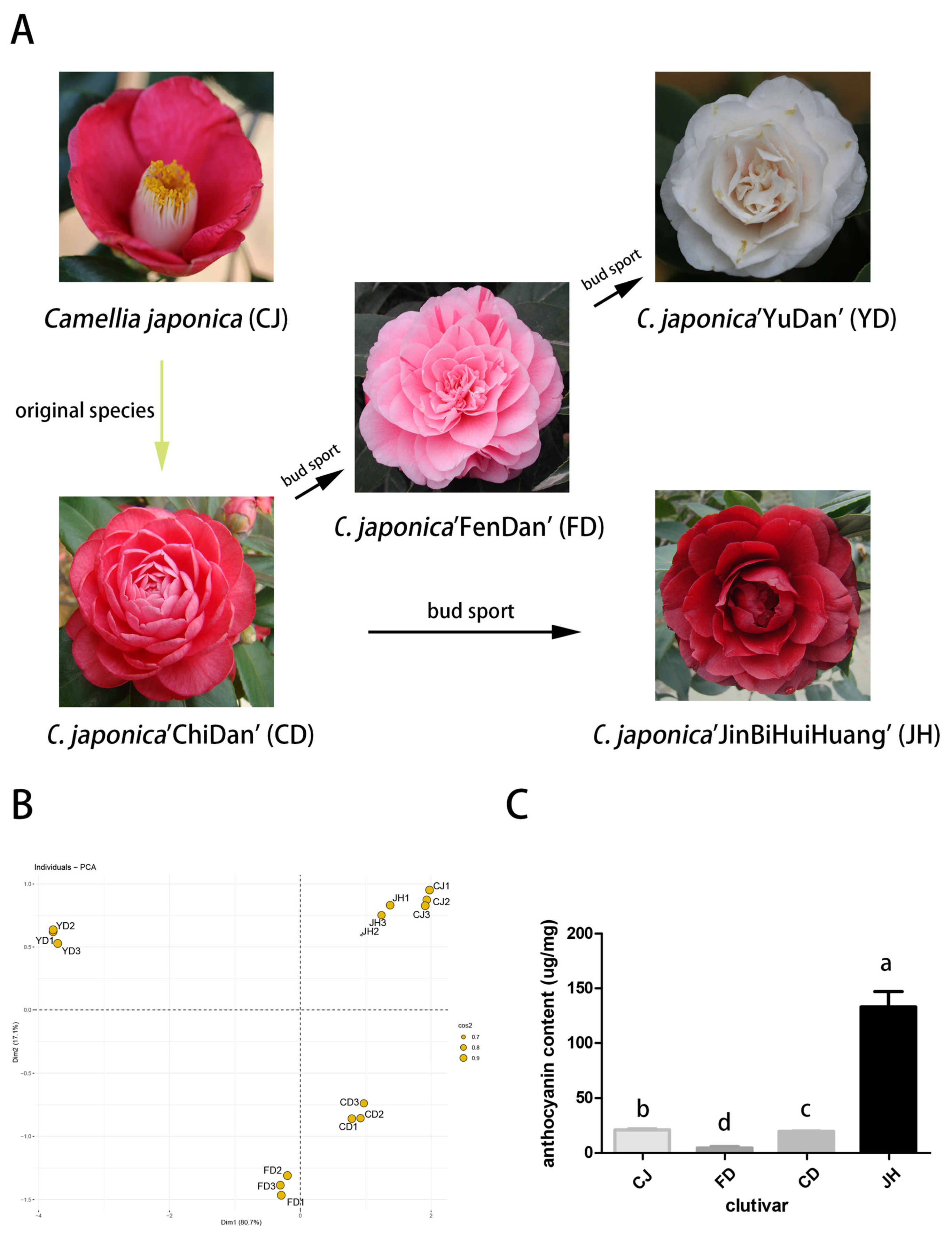
3.1. The Chroma Values of the Different Petals
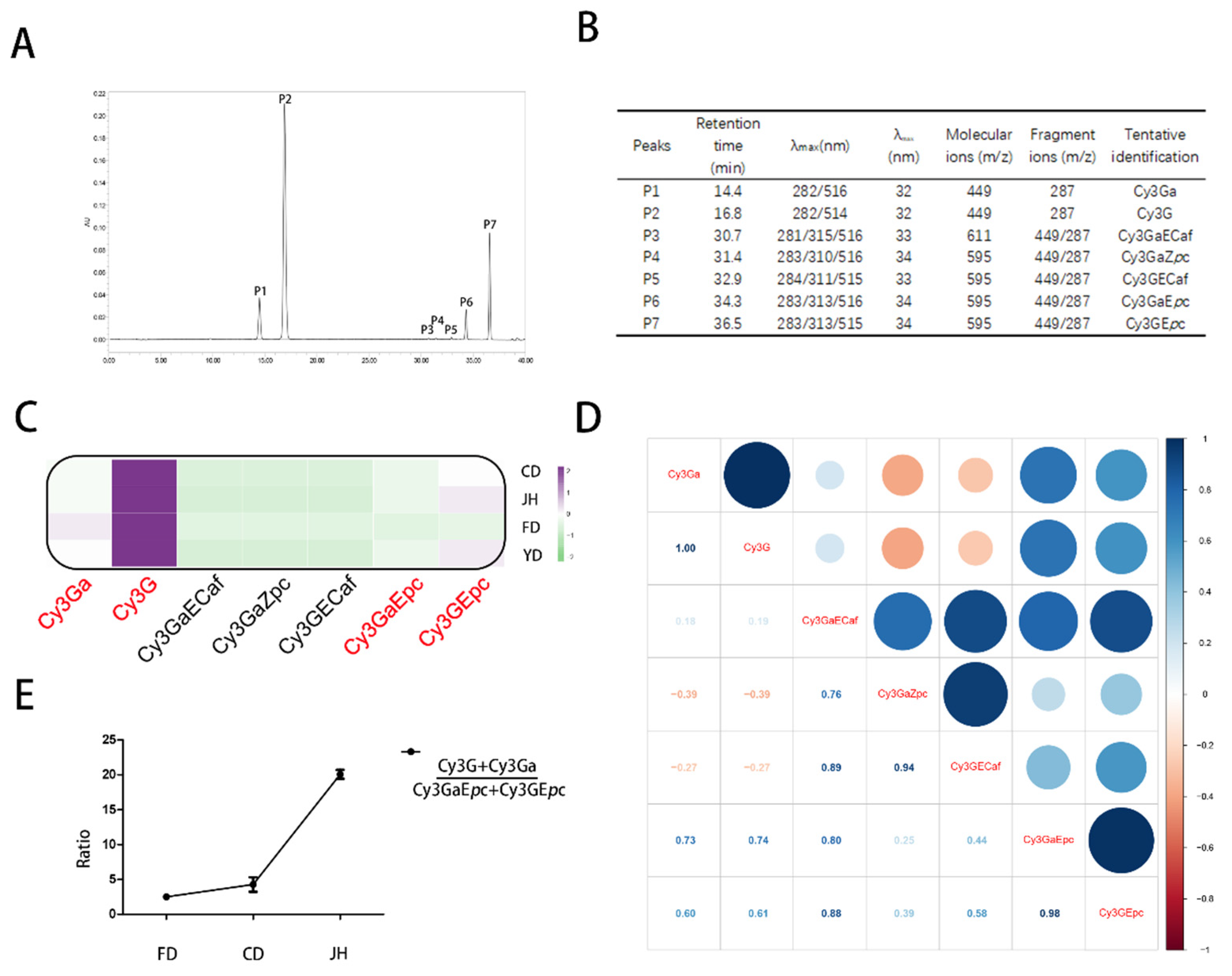
3.2. Qualitative and Quantitative Analyses of the Anthocyanins
3.3. Overview of the Transcriptome Data
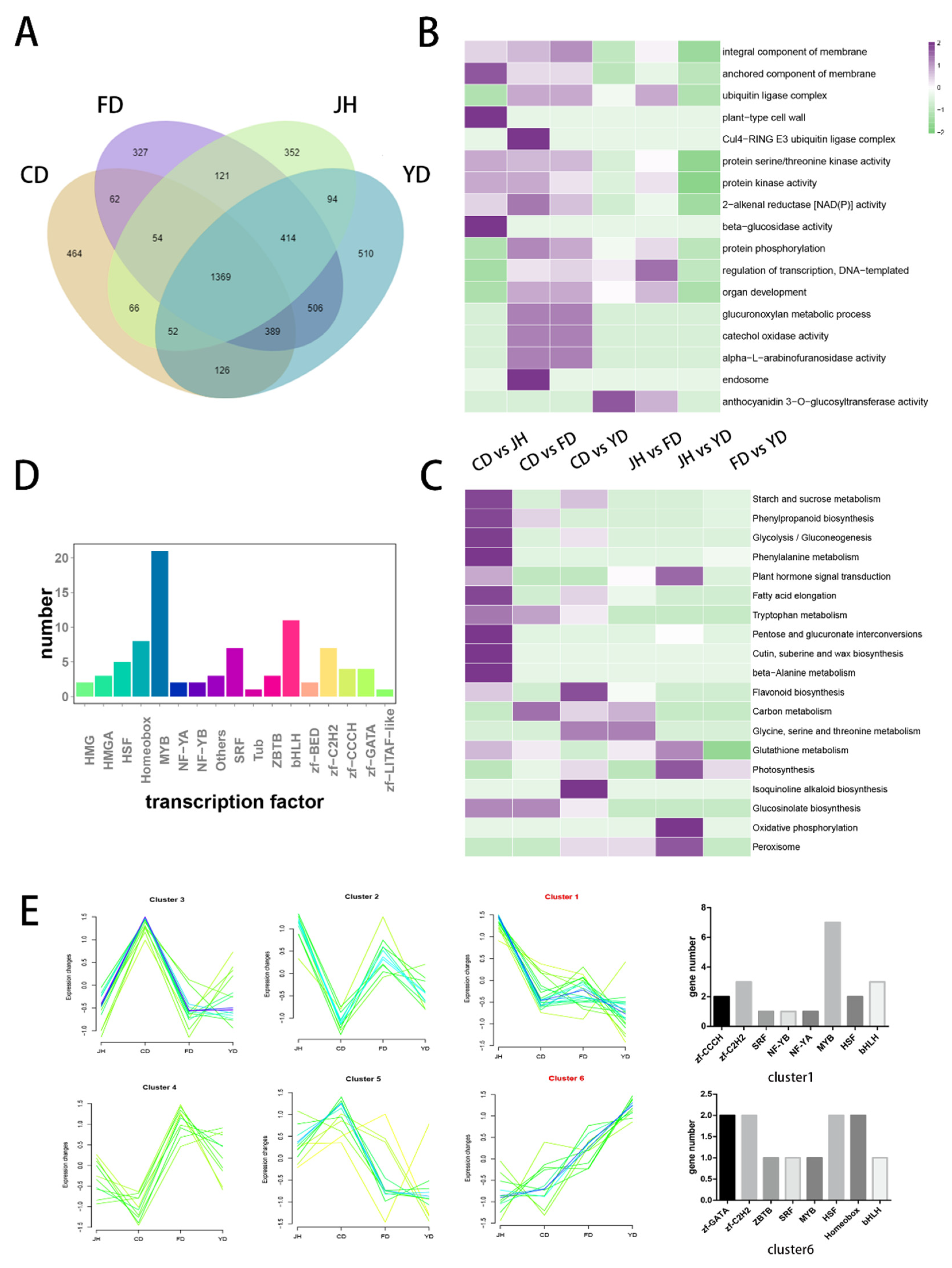
3.4. Identification of DEGs among Camellias of Different Flower Colors
3.5. Identification and Cluster Analysis of the Transcription Factors
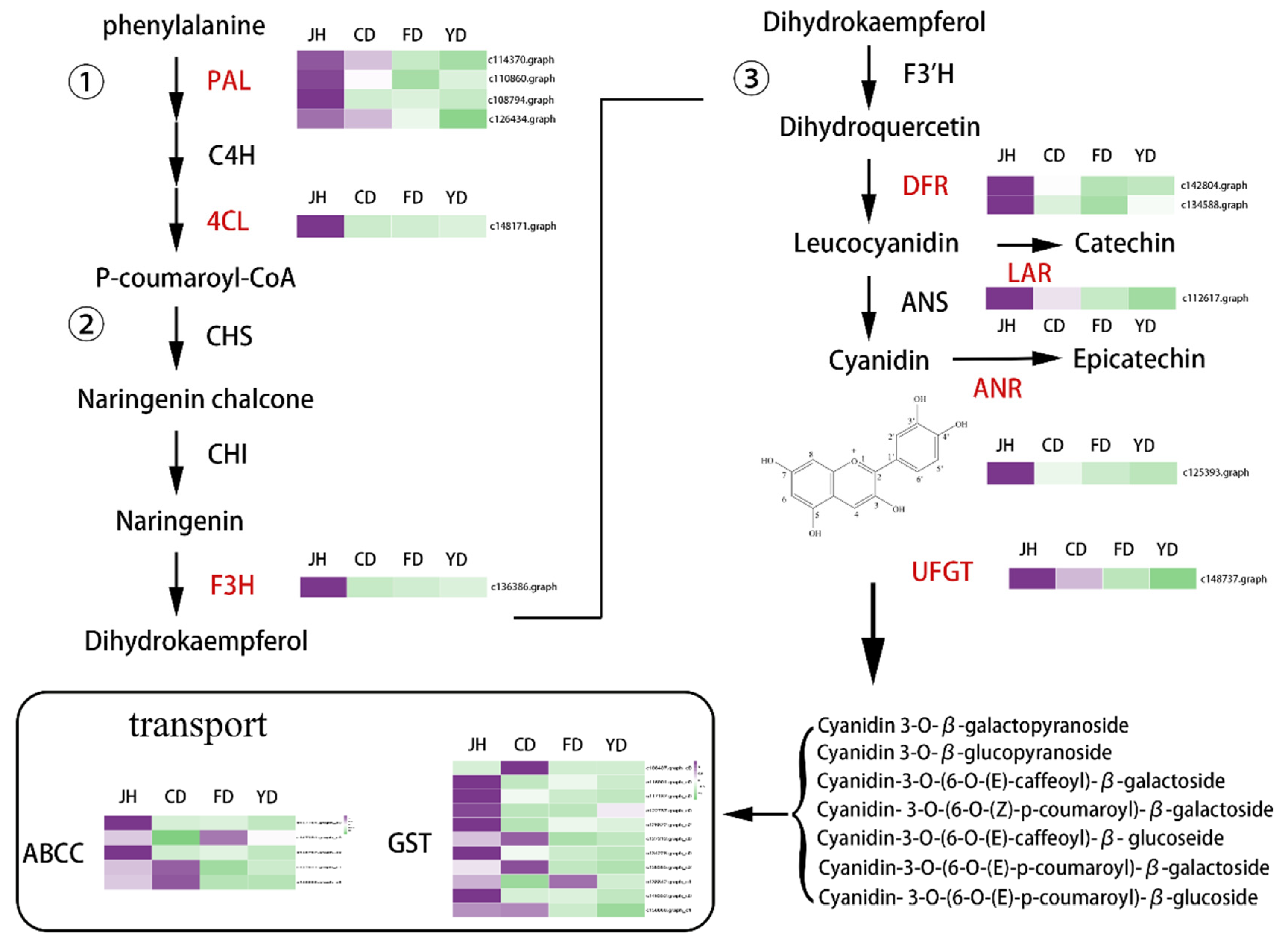
3.6. Identification of Candidate Genes Involved in Anthocyanin Biosynthesis
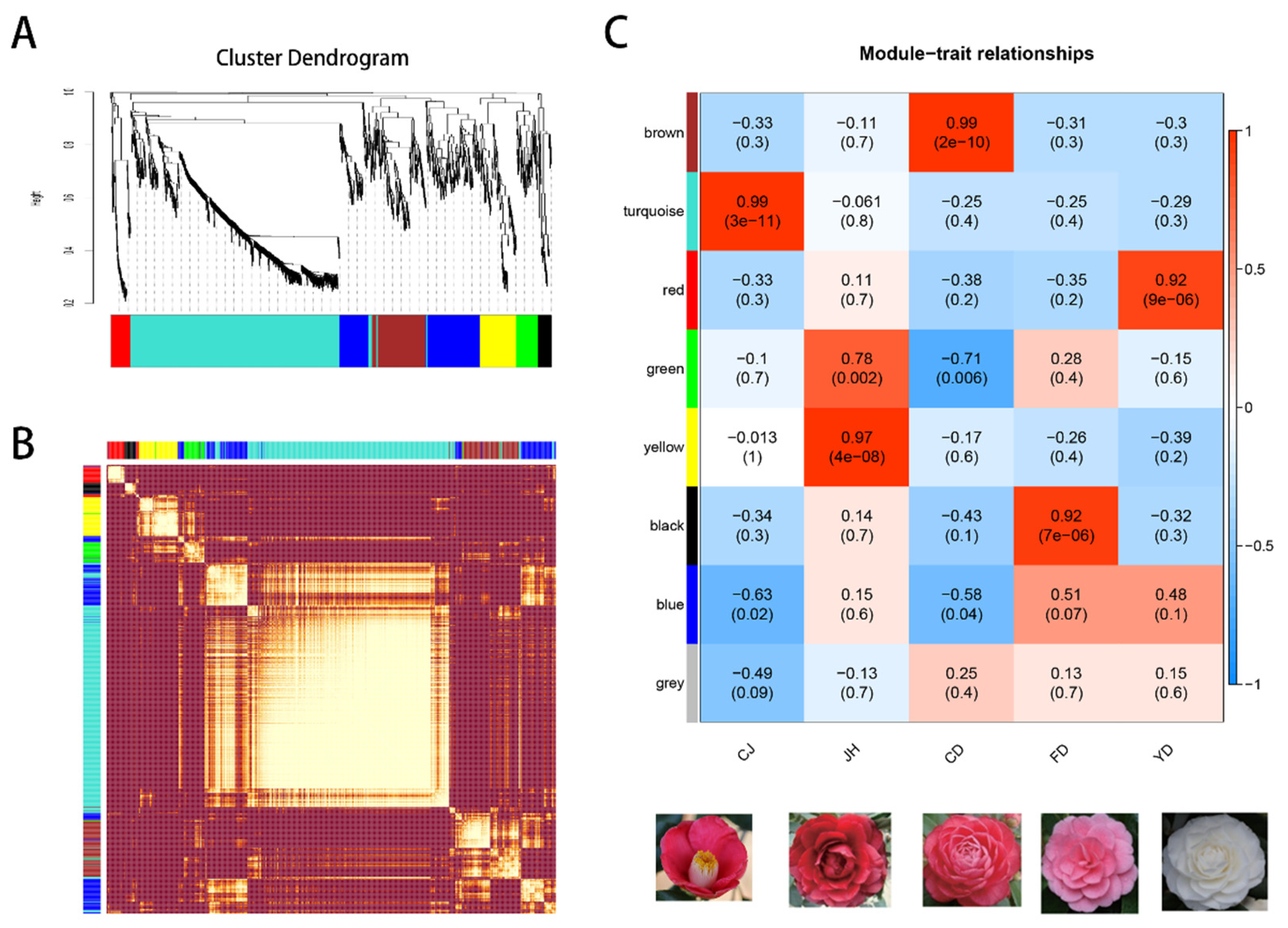
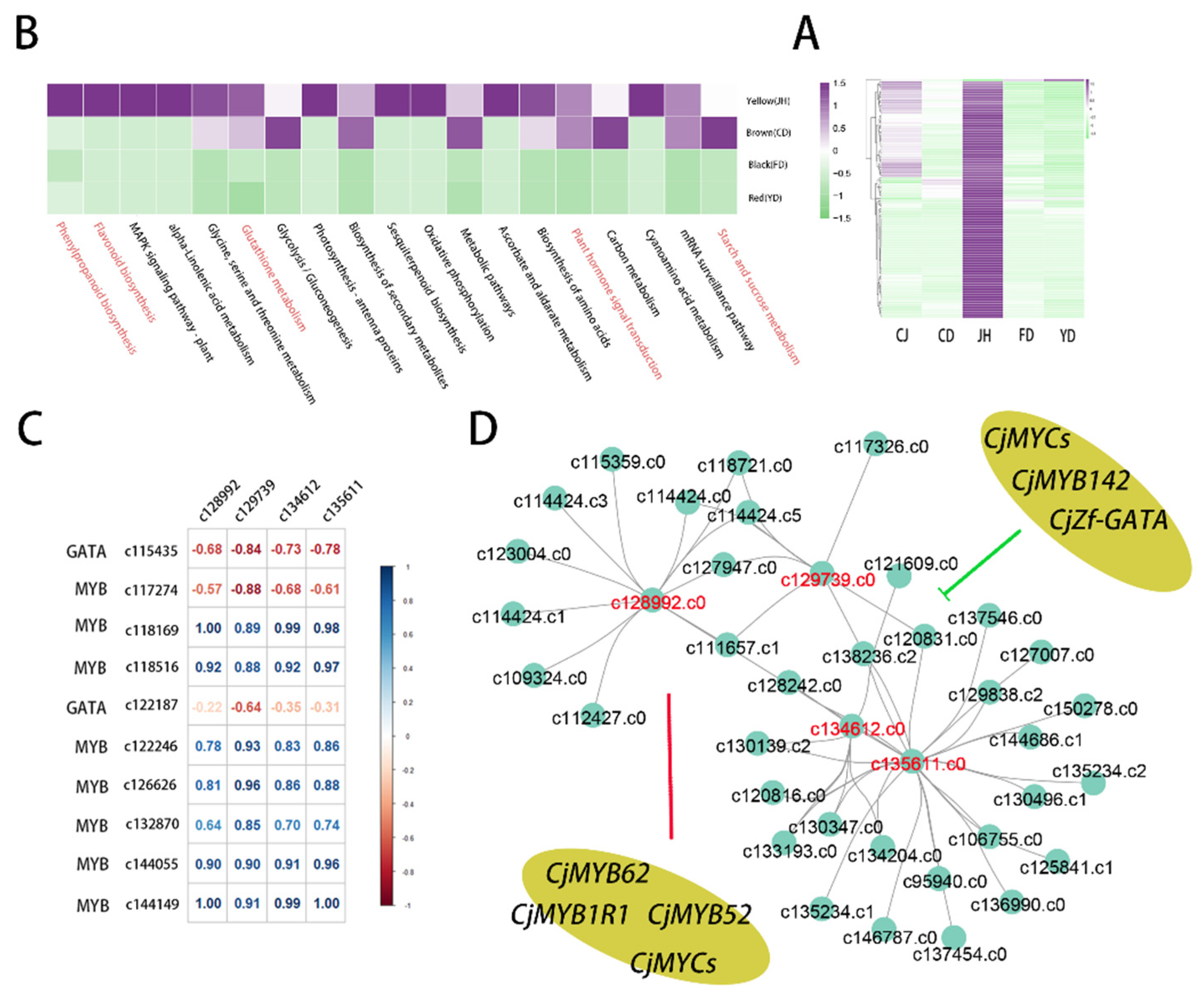
3.7. The Co-Expression Modules Related to High-Anthocyanin Accumulation in Petals
3.8. Quantitative Real-Time PCR Verification of the Transcriptome Data
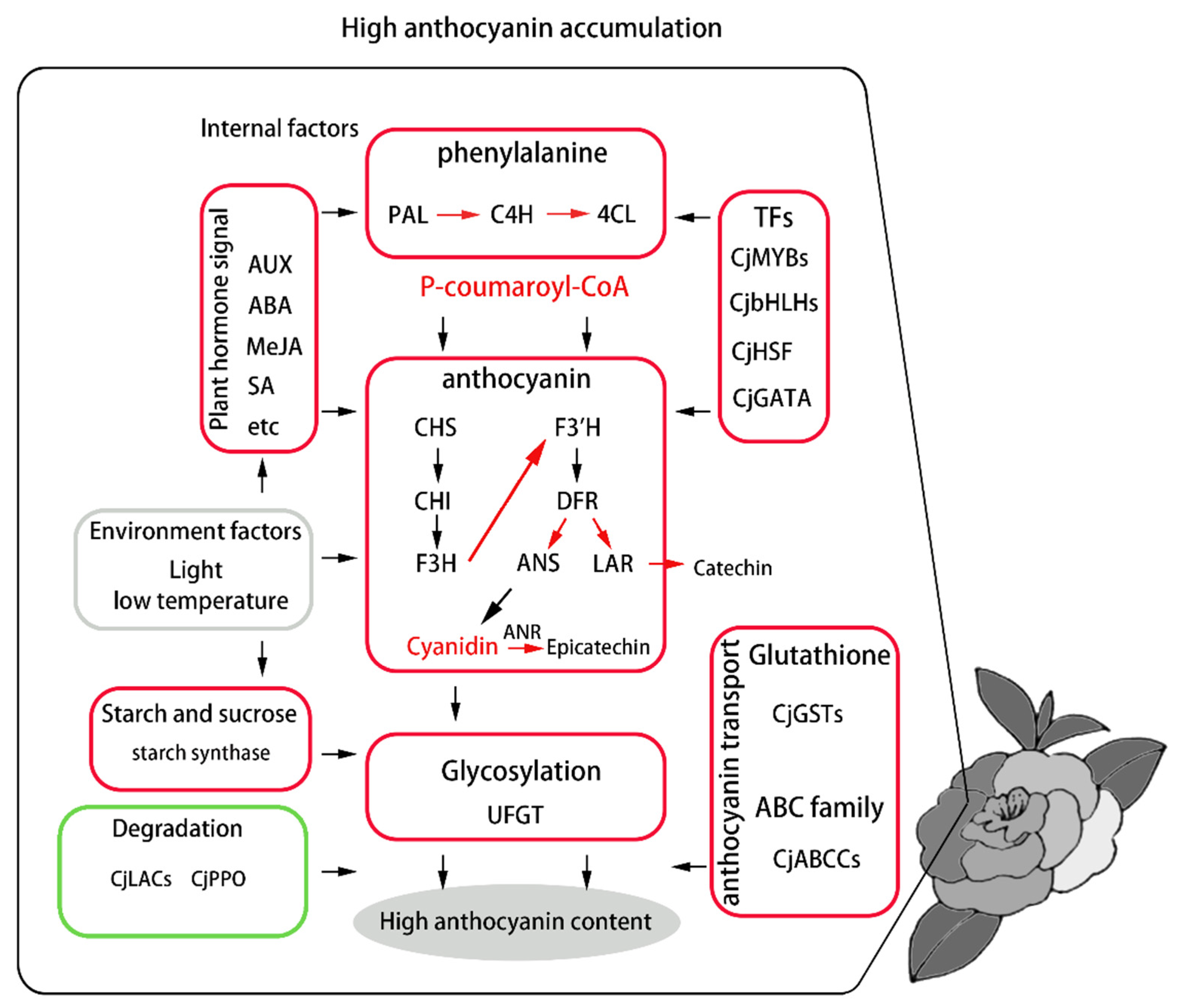
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Farcuh, M.; Toubiana, D.; Sade, N.; Rivero, R.M.; Doron-Faigenboim, A.; Nambara, E.; Sadka, A.; Blumwald, E. Hormone balance in a climacteric plum fruit and its non-climacteric bud mutant during ripening. Plant Sci. 2019, 280, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.F.; Pu, Y.; Liu, Y.T.; Li, Y.J.; Qu, J.P.; Huang, H.; Dai, S.L. Comparative transcriptomics and weighted gene co-expression correlation network analysis (WGCNA) reveal potential regulation mechanism of carotenoid accumulation in Chrysanthemum × morifolium. Plant Physiol. Biochem. 2019, 142, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, H.; Narumi-Kawasaki, T.; Takamura, T.; Fukai, S. Flower color mutation caused by spontaneous cell layer displacement in carnation (Dianthus caryophyllus). Plant Sci. 2020, 299, 110598. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Mao, Y.; Liu, H.; Yu, F.; Li, S.; Yin, T. Transcriptome analysis of differentially expressed genes relevant to variegation in peach flowers. PLoS ONE 2014, 9, e90842. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhao, X.; Zhao, S.; Han, N. A novel bud sport from the ‘Benitaka’ table grape cultivar (Vitis vinifera L.) improves sugar and anthocyanin accumulation at the berry ripening stage. S. Afr. J. Bot. 2015, 97, 111–116. [Google Scholar] [CrossRef]
- Qian, M.; Sun, Y.; Allan, A.C.; Teng, Y.; Zhang, D. The red sport of ‘Zaosu’ pear and its red-striped pigmentation pattern are associated with demethylation of the PyMYB10 promoter. Phytochemistry 2014, 107, 16–23. [Google Scholar] [CrossRef]
- Wessinger, C.A.; Hileman, L.C.; Rausher, M.D. Identification of major quantitative trait loci underlying floral pollination syndrome divergence in Penstemon. Philos. Trans. R. Soc. B Biol. Sci. 2014, 369, 20130349. [Google Scholar] [CrossRef]
- Zhao, D.Q.; Wei, M.R.; Liu, D.; Tao, J. Anatomical and biochemical analysis reveal the role of anthocyanins in flower coloration of herbaceous peony. Plant Physiol. Biochem. 2016, 102, 97–106. [Google Scholar] [CrossRef]
- Li, X.; Wang, J.; Yin, H.; Fan, Z.; Li, J. Variation of flower colors and their relationships with anthocyanins in cultivars of camellia japonica. J. Ecol. Rural. Environ. 2019, 35, 1307–1313. [Google Scholar]
- Xie, R.; Zheng, L.; He, S.; Zheng, Y.; Deng, L. Anthocyanin biosynthesis in fruit tree crops: Genes and their regulation. Afr. J. Biotechnol. 2014, 10, 19890–19897. [Google Scholar] [CrossRef]
- Rodrigues, J.A.; Espley, R.V.; Allan, A.C. Genomic analysis uncovers functional variation in the C-terminus of anthocyanin-activating MYB transcription factors. Hortic. Res. 2021, 8, 77. [Google Scholar] [CrossRef] [PubMed]
- Li, J.B.; Hashimoto, F.; Shimizu, K.; Sakata, Y. A new acylated anthocyan inform the red flowers of Camellia hongkongensis and characterization of anthocyanins in the Section Camellia species. J. Plant Ecol. 2009, 51, 545–552. [Google Scholar] [CrossRef]
- Li, J.B.; Hashimoto, F.; Shimizu, K.; Sakata, Y. Anthocyanins from the red flowers of Camellia saluenensis Stapf ex Bean. J. Jpn. Soc. Hortic. Sci. 2008, 77, 75–79. [Google Scholar] [CrossRef][Green Version]
- Wang, L.S.; Shiraishi, A.; Hashimoto, F.; Aoki, N.; Shimizu, K.; Sakata, Y. Analysis of petal anthocyanins to investigate flower coloration of Zhongyuan (Chinese) and Daikon Island (Japanese) tree peony cultivars. J. Plant Res. 2001, 114, 33–43. [Google Scholar] [CrossRef]
- Huang, B.; Rong, H.; Ye, Y.; Ni, Z.; Xu, M.; Zhang, W.; Xu, L.A. Transcriptomic analysis of flower color variation in the ornamental crabapple (Malus spp.) half-sib family through Illumina and PacBio Sequel sequencing. Plant Physiol. Biochem. 2020, 149, 27–35. [Google Scholar] [CrossRef]
- Deng, Y.; Li, J.; Wu, S.; Zhu, Y.; Chen, Y.; He, F. Integrated NR Database in Protein Annotation System and Its Localization. Comput. Eng. 2006, 32, 71–74. [Google Scholar]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J.; et al. Pfam: The protein families database. Nucleic Acids Res. 2014, 42, D222-30. [Google Scholar] [CrossRef]
- Koonin, E.V.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Krylov, D.M.; Makarova, K.S.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N.; Rao, B.S.; et al. A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes. Genome Biol. 2014, 5, R7. [Google Scholar] [CrossRef] [PubMed]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A.; Koonin, E.V. The COG database: A tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef]
- Apweiler, R.; Bairoch, A.; Wu, C.H.; Barker, W.C.; Boeckmann, B.; Ferro, S.; Gasteiger, E.; Huang, H.; Lopez, R.; Magrane, M.; et al. UniProt: The Universal Protein knowledgebase. Nucleic Acids Res. 2004, 32, D115–D119. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277-80. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Jiao, C.; Sun, H.; Rosli, H.G.; Pombo, M.A.; Zhang, P.; Banf, M.; Dai, X.; Martin, G.B.; Giovannoni, J.J.; et al. iTAK: A Program for Genome-wide Prediction and Classification of Plant Transcription Factors, Transcriptional Regulators, and Protein Kinases. Mol. Plant 2016, 9, 1667–1670. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef]
- Doyle, J.J.; Doyle, J.L. A rapid DNA isolation procedure from small quantities of fresh leaf tissues. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar]
- Lescot, M.; Déhais, P.; Thijs, G.; Marchal, K.; Moreau, Y.; Van de Peer, Y.; Rouzé, P.; Rombauts, S. Plant CARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002, 30, 325–327. [Google Scholar] [CrossRef]
- Lopes-da-Silva, F.; Escribano-Bailón, M.T.; Pérez-Alonso, J.J.; Perez-Alonso, J.J.; Santos-Buelga, C. Anthocyanin pigments in strawberry. LWT Food Sci. Technol. 2007, 40, 374–382. [Google Scholar] [CrossRef]
- Harborne, J.B. Spectral methods of characterizing anthocyanins. Biochem. J. 1958, 70, 22–28. [Google Scholar] [CrossRef]
- Fan, M.; Yang, K.; Zhou, R.; Liu, Q.; Guo, X.; Sun, Y. Temporal transcriptome profiling reveals candidate genes involved in cold acclimation of Camellia japonica (Naidong). Plant Physiol. Biochem. 2021, 167, 795–805. [Google Scholar] [CrossRef]
- Li, J.B.; Hashimoto, F.; Shimizu, K.; Sakata, Y. Anthocyanins from red flowers of Camellia cultivar ‘Dalicha’. Phytochemistry 2008, 69, 3166–3171. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Feng, Y.; Yu, S.; Fan, Z.; Li, X.; Li, J.; Yin, H. The Flavonoid Biosynthesis Network in Plants. Int. J. Mol. Sci. 2021, 22, 12824. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zhang, L.Y.; Wang, S.L.; Niu, X.Y. Analysis of anthocyanins and flavonols in petals of 10 Rhododendron species from the Sygera Mountains in Southeast Tibet. Plant Physiol. Biochem. 2016, 104, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Chen, M.; Liu, Y.; Ding, H.; Du, H.; Sun, Z.; Zhu, J. Metabolome and Transcriptome Analyses Reveal Different Flavonoid Biosynthesis and Chlorophyll Metabolism Profiles between Red Leaf and Green Leaf of Eucommia ulmoides. Forests 2021, 12, 1260. [Google Scholar] [CrossRef]
- Li, Q.; Kou, M.; Li, C.; Zhang, Y.G. Comparative transcriptome analysis reveals candidate genes involved in anthocyanin biosynthesis in sweetpotato (Ipomoea batatas L.). Plant Physiol. Biochem. 2021, 158, 508–517. [Google Scholar] [CrossRef] [PubMed]
- Boss, P.K.; Davies, C.; Robinson, S.P. Expression of anthocyanin biosynthesis pathway genes in red and white grapes. Plant Mol. Biol. 1996, 32, 565–569. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Yuan, H.H.; Sun, X.B. Preliminary studies on the changes of flower color during the flowering period in two tree peony cultivars. Acta Hortic. Sin. 2015, 42, 930–938. [Google Scholar] [CrossRef]
- Byamukama, R.; Jordheim, M.; Kiremire, B.; Namukobe, J.; Andersen, Ø.M. Anthocyanins from flowers of Hippeastrum cultivars. Sci. Hortic. 2006, 109, 262–266. [Google Scholar] [CrossRef]
- Brugliera, F.; Tao, G.Q.; Tems, U.; Kalc, G.; Mouradova, E.; Price, K.; Stevenson, K.; Nakamura, N.; Stacey, I.; Katsumoto, Y.; et al. Violet/Blue Chrysanthemums—Metabolic Engineering of the Anthocyanin Biosynthetic Pathway Results in Novel Petal Colors. Plant and Cell Physiology. 2013, 54, 1696–1710. [Google Scholar] [CrossRef]
- Khoo, H.E.; Azlan, A.; Tang, S.T.; Lim, S.M. Anthocyanidins and anthocyanins: Colored pigments as food, pharmaceutical ingredients, and the potential health benefits. Food Nutr. Res. 2017, 61, 1361779. [Google Scholar] [CrossRef]
- Oh, Y.S.; Lee, J.H.; Yoon, S.H.; Oh, C.H.; Choi, D.S.; Choe, E.; Jung, M.Y. Characterization and quantification of anthocyanins in grape juices obtained from the grape cultivated in Korea. J. Food Sci. 2008, 73, 378–389. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Wei, G.; Zhou, H.; Gu, C.; Vimolmangkang, S.; Liao, L.; Han, Y. Unraveling the mechanism underlying the glycosylation and methylation of anthocyanins in peach. Plant Physiol. 2014, 166, 1044–1058. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhou, L.J.; Wang, Y.; Liu, S.; Geng, Z.; Song, A.; Jiang, J.; Chen, S.; Chen, F. Functional identification of a flavone synthase and a flavonol synthase genes affecting flower color formation in Chrysanthemum morifolium. Plant Physiol. Biochem. 2021, 166, 1109–1120. [Google Scholar] [CrossRef] [PubMed]
- Oren-Shamir, M. Does anthocyanin degradation play a significant role in determining pigment concentration in plants? Plant Sci. 2009, 177, 310–316. [Google Scholar] [CrossRef]
- Han, M.L.; Yin, J.; Zhao, Y.H.; Sun, X.W.; Meng, J.X.; Zhou, J.; Shen, T.; Li, H.H.; Zhang, F. How the Color Fades from Malus halliana Flowers: Transcriptome Sequencing and DNA Methylation Analysis. Front. Plant Sci. 2020, 11, 576054. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.P.; Wang, Y.J.; Teixeira da Silva, J.A.; Fan, Y.M.; Yu, X.N. Transcriptome and chemical analysis reveal putative genes involved in flower color change in Paeonia ‘Coral Sunset’. Plant Physiol. Biochem. 2019, 138, 130–139. [Google Scholar] [CrossRef]
- Zou, H.; Ma, Y.; Liao, X.; Wang, Y. Effects of high-pressure processing on the copigmentation reaction of pelargonidin-3-glucoside and catechin. LWT 2019, 108, 240–246. [Google Scholar] [CrossRef]
- Grzesik, M.; Naparło, K.; Bartosz, G.; Sadowska-Bartosz, I. Antioxidant properties of catechins: Comparison with other antioxidants. Food Chem. 2018, 241, 480–492. [Google Scholar] [CrossRef]
- Mueller, L.A.; Goodman, C.D.; Silady, R.A.; Walbot, V. AN9, a petunia glutathione S-transferase required for anthocyanin sequestration, is a flavonoid-binding protein. Plant Physiol. 2000, 123, 1561–1570. [Google Scholar] [CrossRef]
- Zipor, G.; Duarte, P.; Carqueijeiro, I.M.; Shahar, L.; Ovadia, R.; Teper-bamnolker, P.; Eshel, D.; Levin, Y.; Faigenboim, A.; Sottomayor, M.; et al. In planta anthocyanin degradation by a vacuolar class III peroxidase in Brunfelsia calycina flowers. New Phytol. 2015, 205, 653–665. [Google Scholar] [CrossRef]
- Fang, F.; Zhang, X.; Luo, H.; Zhou, J.; Gong, Y.; Li, W.; Shi, Z.; He, Q.; Wu, Q.; Li, L.; et al. Anintracellular laccase is responsible for the epicatechin mediated anthocyanin degradation in litchi fruit pericarp. Plant Physiol. 2015, 169, 2391–2408. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.H.; Zhang, R.; Wang, N.; Yang, L.; Chen, X. Transcriptome profiling reveals auxin suppressed anthocyanin biosynthesis in red-fleshed apple callus (Malus sieversii f. niedzwetzkyana). Plant Cell Tissue Organ Cult. 2015, 123, 389–404. [Google Scholar] [CrossRef]
- Katja, K.; Pinja, T.; Hely, H.; Laura, J. Abscisic Acid Regulates Anthocyanin Biosynthesis and Gene Expression Associated with Cell Wall Modification in Ripening Bilberry (Vaccinium myrtillus L.) Fruits. Front. Plant Sci. 2018, 9, 1259. [Google Scholar] [CrossRef]
- Liu, Y.J.; Li, M.; Li, T.; Chen, Y.J.; Zhang, L.J.; Zhao, G.F.; Zhuang, G.H.; Zhao, W.Y.; Gao, L.P.; Xia, T. Airborne fungus-induced biosynthesis of anthocyanins in Arabidopsis thaliana via jasmonic acid and salicylic acid signaling. Plant Sci. 2020, 300, 110635. [Google Scholar] [CrossRef]
- Xu, W.; Dubos, C.; Lepiniec, L. Transcriptional control of flavonoid biosynthesis by MYB-bHLH-WDR complexes. Trends Plant Sci. 2014, 20, 176–185. [Google Scholar] [CrossRef]
- Stracke, R.; Werber, M.; Weisshaar, B. The r2r3-myb gene family in Arabidopsis thaliana. Curr. Opin. Plant Biol. 2011, 4, 447–456. [Google Scholar] [CrossRef]
- Mehrtens, F.; Kranz, H.; Bednarek, P.; Weisshaar, B. The Arabidopsis transcription factor MYB12 is a flavonol-specific regulator of phenylpropanoid biosynthesis. Plant Physiol. 2005, 138, 1083–1096. [Google Scholar] [CrossRef]
- Espley, R.V.; Hellens, R.P.; Putterill, J.; Stevenson, D.E.; Kutty-Amma, S.; Allan, A.C. Red colouration in apple fruit is due to the activity of the MYB transcription factor, MdMYB10. Plant J. 2006, 49, 414–427. [Google Scholar] [CrossRef]
- Schaart, J.G.; Dubos, C.; Romero De La Fuente, I.; van Houwelingen, A.M.M.L.; de Vos, R.C.H.; Jonker, H.H.; Xu, W.; Routaboul, J.M.; Lepiniec, L.; Bovy, A.G. Identification and characterization of MYB-bHLH-WD40 regulatory complexes controlling proanthocyanidin biosynthesis in strawberry (Fragaria × ananassa) fruits. New Phytol. 2013, 197, 454–467. [Google Scholar] [CrossRef]
- Albert, N.W.; Butelli, E.; Moss, S.M.A.; Piazza, P.; Waite, C.N.; Schwinn, W.K.E.; Davies, K.M.; Martin, C. Discrete bHLH transcription factors play functionally overlapping roles in pigmentation patterning in flowers of Antirrhinum majus. New Phytol. 2020, 231, 849–863. [Google Scholar] [CrossRef]
- Jing, N.; Song, J.; Liu, Z.; Wang, L.; Jiang, G. Glycosylation of anthocyanins enhances the apoptosis of colon cancer cells by handicapping energy metabolism. BMC Complement. Med. Ther. 2020, 20, 312. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Li, J.; Shang, H.; Meng, X. Effect of methyl jasmonate on the anthocyanin content and antioxidant activityof blueberries during cold storage. J. Sci. Food Agric. 2015, 95, 337–343. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fan, M.; Zhang, Y.; Yang, M.; Wu, S.; Yin, H.; Li, J.; Li, X. Transcriptomic and Chemical Analyses Reveal the Hub Regulators of Flower Color Variation from Camellia japonica Bud Sport. Horticulturae 2022, 8, 129. https://doi.org/10.3390/horticulturae8020129
Fan M, Zhang Y, Yang M, Wu S, Yin H, Li J, Li X. Transcriptomic and Chemical Analyses Reveal the Hub Regulators of Flower Color Variation from Camellia japonica Bud Sport. Horticulturae. 2022; 8(2):129. https://doi.org/10.3390/horticulturae8020129
Chicago/Turabian StyleFan, Menglong, Ying Zhang, Meiying Yang, Si Wu, Hengfu Yin, Jiyuan Li, and Xinlei Li. 2022. "Transcriptomic and Chemical Analyses Reveal the Hub Regulators of Flower Color Variation from Camellia japonica Bud Sport" Horticulturae 8, no. 2: 129. https://doi.org/10.3390/horticulturae8020129
APA StyleFan, M., Zhang, Y., Yang, M., Wu, S., Yin, H., Li, J., & Li, X. (2022). Transcriptomic and Chemical Analyses Reveal the Hub Regulators of Flower Color Variation from Camellia japonica Bud Sport. Horticulturae, 8(2), 129. https://doi.org/10.3390/horticulturae8020129