
Construction of a High-Density Genetic Map for Pitaya Using the Whole Genome Resequencing Approach

 and
and
Abstract
:1. Introduction
2. Materials and Methods
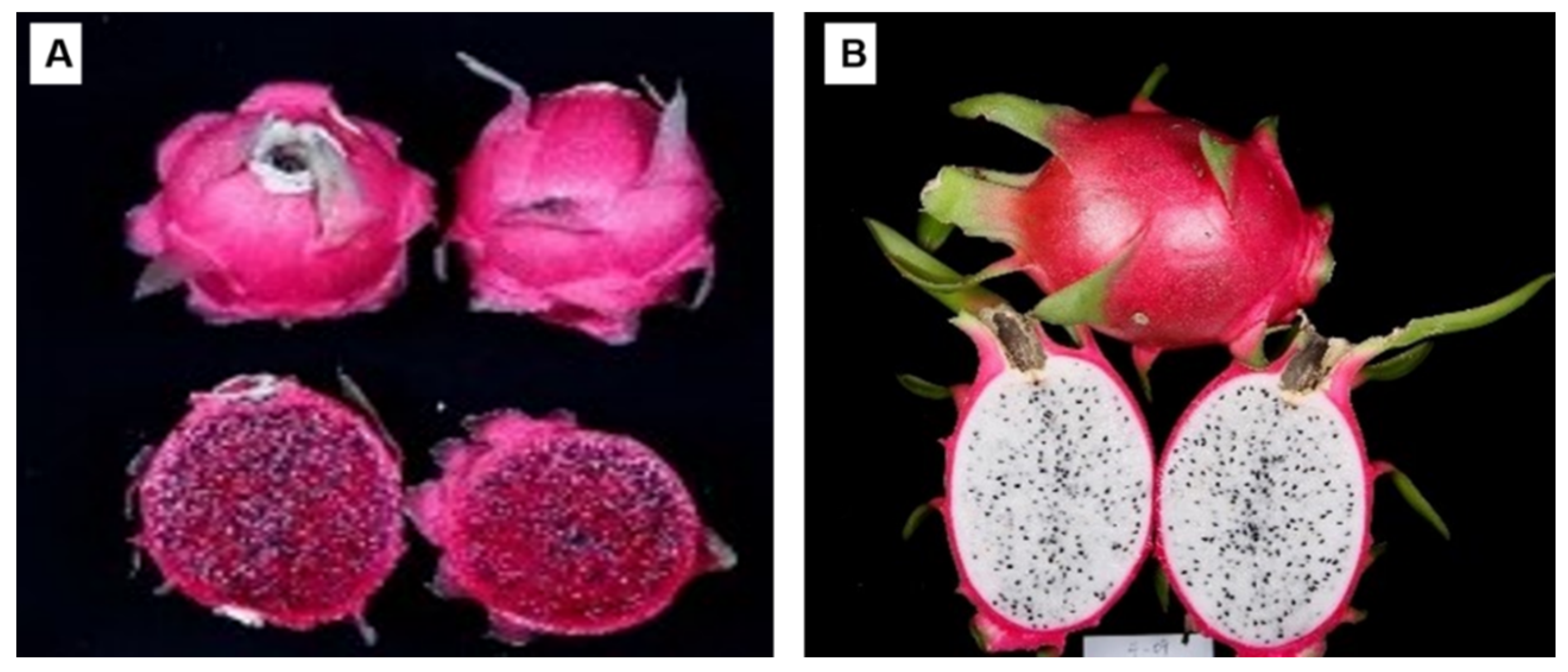
2.1. Materials
2.2. Genomic DNA Extraction and Genotyping of Population by Whole Genome Resequencing
2.3. SNP Calling and Filtering
2.4. Analysis of Linkage and Construction of Genetic Map
3. Results
3.1. Genotyping by Whole Genome Resequencing and Sequencing Quality Assessment
3.2. SNP Markers Detection and Evaluation
3.3. Genetic Linkage Analysis and Construction of Maps
3.4. Consensus Map and Collinearity Analysis of Genetic and Physical Map
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fan, Q.; Yan, F.; Qiao, G.; Zhang, B.; Wen, X. Identification of differentially-expressed genes potentially implicated in drought response in pitaya (Hylocereus undatus) by suppression subtractive hybridization and cDNA microarray analysis. Gene 2014, 533, 322–331. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.J.; An, D.; Nguyen, C.T.; Patil, B.S.; Kim, J.; Yoo, K.S. Betalain and betaine composition of greenhouse-or field-produced beetroot (Beta vulgaris L.) and inhibition of HepG2 cell proliferation. J. Agric. Food Chem. 2014, 62, 1324–1331. [Google Scholar] [CrossRef] [PubMed]
- Suh, D.H.; Lee, S.; Heo, D.Y.; Kim, Y.-S.; Cho, S.K.; Lee, S.; Lee, C.H. Metabolite profiling of red and white pitayas (Hylocereus polyrhizus and Hylocereus undatus) for comparing betalain biosynthesis and antioxidant activity. J. Agric. Food Chem. 2014, 62, 8764–8771. [Google Scholar] [CrossRef]
- da Silva, D.V.T.; dos Santos Baião, D.; de Oliveira Silva, F.; Alves, G.; Perrone, D.; Del Aguila, E.M.; Paschoalin, V.M.F. Betanin, a natural food additive: Stability, bioavailability, antioxidant and preservative ability assessments. Molecules 2019, 24, 458. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Xie, F.; Cui, Y.; Chen, C.; Lu, W.; Hu, X.; Hua, Q.; Zhao, J.; Wu, Z.; Gao, D.; et al. A chromosome-scale genome sequence of pitaya (Hylocereus undatus) provides novel insights into the genome evolution and regulation of betalain biosynthesis. Hortic. Res. 2021, 8, 164. [Google Scholar] [CrossRef]
- Zhuang, Y.; Zhang, Y.; Sun, L. Characteristics of fibre-rich powder and antioxidant activity of pitaya (Hylocereus undatus) peels. Int. J. Food Sci. Technol. 2012, 47, 1279–1285. [Google Scholar] [CrossRef]
- Cai, Y.; Xiang, Q.; Chen, J.; Peng, Y.; Wang, B. Analysis of nutritional components in pitaya fruit. Nonwood For. Res. 2008, 26, 253–256. [Google Scholar]
- Hua, Q.; Chen, C.; Chen, Z.; Chen, P.; Ma, Y.; Wu, J.; Zheng, J.; Hu, G.; Zhao, J.; Qin, Y. Transcriptomic analysis reveals key genes related to betalain biosynthesis in pulp coloration of Hylocereus polyrhizus. Front. Plant Sci. 2016, 6, 1179. [Google Scholar]
- Adnan, L.; Osman, A.; Abdul Hamid, A. Antioxidant activity of different extracts of red pitaya (Hylocereus polyrhizus) seed. Int. J. Food Prop. 2011, 14, 1171–1181. [Google Scholar] [CrossRef] [Green Version]
- Taguiam, J.D.; Evallo, E.; Bengoa, J.; Maghirang, R.; Balendres, M.A. Pathogenicity of Epicoccum sorghinum towards dragon fruits (Hylocereus species) and in vitro evaluation of chemicals with antifungal activity. J. Phytopathol. 2020, 168, 303–310. [Google Scholar] [CrossRef]
- Stintzing, F.C.; Schieber, A.; Carle, R. Identification of betalains from yellow beet (Beta vulgaris L.) and cactus pear [Opuntia ficus-indica (L.) Mill.] by high-performance liquid chromatography−electrospray ionization mass spectrometry. J. Agric. Food Chem. 2002, 50, 2302–2307. [Google Scholar] [CrossRef] [PubMed]
- Pang, X.; Li, X.; Liu, X.; Cai, L.; Li, B.; Li, X. Transcriptomic analysis reveals Cu/Zn SODs acting as hub genes of SODs in Hylocereus undatus induced by trypsin during storage. Antioxidants 2020, 9, 162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Xu, J.; He, Y.; Shi, M.; Han, X.; Li, W.; Zhang, X.; Wen, X. Metabolic profiling of pitaya (Hylocereus polyrhizus) during fruit development and maturation. Molecules 2019, 24, 1114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freitas, S.T.d.; Mitcham, E.J. Quality of pitaya fruit (Hylocereus undatus) as influenced by storage temperature and packaging. Sci. Agric. 2013, 70, 257–262. [Google Scholar] [CrossRef] [Green Version]
- Zheng, J.; Meinhardt, L.W.; Goenaga, R.; Zhang, D.; Yin, Y. The chromosome-level genome of dragon fruit reveals whole-genome duplication and chromosomal co-localization of betacyanin biosynthetic genes. Hortic. Res. 2021, 8, 63. [Google Scholar] [CrossRef] [PubMed]
- Cormier, F.; Lawac, F.; Maledon, E.; Gravillon, M.-C.; Nudol, E.; Mournet, P.; Vignes, H.; Arnau, G. A reference high-density genetic map of greater yam (Dioscorea alata L.). Theor. Appl. Genet. 2019, 132, 1733–1744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Rashid, M.A.R.; Li, X.; Yao, C.; Lu, L.; Bai, J.; Li, Y.; Xu, N.; Yang, Q.; Zhang, L. Collection and evaluation of genetic diversity and population structure of potato landraces and varieties in China. Front. Plant Sci. 2019, 10, 139. [Google Scholar] [CrossRef] [Green Version]
- Rashid, M.A.R.; Zhao, Y.; Zhang, H.; Li, J.; Li, Z. Nucleotide diversity, natural variation, and evolution of Flexible culm-1 and Strong culm-2 lodging resistance genes in rice. Genome 2016, 59, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Nashima, K.; Hosaka, F.; Shimajiri, Y.; Matsumura, M.; Tarora, K.; Urasaki, N.; Shoda, M.; Nishitani, C.; Sawamura, Y.; Yamamoto, T. SSR marker development and genetic identification of pitaya (Hylocereus spp.) collected in Okinawa Prefecture, Japan. Hortic. J. 2021, 90, 23–30. [Google Scholar] [CrossRef]
- Fan, Q.; Zheng, S.; Yan, F.; Zhang, B.; Qiao, G.; Wen, X. Efficient regeneration of dragon fruit (Hylocereus undatus) and an assessment of the genetic fidelity of in vitro-derived plants using ISSR markers. J. Hortic. Sci. Biotechnol. 2013, 88, 631–637. [Google Scholar] [CrossRef]
- Tao, J.; Qiao, G.; Wen, X.; Gao, G.; Liu, T.; Peng, Z.; Cai, Y.; Chen, N.; Yan, F.; Zhang, B. Characterization of genetic relationship of dragon fruit accessions (Hylocereus spp.) by morphological traits and ISSR markers. Sci. Hortic. 2014, 170, 82–88. [Google Scholar] [CrossRef]
- Rifat, T.; Khan, K.; Islam, M.S. Genetic diversity in dragon fruit (Hylocereus sp.) germplasms revealed by RAPD marker. J. Anim. Plant Sci. 2019, 29, 809–818. [Google Scholar]
- Tel-Zur, N.; Abbo, S.; Bar-Zvi, D.; Mizrahi, Y. Clone identification and genetic relationship among vine cacti from the genera Hylocereus and Selenicereus based on RAPD analysis. Sci. Hortic. 2004, 100, 279–289. [Google Scholar] [CrossRef]
- Poland, J.A.; Brown, P.J.; Sorrells, M.E.; Jannink, J.-L. Development of high-density genetic maps for barley and wheat using a novel two-enzyme genotyping-by-sequencing approach. PLoS ONE 2012, 7, e32253. [Google Scholar]
- Ward, J.A.; Bhangoo, J.; Fernández-Fernández, F.; Moore, P.; Swanson, J.; Viola, R.; Velasco, R.; Bassil, N.; Weber, C.A.; Sargent, D.J. Saturated linkage map construction in Rubus idaeus using genotyping by sequencing and genome-independent imputation. BMC Genom. 2013, 14, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herten, K.; Hestand, M.S.; Vermeesch, J.R.; Van Houdt, J.K. GBSX: A toolkit for experimental design and demultiplexing genotyping by sequencing experiments. BMC Bioinform. 2015, 16, 73. [Google Scholar] [CrossRef] [Green Version]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Garsmeur, O.; Droc, G.; Antonise, R.; Grimwood, J.; Potier, B.; Aitken, K.; Jenkins, J.; Martin, G.; Charron, C.; Hervouet, C. A mosaic monoploid reference sequence for the highly complex genome of sugarcane. Nat. Commun. 2018, 9, 2638. [Google Scholar] [CrossRef] [PubMed]
- Knaus, B.J.; Grünwald, N.J. VCFR: A package to manipulate and visualize variant call format data in R. Mol. Ecol. Resour. 2017, 17, 44–53. [Google Scholar] [CrossRef]
- RC Team. R: A Language and Environment for Statistical Computing; RC Team: Diepoldsau, Switzerland, 2013. [Google Scholar]
- van Ooijen, J.W. JoinMap® 4: Software for the Calculation of Genetic Linkage Maps in Experimental Populations; Scientific Reserch: Wagening, Switzerland, 2006; Volume 93, p. 7778Walling. [Google Scholar]
- Yu, Y.; Zhang, X.; Yuan, J.; Li, F.; Chen, X.; Zhao, Y.; Huang, L.; Zheng, H.; Xiang, J. Genome survey and high-density genetic map construction provide genomic and genetic resources for the Pacific White Shrimp Litopenaeus vannamei. Sci. Rep. 2015, 5, 15612. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Liu, D.; Zhang, X.; Li, W.; Liu, H.; Hong, W.; Jiang, C.; Guan, N.; Ma, C.; Zeng, H. SLAF-seq: An efficient method of large-scale de novo SNP discovery and genotyping using high-throughput sequencing. PLoS ONE 2013, 8, e58700. [Google Scholar]
- Campos-Ramos, R. Chromosome studies on the marine shrimps Penaeus Uannamei and P. Californiensis (Decapoda). J. Crustacean Biol. 1997, 17, 666–673. [Google Scholar] [CrossRef]
- Ren, Y.; Zhao, H.; Kou, Q.; Jiang, J.; Guo, S.; Zhang, H.; Hou, W.; Zou, X.; Sun, H.; Gong, G. A high resolution genetic map anchoring scaffolds of the sequenced watermelon genome. PLoS ONE 2012, 7, e29453. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Yang, C.; Zhang, Y.; Li, L.; Zhang, X.; Zhang, Q.; Xiang, J. A genetic linkage map of Pacific white shrimp (Litopenaeus vannamei): Sex-linked microsatellite markers and high recombination rates. Genetica 2007, 131, 37–49. [Google Scholar] [CrossRef]
- Du, Z.Q.; Ciobanu, D.; Onteru, S.; Gorbach, D.; Mileham, A.; Jaramillo, G.; Rothschild, M. A gene-based SNP linkage map for pacific white shrimp, Litopenaeus vannamei. Anim. Genet. 2010, 41, 286–294. [Google Scholar] [CrossRef]
- Pérez, F.; Erazo, C.; Zhinaula, M.; Volckaert, F.; Calderón, J. A sex-specific linkage map of the white shrimp Penaeus (Litopenaeus) vannamei based on AFLP markers. Aquaculture 2004, 242, 105–118. [Google Scholar] [CrossRef] [Green Version]
- Andriantahina, F.; Liu, X.; Huang, H. Correction: Genetic map construction and quantitative trait locus (QTL) detection of growth-related traits in Litopenaeus vannamei for selective breeding applications. PLoS ONE 2013, 8, e75206. [Google Scholar]
- Alcivar-Warren, A.; Meehan-Meola, D.; Park, S.W.; Xu, Z.; Delaney, M.; Zuniga, G. ShrimpMap: A low-density, microsatellite-based linkage map of the pacific whiteleg shrimp, Litopenaeus vannamei: Identification of sex-linked markers in linkage group 4. J. Shellfish Res. 2007, 26, 1259–1277. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Male Parent, H. undatus | Female Parent, H. monacanthus | F1 Progeny | |||
|---|---|---|---|---|---|
| Average | Maximum | Minimum | |||
| Raw bases (bp) | 28,362,977,400 | 29,982,174,300 | 7,853,867,311 | 11,374,635,600 | 6,902,555,400 |
| Clean bases (bp) | 28,167,050,700 | 29,898,491,400 | 7,827,082,255 | 11,338,035,600 | 6,879,546,300 |
| Clean reads | 187,780,338 | 199,323,276 | 52,180,548.36 | 75,586,904 | 45,863,642 |
| Mapped reads | 185,486,093 | 194,135,772 | 51,026,958.15 | 73,621,450 | 44,944,080 |
| Effective rate (%) | 99.31 | 99.72 | 100 | 99.86 | 99.04 |
| Error rate (%) | 0.03 | 0.03 | 0 | 0.04 | 0.03 |
| Q20 (%) | 96.02 | 96.89 | 97 | 97.84 | 95.37 |
| Q30 (%) | 89.82 | 91.78 | 91 | 93.62 | 88.17 |
| GC content (%) | 38.81 | 38.63 | 38 | 40.31 | 37.28 |
| Mapping rate (%) | 98.78 | 97.40 | 98 | 0.9838 | 0.9652 |
| Average mapping depth | 15.36 | 17.65 | 5.102 | 7.5 | 4.43 |
| Coverage 1X | 89.45 | 90.26 | 82.468 | 86.32 | 77.87 |
| Coverage 4X | 83.74 | 83.97 | 39.579 | 60.44 | 28.64 |
| Total SNPs | 12,343,419 | 2,962,195 | 4,161,067.303 | 8,070,891 | 2,369,880 |
| Homozygous SNP | 570,172 | 338,723 | 26,448.22222 | 85,498 | 15,662 |
| Heterozygous SNP | 11,773,247 | 2,623,472 | 4,134,619.081 | 8,025,401 | 2,349,592 |
| Heterozygosity rate (%) | 95.3808 | 88.5651 | 99.36101263 | 99.5461 | 98.3093 |
| Segregant Type | Cross Type | Count | % † | Female Parent | Male Parent | Alleles Detectable | F1 Ratio Detectable |
|---|---|---|---|---|---|---|---|
| Type 1 | lmxll | 3,001,321 | 65.15 | Heterozygous | Homozygous | 2 | 1:1 |
| Type 2 | nnxnp | 482,398 | 10.47 | Homozygous | Heterozygous | 2 | 1:1 |
| Type 3 | hkxhk | 1,117,173 | 24.25 | Heterozygous | Heterozygous | 2 | 1:2:1, 3:1, 1:3 |
| Type 4a | abxcd | 1 | 0.00002 | Heterozygous | Heterozygous | 4 | 1:1:1:1 |
| Type 4b | efxeg | 746 | 0.02 | Heterozygou | Heterozygous | 3 | 1:1:1:1 |
| Type 5 | aaxbb | 5,146 | 0.11 | Homozygous | Homozygous | 2 | |
| Type 6 | abxcc | 186 | 0.0040 | Heterozygous | Homozygous | 3 | 2:1:1, 1:1 |
| Type 7 | ccxab | 15 | 0.0003 | Homozygous | Heterozygous | 3 | 2:1:1, 1:1 |
| Linkage Group | Marker Number | Length Covered | Average Length | Maximum Gap | Gap < 5 cM | Gap 5 to 10 cM | Gap 10 to 20 cM | Gap > 20 cM | Ratio |
|---|---|---|---|---|---|---|---|---|---|
| LG01 | 708 | 2070.07 | 2.92 | 23.05 | 568 | 100 | 39 | 1 | 80.23 |
| LG02 | 483 | 987.5 | 2.04 | 33.31 | 430 | 39 | 13 | 1 | 89.03 |
| LG03 | 667 | 1828.85 | 2.74 | 20.05 | 540 | 94 | 32 | 1 | 80.96 |
| LG04 | 1341 | 1986.08 | 1.48 | 12.84 | 1233 | 103 | 5 | 0 | 91.95 |
| LG05 | 566 | 1459.75 | 2.58 | 14.12 | 448 | 100 | 18 | 0 | 79.15 |
| LG06 | 255 | 864.52 | 3.39 | 15.44 | 184 | 59 | 12 | 0 | 72.16 |
| LG07 | 836 | 1954.33 | 2.34 | 15.35 | 694 | 97 | 45 | 0 | 83.01 |
| LG08 | 159 | 255.81 | 1.61 | 12.45 | 150 | 8 | 1 | 0 | 94.34 |
| LG09 | 457 | 1049.96 | 2.3 | 26.89 | 382 | 58 | 15 | 2 | 83.59 |
| LG10 | 390 | 255.1 | 0.65 | 7.46 | 382 | 8 | 0 | 0 | 97.95 |
| LG11 | 572 | 1416.76 | 2.48 | 26.08 | 485 | 67 | 17 | 3 | 84.79 |
| Total | 6434 | 14128.7 | 2.2 | 33.31 | 5496 | 733 | 197 | 8 | 85.42 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, Z.; Deng, H.; Liang, G.; Ye, X.; Qin, Y.; Huang, L. Construction of a High-Density Genetic Map for Pitaya Using the Whole Genome Resequencing Approach. Horticulturae 2021, 7, 534. https://doi.org/10.3390/horticulturae7120534
Wu Z, Deng H, Liang G, Ye X, Qin Y, Huang L. Construction of a High-Density Genetic Map for Pitaya Using the Whole Genome Resequencing Approach. Horticulturae. 2021; 7(12):534. https://doi.org/10.3390/horticulturae7120534
Chicago/Turabian StyleWu, Zhijiang, Haiyan Deng, Guidong Liang, Xiaoying Ye, Yonghua Qin, and Lifang Huang. 2021. "Construction of a High-Density Genetic Map for Pitaya Using the Whole Genome Resequencing Approach" Horticulturae 7, no. 12: 534. https://doi.org/10.3390/horticulturae7120534
APA StyleWu, Z., Deng, H., Liang, G., Ye, X., Qin, Y., & Huang, L. (2021). Construction of a High-Density Genetic Map for Pitaya Using the Whole Genome Resequencing Approach. Horticulturae, 7(12), 534. https://doi.org/10.3390/horticulturae7120534