
Characteristics and Phylogenetic Analysis of the Complete Plastomes of Anthogonium gracile and Eleorchis japonica (Epidendroideae, Orchidaceae)

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Taxon Sampling, DNA Extraction and Sequencing
2.2. Plastome Assembly and Annotation
2.3. Repeat Sequence and Comparative Analyses
2.4. Codon Usage and Sequence Divergence Analyses
2.5. Selection Pressure Analyses
2.6. Phylogenetic Analyses
3. Results
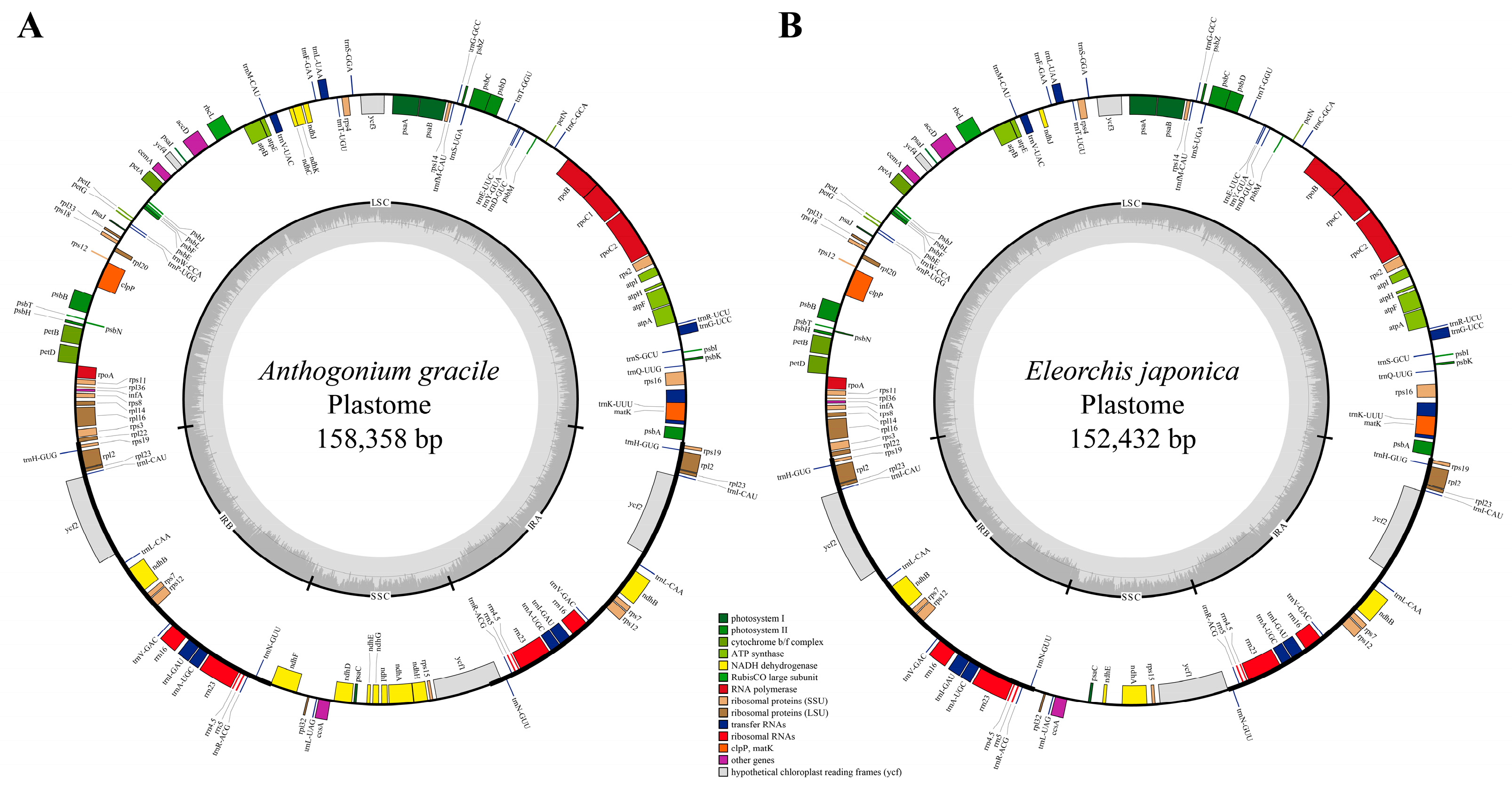
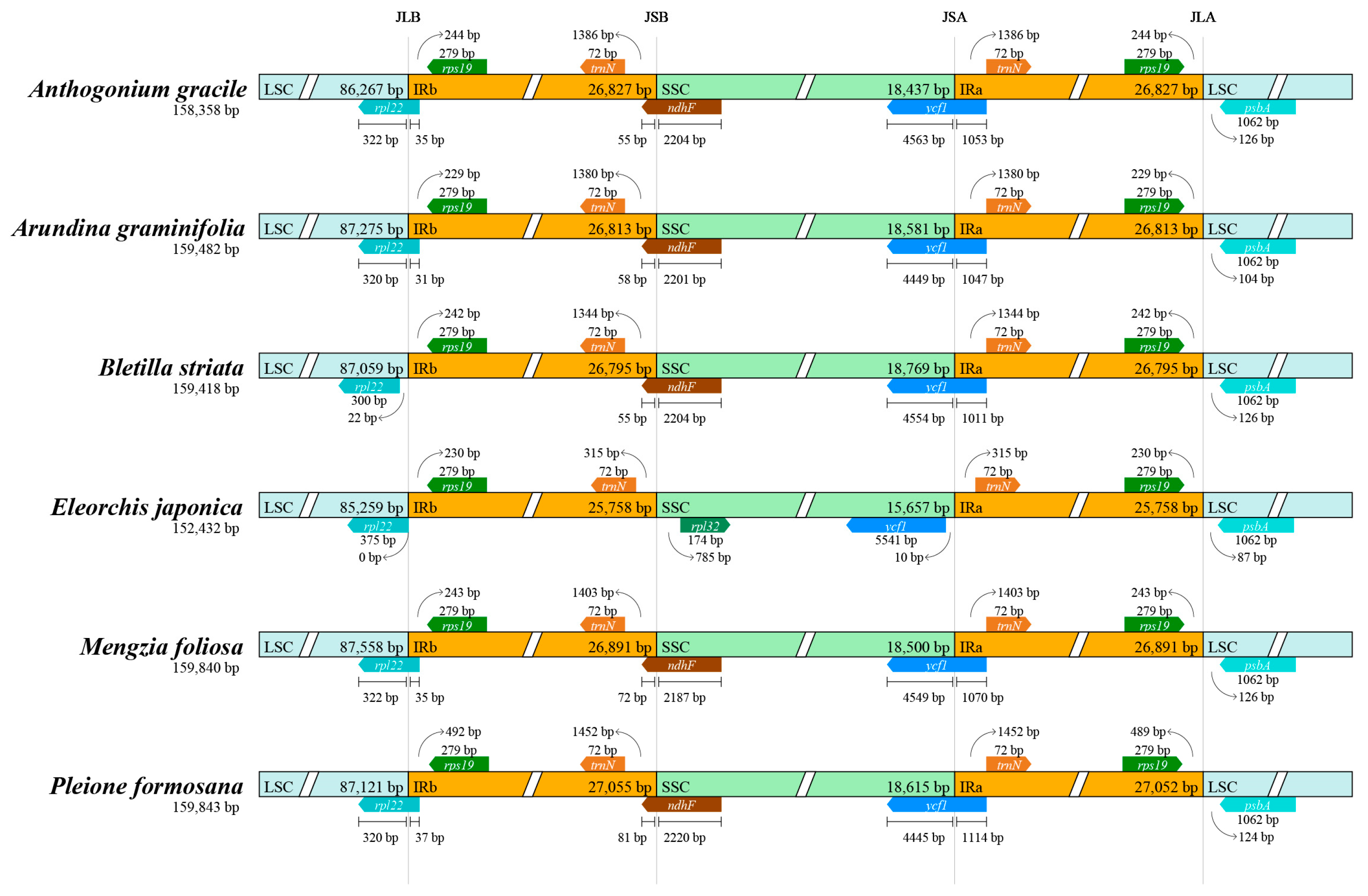
3.1. Plastome Features and Comparison Analysis
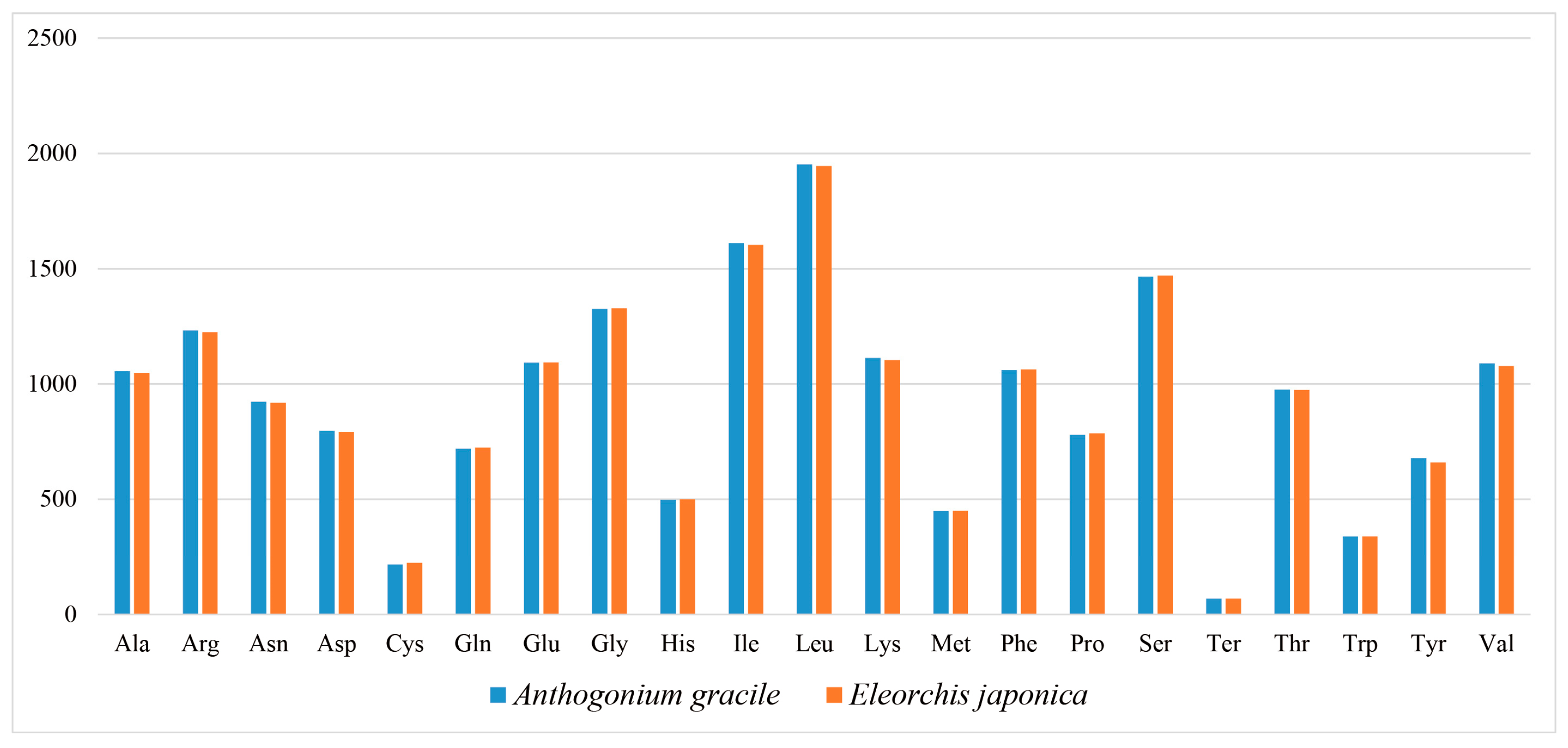
3.2. Codon Usage Analysis
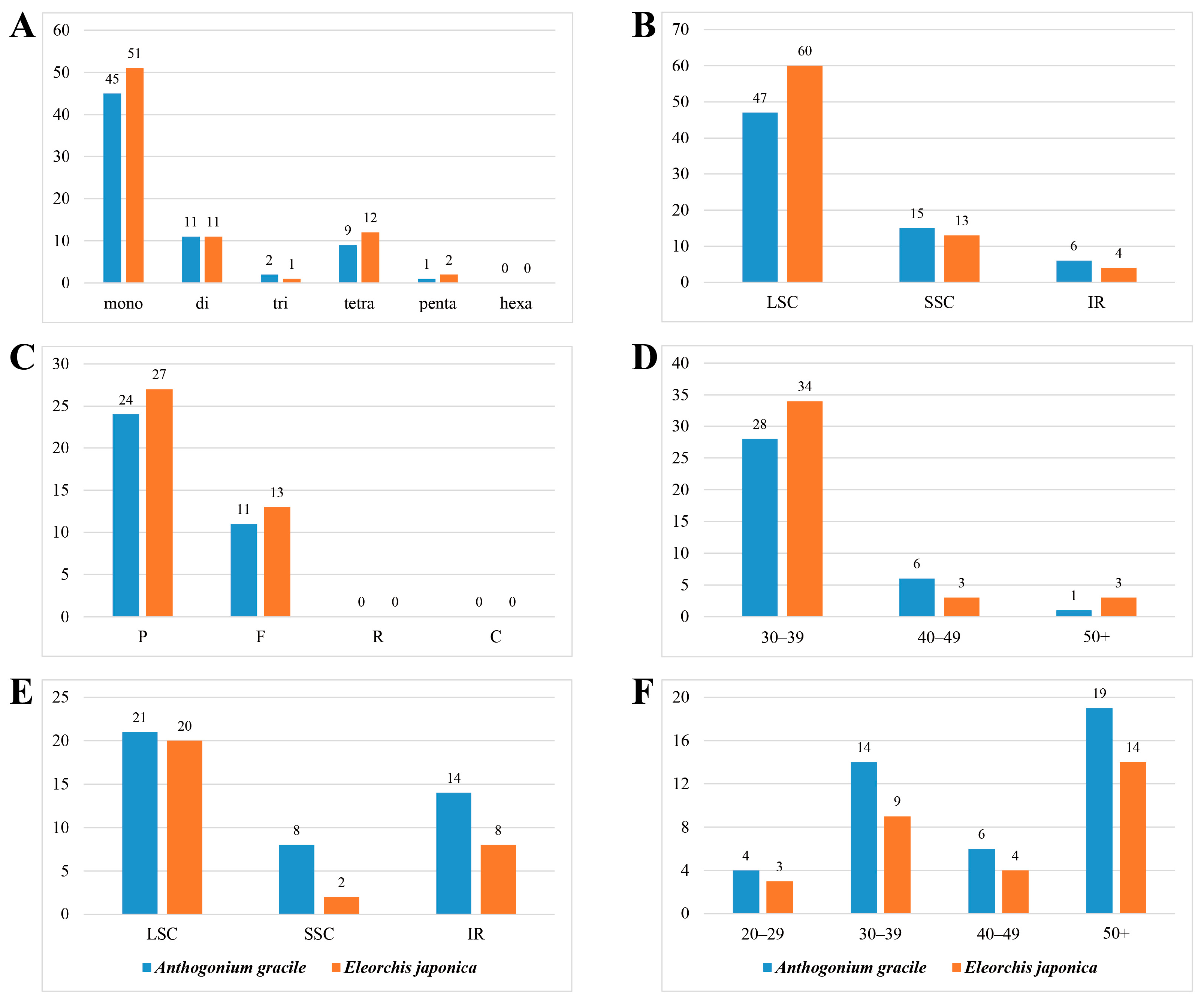
3.3. Repeat Sequence Analysis
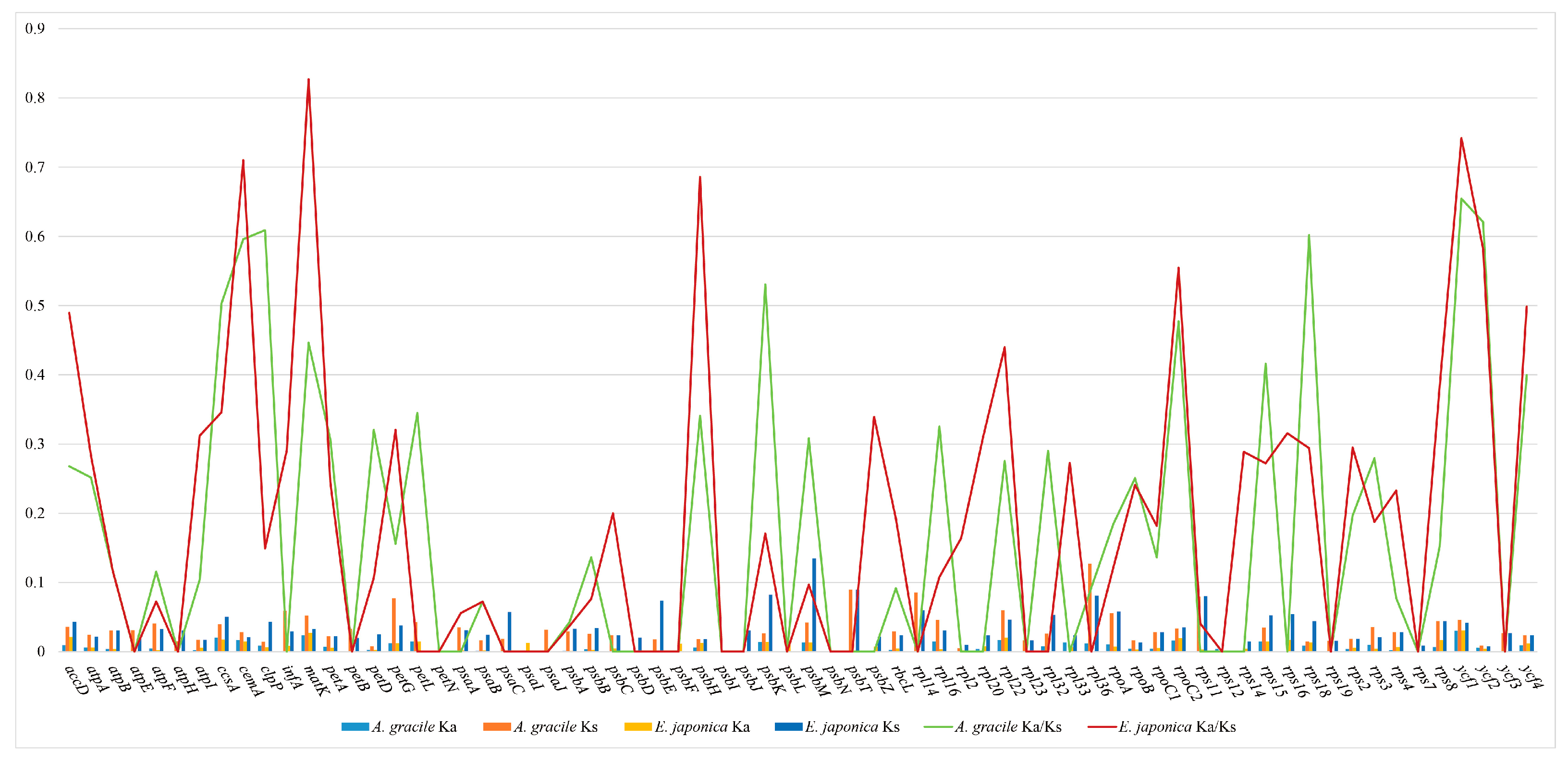
3.4. Selective Pressure Analysis
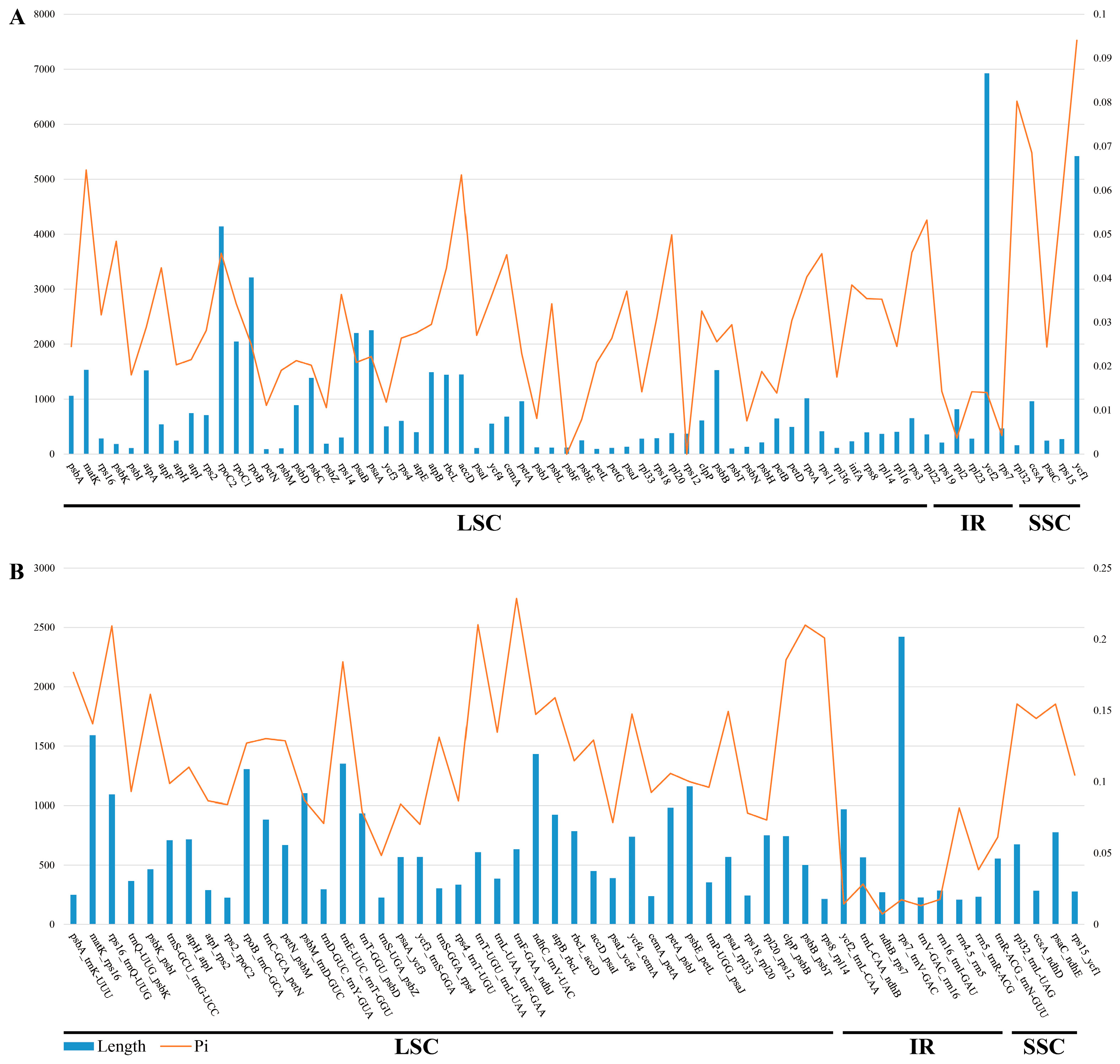
3.5. Sequence Divergence Analysis
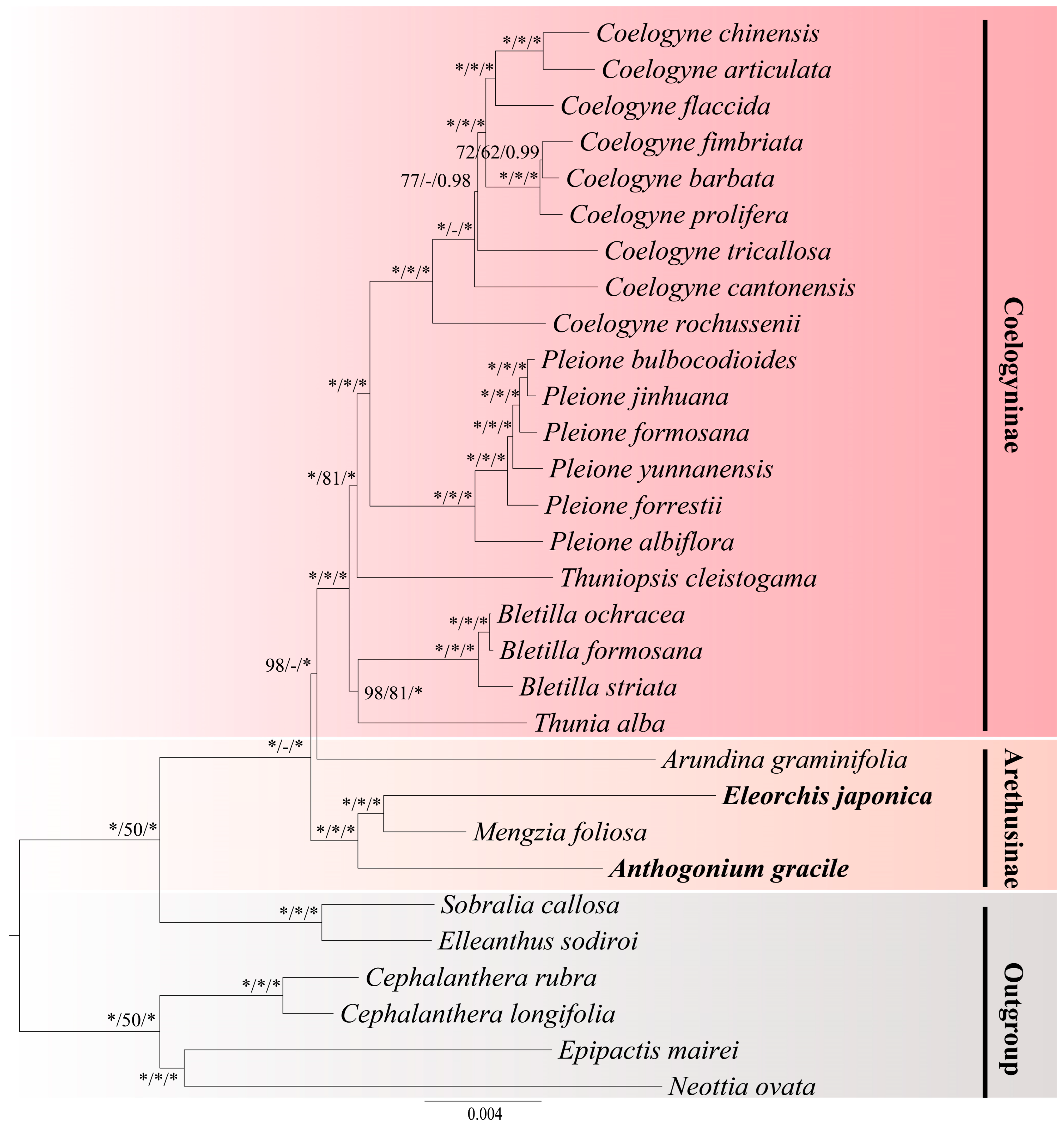
3.6. Phylogenetic Analysis
4. Discussion
4.1. Plastome Characteristics and Comparison
4.2. Codon Usage and Repeat Sequences Analysis
4.3. Hypervariable Regions Analysis
4.4. Phylogenetic Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chase, M.W.; Cameron, K.M.; Freudestein, J.V.; Pridgeon, A.M.; Salazar, G.; Van den Berg, C.; Schuiteman, A. An update classification of Orchidaceae. Bot. J. Linn. Soc. 2015, 177, 151–174. [Google Scholar] [CrossRef]
- Chase, M.W.; Gravendeel, B.; Sulistyo, B.P.; Wti, R.K.; Schuiteman, A. Expansion of the orchid genus Coelogyne (Arethuseae; Epidendroideae) to include Bracisepalum, Bulleyia, Chelonistele, Dendrochilum, Dickasonia, Entomophobia, Geesinkorchis, Gynoglottis, Ischnogyne, Nabaluia, Neogyna, Otochilus, Panisea and Pholidota. Phytotaxa 2021, 510, 94–134. [Google Scholar] [CrossRef]
- Li, L.; Ye, D.P.; Niu, M.; Yan, H.F.; Wen, T.L.; Li, S.J. Thuniopsis: A new orchid genus and phylogeny of the tribe Arethuseae (Orchidaceae). PLoS ONE 2015, 10, e0132777. [Google Scholar] [CrossRef]
- Huang, W.C.; Liu, Z.J.; Jiang, K.; Luo, Y.B.; Jin, X.H.; Zhang, Z.; Xu, R.H.; Muchuku, J.K.; Musungwa, S.S.; Yukawa, T.; et al. Phylogenetic analysis and character evolution of tribe Arethuseae (Orchidaceae) reveal a new genus Mengzia. Mol. Phylogenet. Evol. 2022, 167, 107362. [Google Scholar] [CrossRef]
- Pridgeon, A.M.; Cribb, P.J.; Chase, M.W.; Rasmussen, F.N. Genera Ochidacearum Volume 4: Epidendroideae (Part One); Oxford University Press: Oxford, UK, 2005. [Google Scholar]
- Bentham, G. Notes on Orchideae. Biol. J. Linn. Soc. 1881, 18, 281–360. [Google Scholar] [CrossRef]
- Dressler, R.L. The Orchids: Natural History and Classification; Harvard University Press: Cambridge, MA, USA, 1981. [Google Scholar]
- Dressler, R.L. Phylogeny and Classification of the Orchid Family; Cambridge University Press: Cambridge, UK, 1993. [Google Scholar]
- Szlachetko, D.L. Systema orchidalium. Fragm. Florist. Geobot. Pol. 1995, 3, 1–152. [Google Scholar]
- Goldman, D.H.; Freudenstein, J.V.; Kores, P.J.; Molvray, M.; Jarrell, D.C.; Whitten, W.M.; Cameron, K.M.; Jansen, R.K.; Chase, M.W. Phylogenetics of Arethuseae (Orchidaceae) based on plastid matK and rbcL sequences. Syst. Bot. 2001, 37, 670–695. [Google Scholar]
- Górniak, M.; Paun, O.; Chase, M.W. Phylogenetic relationships within Orchidaceae based on a low-copy nuclear coding gene, Xdh: Congruence with organellar and nuclear ribosomal DNA results. Mol. Phylogenet. Evol. 2010, 56, 784–795. [Google Scholar] [CrossRef]
- Freudenstein, J.V.; Chase, M.W. Phylogenetic relationships in Epidendroideae (Orchidaceae), one of the great flowering plant radiations: Progressive specialization and diversification. Ann. Bot. 2015, 115, 665–681. [Google Scholar] [CrossRef]
- Li, Y.X.; Li, Z.H.; Schuiteman, A.; Chase, M.W.; Li, J.W.; Huang, W.C.; Hidayat, A.; Wu, S.S.; Jin, X.H. Phylogenomics of Orchidaceae based on plastid and mitochondrial genomes. Mol. Phylogenet. Evol. 2019, 139, 106540. [Google Scholar] [CrossRef]
- Van den Berg, C.; Goldman, D.H.; Freudenstein, J.V.; Pridgeon, A.M.; Cameron, K.M.; Chase, M.W. An overview of the phylogenetic relationships within Epidendroideae inferred from multiple DNA regions and recircumscription of Epidendreae and Arethuseae (Orchidaceae). Am. J. Bot. 2005, 92, 613–624. [Google Scholar] [CrossRef]
- Jansen, R.K.; Ruhlman, T.A. Plastid Genomes of Seed Plants. In Genomics of Chloroplasts and Mitochondria; Springer: Dordrecht, The Netherlands, 2012. [Google Scholar]
- Li, L.; Wu, Q.P.; Zhai, J.W.; Wu, K.L.; Fang, L.; Li, M.Z.; Zeng, S.J.; Li, S.J. Comparative chloroplast genomics of 24 species shed light on the genome evolution and phylogeny of subtribe Coelogyninae (Orchidaceae). BMC Plant Biol. 2024, 24, 31. [Google Scholar] [CrossRef]
- Liu, D.K.; Tu, X.D.; Zhao, Z.; Zeng, M.Y.; Zhang, S.; Ma, L.; Zhang, G.Q.; Wang, M.M.; Liu, Z.J.; Lan, S.R.; et al. Plastid phylogenomic data yield new and robust insights into the phylogeny of Cleisostoma–Gastrochilus clades (Orchidaceae, Aeridinae). Mol. Phylogenet. Evol. 2020, 145, 106729. [Google Scholar] [CrossRef]
- Serna-Sánchez, M.A.; Pérez-Escobar, O.A.; Bogarín, D.; Torres-Jimenez, M.F.; Alvarez-Yela, A.C.; Arcila-Galvis, J.E.; Hall, C.F.; de Barros, F.; Pinheiro, F.; Dodsworth, S.; et al. Plastid phylogenomics resolves ambiguous relationships within the orchid family and provides a solid timeframe for biogeography and macroevolution. Sci. Rep. 2021, 11, 6858. [Google Scholar]
- Tu, X.D.; Liu, D.K.; Xu, S.W.; Zhou, C.Y.; Gao, X.Y.; Zeng, M.Y.; Zhang, S.; Chen, J.L.; Ma, L.; Zhou, Z.; et al. Plastid phylogenomics improves resolution of phylogenetic relationship in the Cheirostylis and Goodyera clades of Goodyerinae (Orchidoideae, Orchidaceae). Mol. Phylogenet. Evol. 2021, 164, 107269. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Li, R.; Tang, S.; Chen, Y.; Yan, Y.; Gao, X.; Zhuo, X. Plastomes of Seven Coelogyne s.l. (Arethuseae, Orchidaceae) Species: Comparative Analysis and Phylogenetic Relationships. Horticulturae 2025, 11, 144. [Google Scholar] [CrossRef]
- Ai, Y.; Xie, T.-X.; Liu, D.-K.; Tu, X.-D.; Zhou, J.; Liu, Z.-J. Complete chloroplast genome of Arundina graminifolia (Orchidaceae). Mitochondrial DNA Part B 2019, 4, 2898–2899. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.-T.; Chao, W.-C.; Huang, C.-L.; Lan, S.-R.; Liu, Z.-J.; Wu, S.-S. The complete chloroplast genome of Pleione formosana (Orchidaceae). Mitochondrial DNA Part B 2019, 4, 1044–1046. [Google Scholar] [CrossRef]
- Zhang, R. Characterization and phylogenetic analysis of the complete plastome sequence of Thunia alba (Lindley) HG Reichenbach (Orchidaceae), a rare wild ornamental orchid. Mitochondrial DNA Part B 2019, 4, 4039–4040. [Google Scholar] [CrossRef]
- Li, L.; Wu, Q.; Fang, L.; Wu, K.; Li, M.; Zeng, S. Comparative chloroplast genomics and phylogenetic analysis of Thuniopsis and closely related genera within Coelogyninae (Orchidaceae). Front. Genet. 2022, 13, 850201. [Google Scholar] [CrossRef]
- Li, J.; Wang, S.; Yu, J.; Wang, L.; Zhou, S. A Modified CTAB Protocol for Plant DNA Extraction. Chin. Bull. Bot. 2013, 48, 72–78. [Google Scholar]
- Chen, S.F.; Zhou, Y.Q.; Chen, Y.R.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, 884–890. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.J.; Yu, W.B.; Yang, J.B.; Song, Y.; dePamphilis, C.W.; Yi, T.S.; Li, D.Z. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020, 21, 241. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Ni, Y.; Li, J.L.; Zhang, C.; Liu, C. Generating Sequencing Depth and Coverage Map for Organelle Genomes. Protocols.io 2023. [Google Scholar] [CrossRef]
- Qu, X.-J.; Moore, M.J.; Li, D.-Z.; Yi, T.-S. PGA: A software package for rapid, accurate, and flexible batch annotation of plastomes. Plant Methods 2019, 15, 50. [Google Scholar] [CrossRef]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef]
- Darling, A.E.; Mau, B.; Perna, N.T. progressiveMauve: Multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE 2010, 5, e11147. [Google Scholar] [CrossRef]
- Brudno, M.; Malde, S.; Poliakov, A.; Do, C.B.; Couronne, O.; Dubchak, I.; Batzoglou, S. Glocal alignment: Finding rearrangements during alignment. Bioinformatics 2003, 19, i54–i62. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Nei, M.; Dudley, J.; Tamura, K. MEGA: A biologist-centric software for evolutionary analysis of DNA and protein sequences. Brief. Bioinform. 2008, 9, 299–306. [Google Scholar] [CrossRef]
- Xia, X. Dambe7: New and improved tools for data analysis in molecular biology and evolution. Mol. Biol. Evol. 2018, 35, 1550–1552. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Glez-Peña, D.; Gómez-Blanco, D.; Reboiro-Jato, M.; Fdez-Riverola, F.; Posada, D. ALTER: Program-oriented format conversion of DNA and protein alignments. Nucleic Acids Res. 2010, 38, W14–W18. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Zhang, Y.; Zhang, Z.; Zhu, J.; Yu, J. KaKs_Calculator 2.0: A toolkit incorporating gamma-series methods and sliding window strategies. Genom. Proteom. Bioinform. 2010, 8, 77–80. [Google Scholar] [CrossRef]
- Capella-Gutierrez, S.; Silla-Martinez, J.M.; Gabaldon, T. Trimal: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. Creating the CIPRES Science Gateway for Inference of Large Phylogenetic Trees. In Proceedings of the 2010 Gateway Computing Environments Workshop, GCE, New Orleans, LA, USA, 14 November 2010. [Google Scholar]
- Tao, L.; Duan, H.; Tao, K.; Luo, Y.; Li, Q.; Li, L. Complete chloroplast genome structural characterization of two Phalaenopsis (Orchidaceae) species and comparative analysis with their alliance. BMC Genom. 2023, 24, 359. [Google Scholar] [CrossRef]
- Wu, Y.; Zeng, M.-Y.; Wang, H.-X.; Lan, S.; Liu, Z.-J.; Zhang, S.; Li, M.-H.; Guan, Y. The Complete Chloroplast Genomes of Bulbophyllum (Orchidaceae) Species: Insight into Genome Structure Divergence and Phylogenetic Analysis. Int. J. Mol. Sci. 2024, 25, 2665. [Google Scholar] [CrossRef]
- Lin, C.S.; Chen, J.J.W.; Chiu, C.C.; Hsiao, H.C.W.; Yang, C.J.; Jin, X.H.; Leebens-Mack, J.; de Pamphilis, C.W.; Huang, Y.T.; Yang, L.H.; et al. Concomitant loss of NDH complex-related genes within chloroplast and nuclear genomes in some orchids. Plant J. 2017, 90, 994–1006. [Google Scholar] [CrossRef]
- Zavala-Páez, M.; Vieira, L.D.N.; Baura, V.A.D.; Balsanelli, E.; Souza, E.M.D.; Cevallos, M.C.; Smidt, E.D.C. Comparative plastid genomics of neotropical Bulbophyllum (Orchidaceae; Epidendroideae). Front. Plant Sci. 2020, 11, 799. [Google Scholar] [CrossRef] [PubMed]
- Xiao, T.; He, L.; Yue, L.; Zhang, Y.; Lee, S.Y. Comparative phylogenetic analysis of complete plastid genomes of Renanthera (Orchidaceae). Front. Genet. 2022, 13, 998575. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.K.; Zhou, C.Y.; Tu, X.D.; Zhao, Z.; Chen, J.L.; Gao, X.Y.; Xu, S.W.; Zeng, M.Y.; Ma, L.; Ahmad, S.; et al. Comparative and phylogenetic analysis of Chiloschista (Orchidaceae) species and DNA barcoding investigation based on plastid genomes. BMC Genom. 2023, 24, 749. [Google Scholar] [CrossRef]
- Zhao, Z.; Zeng, M.Y.; Wu, Y.W.; Li, J.W.; Zhou, Z.; Liu, Z.J.; Li, M.H. Characterization and comparative analysis of the complete plastomes of five Epidendrum (Epidendreae, Orchidaceae) species. Int. J. Mol. Sci. 2023, 24, 14437. [Google Scholar] [CrossRef]
- Zhou, Z.; Chen, J.; Wang, F.; Wu, X.; Liu, Z.; Peng, D.; Lan, S. The Complete Chloroplast Genome of an Epiphytic Leafless Orchid, Taeniophyllum complanatum: Comparative Analysis and Phylogenetic Relationships. Horticulturae 2024, 10, 660. [Google Scholar] [CrossRef]
- Barrett, C.F.; Kennedy, A.H. Plastid genome degradation in the endangered, mycoheterotrophic, North American orchid Hexalectris warnockii. Genome Biol. Evol. 2018, 10, 1657–1662. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.K.; Jo, S.; Cheon, S.H.; Joo, M.J.; Hong, J.R.; Kwak, M.H.; Kim, K.J. Extensive losses of photosynthesis genes in the plastome of a mycoheterotrophic orchid, Cyrtosia septentrionalis (Vanilloideae: Orchidaceae). Genome Biol. Evol. 2019, 11, 565–571. [Google Scholar] [CrossRef]
- Zeng, M.Y.; Li, M.H.; Lan, S.; Yin, W.L.; Liu, Z.J. Comparative phylogenomic study of Malaxidinae (Orchidaceae) sheds light on plastome evolution and gene divergence. Int. J. Mol. Sci. 2024, 25, 11181. [Google Scholar] [CrossRef]
- Kimura, M. The neutral theory of molecular evolution and the world view of the neutralists. Genome 1989, 31, 24–31. [Google Scholar] [CrossRef]
- Weng, M.L.; Ruhlman, T.A.; Jansen, R.K. Expansion of inverted repeat does not decrease substitution rates in Pelargonium plastid genomes. New Phytol. 2017, 214, 842–851. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wang, W.; Zhang, G.; Wu, K.; Fang, L.; Li, M.; Liu, Z.; Zeng, S. Comparative analyses and phylogenetic relationships of thirteen Pholidota species (Orchidaceae) inferred from complete chloroplast genomes. BMC Plant Biol. 2023, 23, 269. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Wang, R.; Hong, X.; Wu, C.; Zhang, S.; Kan, X. Plastomes of Bletilla (Orchidaceae) and phylogenetic implications. Int. J. Mol. Sci. 2022, 23, 10151. [Google Scholar] [CrossRef]
- Yu, X.; Liu, J.; Li, H.; Liu, B.Y.; Zhao, B.Q.; Ning, Z.Y. Comprehensive analysis of synonymous codon usage bias for complete genomes and E2 gene of atypical porcine pestivirus. Biochem. Genet. 2021, 59, 799–812. [Google Scholar] [CrossRef]
- Jiang, H.; Tian, J.; Yang, J.; Dong, X.; Zhong, Z.; Mwachala, G.; Zhang, C.; Hu, G.; Wang, Q. Comparative and phylogenetic analyses of six Kenya Polystachya (Orchidaceae) species based on the complete chloroplast genome sequences. BMC Plant Biol. 2022, 22, 177. [Google Scholar] [CrossRef]
- Agrama, H.; Tuinstra, M. Phylogenetic diversity and relationships among sorghum accessions using SSRs and RAPDs. Afr. J. Biotechnol. 2003, 2, 334–340. [Google Scholar] [CrossRef]
- Doorduin, L.; Gravendeel, B.; Lammers, Y.; Ariyurek, Y.; Chin-A-Woeng, T.; Vrieling, K. The complete chloroplast genome of 17 individuals of pest species Jacobaea vulgaris: SNPs, microsatellites and barcoding markers for population and phylogenetic studies. DNA Res. 2011, 18, 93–105. [Google Scholar] [CrossRef]
- Yang, A.H.; Zhang, J.J.; Yao, X.H.; Huang, H.W. Chloroplast microsatellite markers in Liriodendron tulipifera (Magnoliaceae) and cross-species amplification in L. chinense. Am. J. Bot. 2011, 98, e123–e126. [Google Scholar] [CrossRef] [PubMed]
- Madhumati, B. Potential and application of molecular markers techniques for plant genome analysis. Int. J. Pure App. Biosci. 2014, 2, 169–188. [Google Scholar]
- Zhou, C.Y.; Zeng, M.Y.; Gao, X.Y.; Zhao, Z.; Li, R.Y.; Wu, Y.H.; Liu, Z.J.; Zhang, D.Y.; Li, M.H. Characteristics and comparative analysis of seven complete plastomes of Trichoglottis s.l. (Aeridinae, Orchidaceae). Int. J. Mol. Sci. 2023, 24, 14544. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Function Classification | Group of Genes | Gene Name | |||
|---|---|---|---|---|---|
| Self-replication | Ribosomal RNA genes | rrn4.5(2) | rrn5(2) | rrn16(2) | rrn23(2) |
| Transfer RNA genes | trnA-UGC(2),# | trnC-GCA | trnD-GUC | trnE-UUC | |
| trnF-GAA | trnfM-CAU | trnG-GCC | trnG-UCC# | ||
| trnH-GUG(2) | trnI-CAU(2) | trnI-GAU(2),# | trnK-UUU# | ||
| trnL-CAA(2) | trnL-UAA# | trnL-UAG | trnM-CAU | ||
| trnN-GUU(2) | trnP-UGG | trnQ-UUG | trnR-ACG(2) | ||
| trnR-UCU | trnS-GCU | trnS-GGA | trnS-UGA | ||
| trnT-GGU | trnT-UGU | trnV-GAC(2) | trnV-UAC# | ||
| trnW-CCA | trnY-GUA | ||||
| RNA polymerase | rpoA | rpoB | rpoC1# | rpoC2 | |
| Small subunits of ribosome | rps2 | rps3 | rps4 | rps7(2) | |
| rps8 | rps11 | rps12(2),##* | rps14 | ||
| rps15 | rps16# | rps18 | rps19(2) | ||
| Lange subunits of ribosome | rpl2(2),# | rpl14 | rpl16# | rpl20 | |
| rpl22 | rpl23(2) | rpl32 | rpl33 | ||
| rpl36 | |||||
| Genes for photosynthesis | Photosystem I | psaA | psaB | psaC | psaI |
| psaJ | |||||
| Photosystem II | psbA | psbB | psbC | psbD | |
| psbE | psbF | psbH | psbI | ||
| psbJ | psbK | psbL | psbM | ||
| psbN | psbT | psbZ | |||
| RubisCO large subunits | rbcL | ||||
| Cytochrome b/f complex | petA | petB# | petD# | petG | |
| petL | petN | ||||
| Subunits of ATP synthase | atpA | atpB | atpE | atpF# | |
| atpH | atpI | ||||
| NADH dehydrogenase | ndhA# | ndhB(2),# | ndhC* | ndhD* | |
| ndhE | ndhF* | ndhG* | ndhH* | ||
| ndhI* | ndhJ | ndhK* | |||
| Other genes | Protease | clpP## | |||
| Maturase | matK | ||||
| Envelope membrane protein | cemA | ||||
| Subunits of acetyl-CoA-carboxylase | accD | ||||
| C-type cytochrome synthesis | ccsA | ||||
| Translational initiation factor | infA | ||||
| Unknown function gene | Hypothetical chloroplast reading frames | ycf1 | ycf2(2) | ycf3## | ycf4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, X.; Chen, Y.; Xu, X.; Chen, H.; Xing, B.; Pan, J.; Li, M.; Zhou, Z. Characteristics and Phylogenetic Analysis of the Complete Plastomes of Anthogonium gracile and Eleorchis japonica (Epidendroideae, Orchidaceae). Horticulturae 2025, 11, 698. https://doi.org/10.3390/horticulturae11060698
Gao X, Chen Y, Xu X, Chen H, Xing B, Pan J, Li M, Zhou Z. Characteristics and Phylogenetic Analysis of the Complete Plastomes of Anthogonium gracile and Eleorchis japonica (Epidendroideae, Orchidaceae). Horticulturae. 2025; 11(6):698. https://doi.org/10.3390/horticulturae11060698
Chicago/Turabian StyleGao, Xuyong, Yuming Chen, Xiaowei Xu, Hongjiang Chen, Bingcong Xing, Jianli Pan, Minghe Li, and Zhuang Zhou. 2025. "Characteristics and Phylogenetic Analysis of the Complete Plastomes of Anthogonium gracile and Eleorchis japonica (Epidendroideae, Orchidaceae)" Horticulturae 11, no. 6: 698. https://doi.org/10.3390/horticulturae11060698
APA StyleGao, X., Chen, Y., Xu, X., Chen, H., Xing, B., Pan, J., Li, M., & Zhou, Z. (2025). Characteristics and Phylogenetic Analysis of the Complete Plastomes of Anthogonium gracile and Eleorchis japonica (Epidendroideae, Orchidaceae). Horticulturae, 11(6), 698. https://doi.org/10.3390/horticulturae11060698