
MLAS: Machine Learning-Based Approach for Predicting Abiotic Stress-Responsive Genes in Chinese Cabbage

 ,
,
Abstract
:1. Introduction
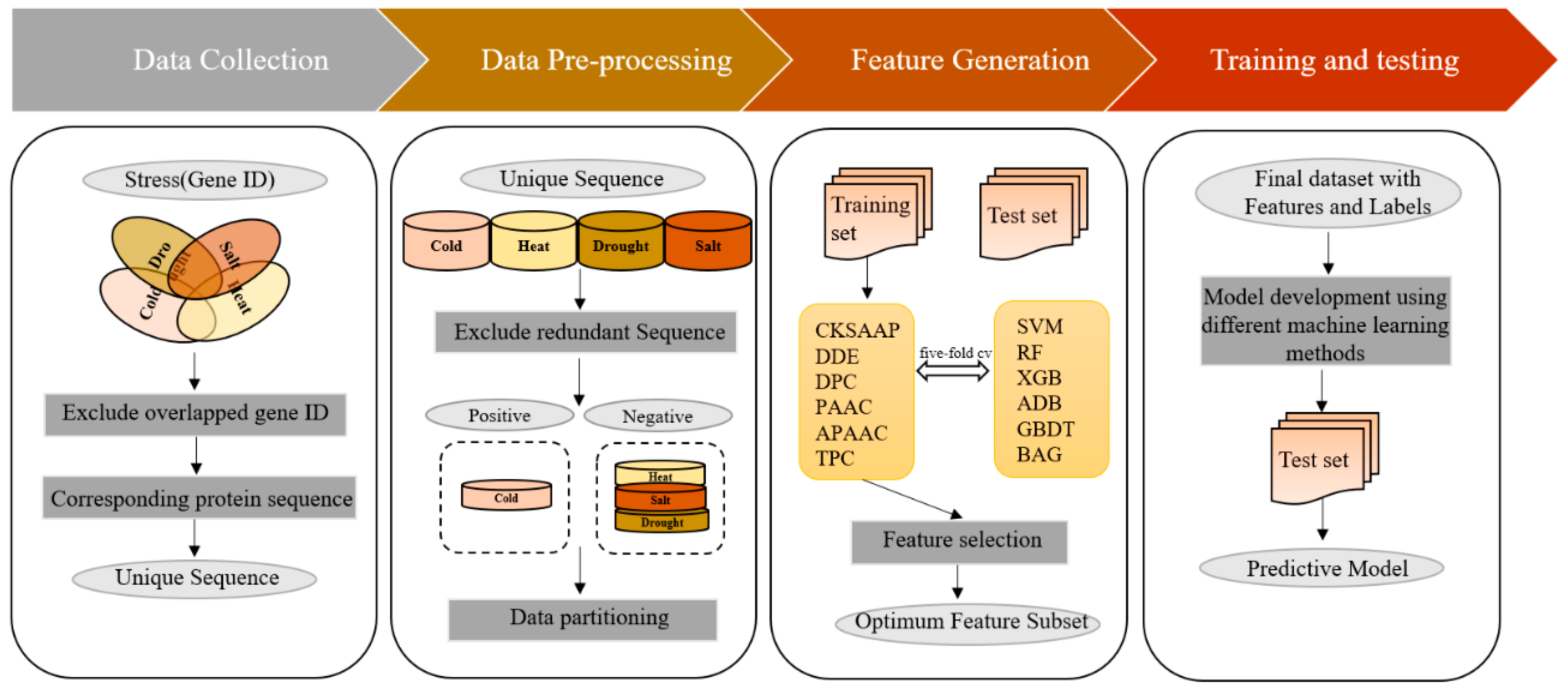
2. Materials and Methods
2.1. Data Collection and Pre-Processing
2.2. Feature Construction and Selection
2.3. Prediction Using Machine-Learning Methods
2.4. Cross Validation and Performance Metrics
3. Results
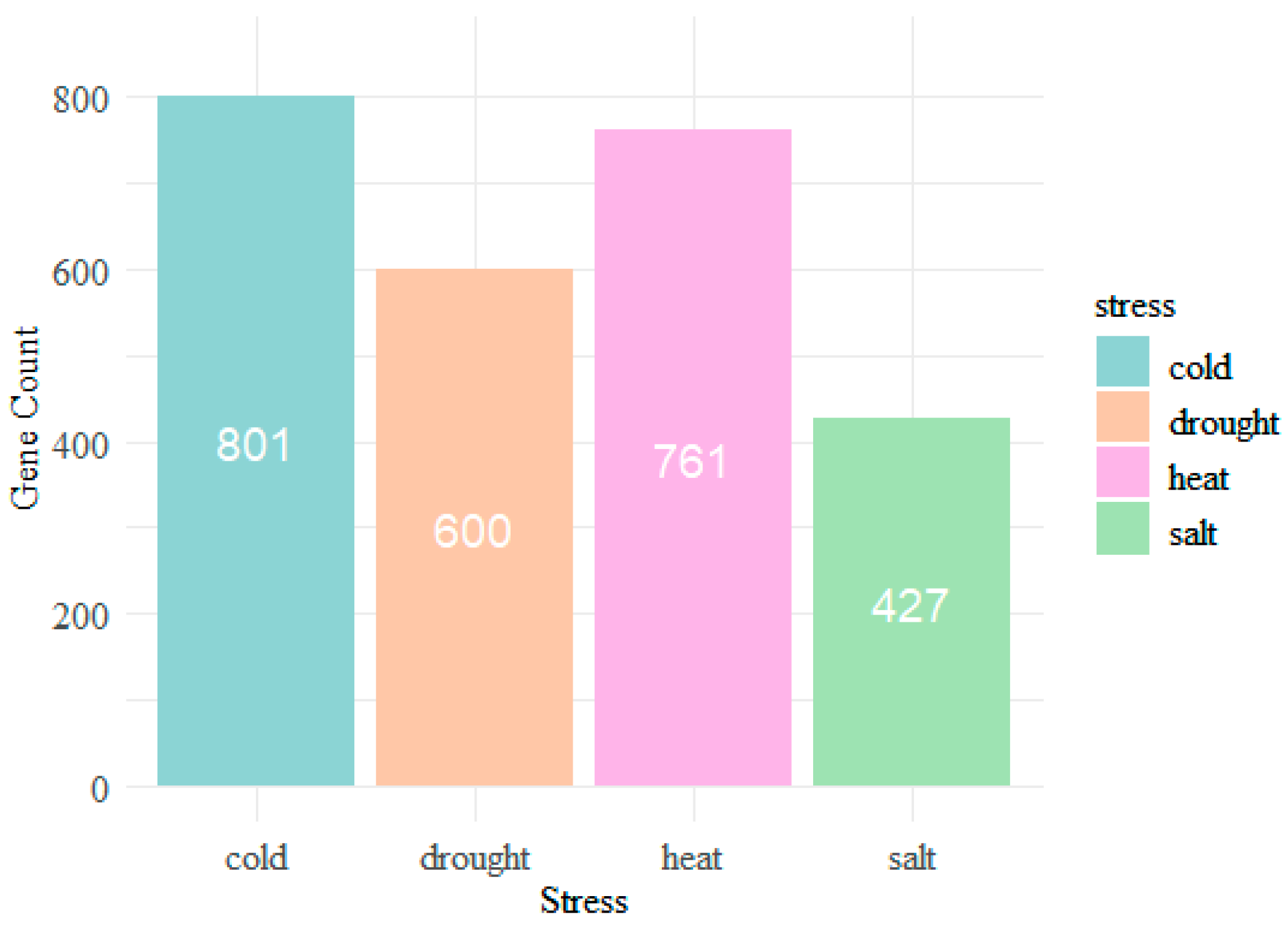
3.1. Preliminary Analysis of the Sequence Data
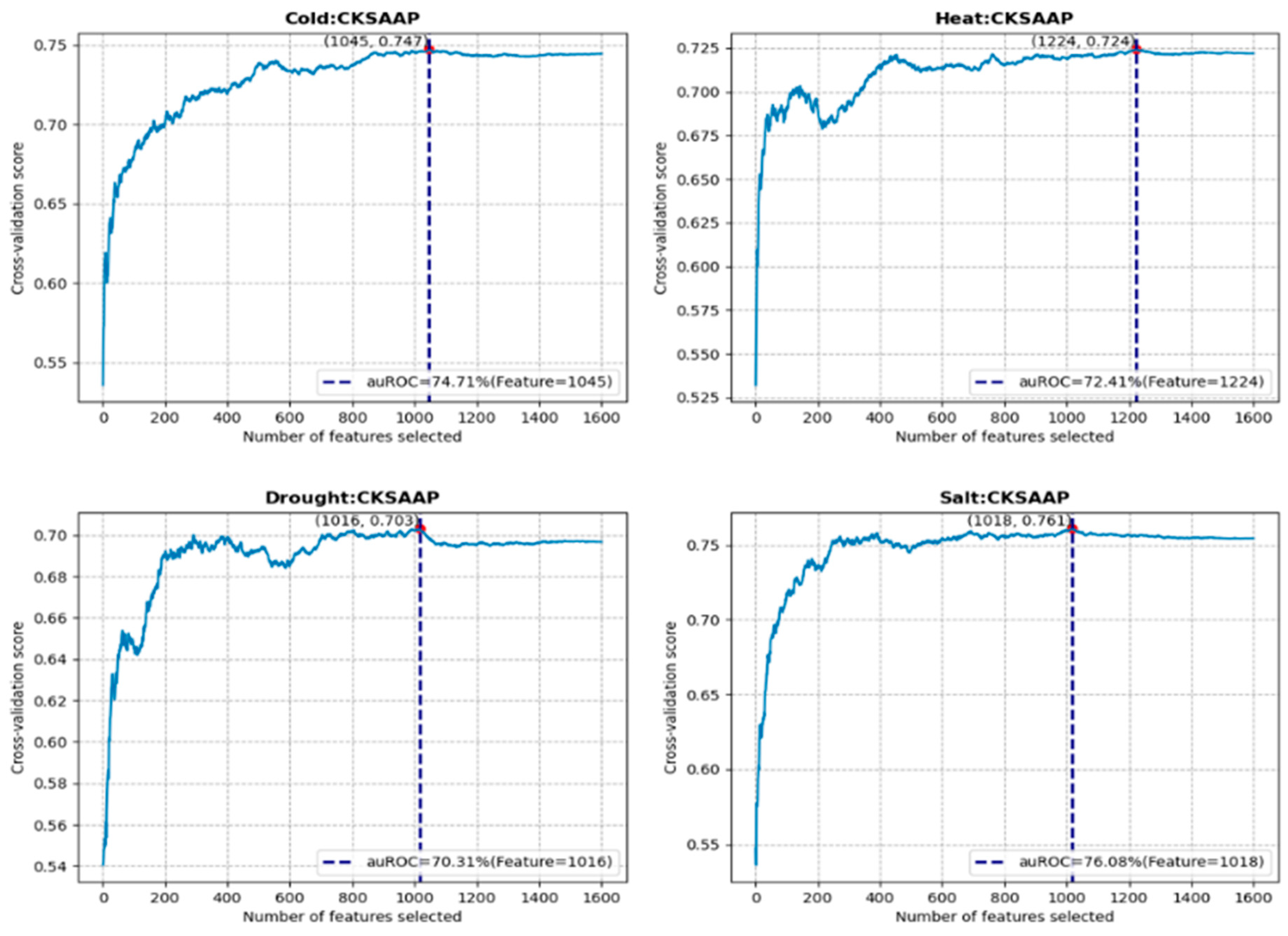
3.2. Feature Construction and Selection Analysis
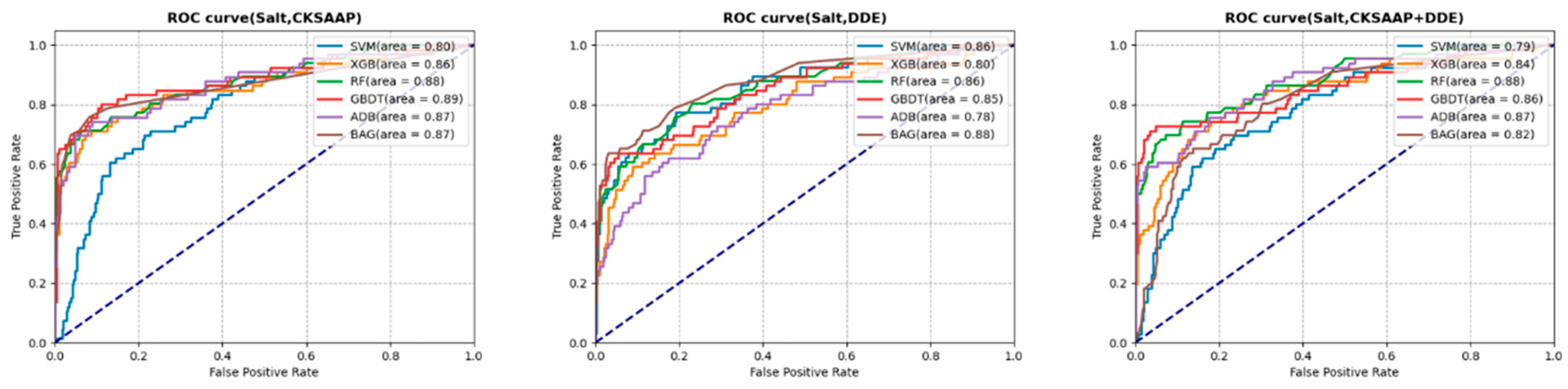
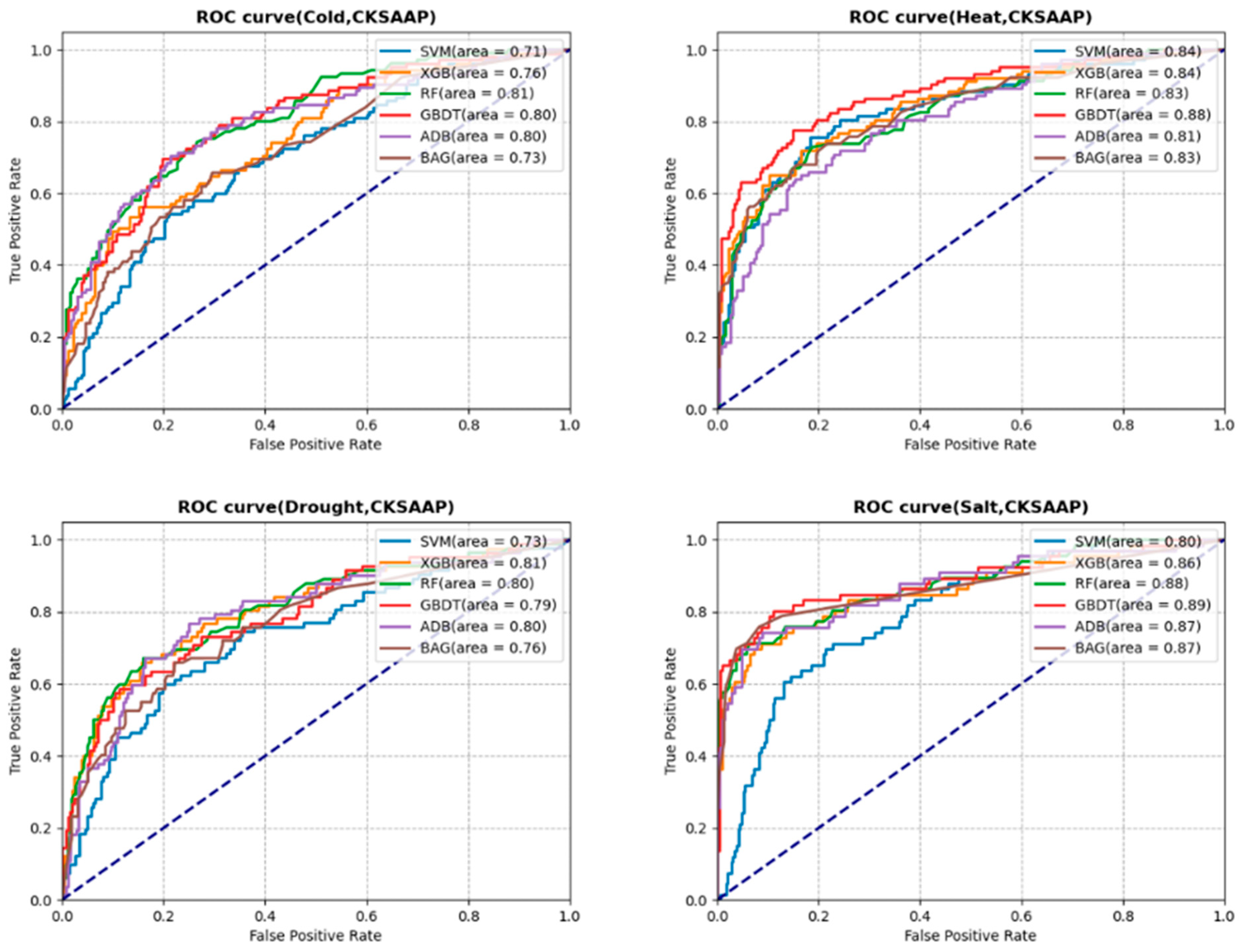
3.3. Prediction Analysis with Selected Features
3.4. Discovery of New Stress-Related Genes in Chinese Cabbage
3.4.1. Cold Stress
3.4.2. Heat Stress
3.4.3. Drought Stress
3.4.4. Salt Stress
3.5. Online Prediction Tool
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Seong, G.-U.; Hwang, I.-W.; Chung, S.-K. Antioxidant Capacities and Polyphenolics of Chinese Cabbage (Brassica rapa L. Ssp. Pekinensis) Leaves. Food Chem. 2016, 199, 612–618. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, D.; Cai, Z.; Wang, L.; Wang, J.; Sun, L.; Fan, X.; Shen, S.; Zhao, J. Spectral Technology and Multispectral Imaging for Estimating the Photosynthetic Pigments and SPAD of the Chinese Cabbage Based on Machine Learning. Comput. Electron. Agric. 2022, 195, 106814. [Google Scholar] [CrossRef]
- Zhang, H.; Zhu, J.; Gong, Z.; Zhu, J.-K. Abiotic Stress Responses in Plants. Nat. Rev. Genet. 2022, 23, 104–119. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.-H.; Lee, G.-S.; Yu, J.-G.; Kim, Y.-H.; Park, Y.-D. Correlation Network Analysis of Abiotic Stress-related Genes Reveals the Coordinated Regulation of Transcription in Chinese Cabbage. HST 2018, 36, 266–279. [Google Scholar] [CrossRef]
- Shaik, R.; Ramakrishna, W. Genes and Co-Expression Modules Common to Drought and Bacterial Stress Responses in Arabidopsis and Rice. PLoS ONE 2013, 8, e77261. [Google Scholar] [CrossRef]
- Ma, Y.; Qin, F.; Tran, L.-S.P. Contribution of Genomics to Gene Discovery in Plant Abiotic Stress Responses. Mol. Plant 2012, 5, 1176–1178. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Wu, X.; Zhang, M.; Yang, L.; Ji, Z.; Chen, R.; Cao, Y.; Huang, J.; Duan, Q. Genome-Wide Identification of BrCMF Genes in Brassica Rapa and Their Expression Analysis under Abiotic Stresses. Plants 2024, 13, 1118. [Google Scholar] [CrossRef]
- Hui, J.; Zhang, M.; Chen, L.; Wang, Y.; He, J.; Zhang, J.; Wang, R.; Jiang, Q.; Lv, B.; Cao, Y. Identification, Classification, and Expression Analysis of Leucine-Rich Repeat Extension Genes from Brassica Rapa Reveals Salt and Osmosis Stress Response Genes. Horticulturae 2024, 10, 571. [Google Scholar] [CrossRef]
- Singh, S.; Chhapekar, S.S.; Ma, Y.; Rameneni, J.J.; Oh, S.H.; Kim, J.; Lim, Y.P.; Choi, S.R. Genome-Wide Identification, Evolution, and Comparative Analysis of B-Box Genes in Brassica rapa, B. oleracea, and B. napus and Their Expression Profiling in B. rapa in Response to Multiple Hormones and Abiotic Stresses. IJMS 2021, 22, 10367. [Google Scholar] [CrossRef]
- Song, X.; Liu, G.; Duan, W.; Liu, T.; Huang, Z.; Ren, J.; Li, Y.; Hou, X. Genome-Wide Identification, Classification and Expression Analysis of the Heat Shock Transcription Factor Family in Chinese Cabbage. Mol. Genet. Genom. 2014, 289, 541–551. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Duan, W.; Riquicho, A.R.; Jing, Z.; Liu, T.; Hou, X.; Li, Y. Genome-Wide Survey and Expression Analysis of the PUB Family in Chinese Cabbage (Brassica rapa Ssp. Pekinesis). Mol. Genet. Genom. 2015, 290, 2241–2260. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Huang, F.; You, X.; Hou, X. Identification and Functional Characterization of a Cold-Related Protein, BcHHP5, in Pak-Choi (Brassica rapa Ssp. Chinensis). IJMS 2018, 20, 93. [Google Scholar] [CrossRef]
- Yang, L.; Zhao, Y.; Wu, X.; Zhang, Y.; Fu, Y.; Duan, Q.; Ma, W.; Huang, J. Genome-Wide Identification and Expression Analysis of BraGLRs Reveal Their Potential Roles in Abiotic Stress Tolerance and Sexual Reproduction. Cells 2022, 11, 3729. [Google Scholar] [CrossRef]
- Ahmed, B.; Haque, M.A.; Iquebal, M.A.; Jaiswal, S.; Angadi, U.B.; Kumar, D.; Rai, A. DeepAProt: Deep Learning Based Abiotic Stress Protein Sequence Classification and Identification Tool in Cereals. Front. Plant Sci. 2023, 13, 1008756. [Google Scholar] [CrossRef]
- Zhang, S.; Fan, R.; Liu, Y.; Chen, S.; Liu, Q.; Zeng, W. Applications of Transformer-Based Language Models in Bioinformatics: A Survey. Bioinform. Adv. 2023, 3, vbad001. [Google Scholar] [CrossRef]
- Bhardwaj, N.; Gerstein, M.; Lu, H. Genome-Wide Sequence-Based Prediction of Peripheral Proteins Using a Novel Semi-Supervised Learning Technique. BMC Bioinform. 2010, 11, S6. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Zhang, H.H.; Wang, X. Machine Learning for Big Data Analytics in Plants. Trends Plant Sci. 2014, 19, 798–808. [Google Scholar] [CrossRef]
- Yan, J.; Wang, X. Machine Learning Bridges Omics Sciences and Plant Breeding. Trends Plant Sci. 2023, 28, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, A.D.J.; Kootstra, G.; Kruijer, W.; De Ridder, D. Machine Learning in Plant Science and Plant Breeding. iScience 2021, 24, 101890. [Google Scholar] [CrossRef] [PubMed]
- Gill, M.; Anderson, R.; Hu, H.; Bennamoun, M.; Petereit, J.; Valliyodan, B.; Nguyen, H.T.; Batley, J.; Bayer, P.E.; Edwards, D. Machine Learning Models Outperform Deep Learning Models, Provide Interpretation and Facilitate Feature Selection for Soybean Trait Prediction. BMC Plant Biol. 2022, 22, 180. [Google Scholar] [CrossRef]
- Asefpour Vakilian, K. Machine Learning Improves Our Knowledge about miRNA Functions towards Plant Abiotic Stresses. Sci. Rep. 2020, 10, 3041. [Google Scholar] [CrossRef]
- Meher, P.K.; Sahu, T.K.; Gupta, A.; Kumar, A.; Rustgi, S. ASRpro: A Machine-learning Computational Model for Identifying Proteins Associated with Multiple Abiotic Stress in Plants. Plant Genom. 2022, 17, e20259. [Google Scholar] [CrossRef]
- Ma, L.; Coulter, J.; Liu, L.; Zhao, Y.; Chang, Y.; Pu, Y.; Zeng, X.; Xu, Y.; Wu, J.; Fang, Y.; et al. Transcriptome Analysis Reveals Key Cold-Stress-Responsive Genes in Winter Rapeseed (Brassica rapa L.). IJMS 2019, 20, 1071. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Wang, J.; Tang, J.; Hou, X. Identification, Evolution and Functional Inference on the Cold-Shock Domain Protein Family in Pak-Choi (Brassica rapa Ssp. Chinensis) and Chinese Cabbage (Brassica rapa Ssp. Pekinensis). J. Plant Interact. 2019, 14, 232–241. [Google Scholar] [CrossRef]
- Yuan, J.; Liu, T.; Yu, Z.; Li, Y.; Ren, H.; Hou, X.; Li, Y. Genome-Wide Analysis of the Chinese Cabbage IQD Gene Family and the Response of BrIQD5 in Drought Resistance. Plant Mol. Biol. 2019, 99, 603–620. [Google Scholar] [CrossRef]
- Chen, Z.; Liu, X.; Zhao, P.; Li, C.; Wang, Y.; Li, F.; Akutsu, T.; Bain, C.; Gasser, R.B.; Li, J.; et al. iFeatureOmega: An Integrative Platform for Engineering, Visualization and Analysis of Features from Molecular Sequences, Structural and Ligand Data Sets. Nucleic Acids Res. 2022, 50, W434–W447. [Google Scholar] [CrossRef]
- Chou, K.-C. Using Amphiphilic Pseudo Amino Acid Composition to Predict Enzyme Subfamily Classes. Bioinformatics 2005, 21, 10–19. [Google Scholar] [CrossRef]
- He, Z.; Li, L.; Huang, Z.; Situ, H. Quantum-Enhanced Feature Selection with Forward Selection and Backward Elimination. Quantum Inf. Process. 2018, 17, 154. [Google Scholar] [CrossRef]
- Huang, M.-L.; Hung, Y.-H.; Lee, W.M.; Li, R.K.; Jiang, B.-R. SVM-RFE Based Feature Selection and Taguchi Parameters Optimization for Multiclass SVM Classifier. Sci. World J. 2014, 2014, 795624. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Liu, J.; Zang, L.; Xiao, T.; Zhang, X.; Li, Z.; Zhu, H.; Gao, W.; Yu, X. Integrated Machine Learning and Bioinformatic Analyses Constructed a Novel Stemness-Related Classifier to Predict Prognosis and Immunotherapy Responses for Hepatocellular Carcinoma Patients. Int. J. Biol. Sci. 2022, 18, 360–373. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Wang, C.; Ding, H.; Zou, Q. Machine Learning and Its Applications in Plant Molecular Studies. Brief. Funct. Genom. 2020, 19, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Cui, P.; Zhong, T.; Wang, Z.; Wang, T.; Zhao, H.; Liu, C.; Lu, H. Identification of Human Circadian Genes Based on Time Course Gene Expression Profiles by Using a Deep Learning Method. Biochim. Et. Biophys. Acta (BBA) Mol. Basis Dis. 2018, 1864, 2274–2283. [Google Scholar] [CrossRef] [PubMed]
- Polanski, K.; Rhodes, J.; Hill, C.; Zhang, P.; Jenkins, D.J.; Kiddle, S.J.; Jironkin, A.; Beynon, J.; Buchanan-Wollaston, V.; Ott, S.; et al. Wigwams: Identifying Gene Modules Co-Regulated across Multiple Biological Conditions. Bioinformatics 2014, 30, 962–970. [Google Scholar] [CrossRef]
- Li, X.; Liu, T.; Tao, P.; Wang, C.; Chen, L. A Highly Accurate Protein Structural Class Prediction Approach Using Auto Cross Covariance Transformation and Recursive Feature Elimination. Comput. Biol. Chem. 2015, 59, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.; Ahn, H.; Lee, S.; Lee, C.-J.; Hur, J.; Jung, W.; Kim, S. StressGenePred: A Twin Prediction Model Architecture for Classifying the Stress Types of Samples and Discovering Stress-Related Genes in Arabidopsis. BMC Genom. 2019, 20, 949. [Google Scholar] [CrossRef] [PubMed]
- Sohail, A.; Arif, F. Supervised and Unsupervised Algorithms for Bioinformatics and Data Science. Prog. Biophys. Mol. Biol. 2020, 151, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Chakraborty, S. Support Vector Machine in Structural Reliability Analysis: A Review. Reliab. Eng. Syst. Saf. 2023, 233, 109126. [Google Scholar] [CrossRef]
- Li, Z. Extracting Spatial Effects from Machine Learning Model Using Local Interpretation Method: An Example of SHAP and XGBoost. Comput. Environ. Urban. Syst. 2022, 96, 101845. [Google Scholar] [CrossRef]
- Wang, H.; Wang, G. Improving Random Forest Algorithm by Lasso Method. J. Stat. Comput. Simul. 2021, 91, 353–367. [Google Scholar] [CrossRef]
- Ngo, G.; Beard, R.; Chandra, R. Evolutionary Bagging for Ensemble Learning. Neurocomputing 2022, 510, 1–14. [Google Scholar] [CrossRef]
- Mienye, I.D.; Sun, Y. A Survey of Ensemble Learning: Concepts, Algorithms, Applications, and Prospects. IEEE Access 2022, 10, 99129–99149. [Google Scholar] [CrossRef]
- Abraham, A. Machine Learning for Neuroimaging with Scikit-Learn. Front. Neuroinform. 2014, 8, 71792. [Google Scholar] [CrossRef] [PubMed]
- Jiang, G.; Wang, W. Error Estimation Based on Variance Analysis of k -Fold Cross-Validation. Pattern Recognit. 2017, 69, 94–106. [Google Scholar] [CrossRef]
- Canbek, G.; Taskaya Temizel, T.; Sagiroglu, S. BenchMetrics: A Systematic Benchmarking Method for Binary Classification Performance Metrics. Neural Comput. Applic 2021, 33, 14623–14650. [Google Scholar] [CrossRef]
- Xiong, H.; Capurso, D.; Sen, Ś.; Segal, M.R. Sequence-Based Classification Using Discriminatory Motif Feature Selection. PLoS ONE 2011, 6, e27382. [Google Scholar] [CrossRef]
- Bailey, T.L.; Johnson, J.; Grant, C.E.; Noble, W.S. The MEME Suite. Nucleic Acids Res. 2015, 43, W39–W49. [Google Scholar] [CrossRef]
- Lu, X.; Cheng, Y.; Gao, M.; Li, M.; Xu, X. Molecular Characterization, Expression Pattern and Function Analysis of Glycine-Rich Protein Genes Under Stresses in Chinese Cabbage (Brassica rapa L. Ssp. Pekinensis). Front. Genet. 2020, 11, 774. [Google Scholar] [CrossRef] [PubMed]
- Kreps, J.A.; Wu, Y.; Chang, H.-S.; Zhu, T.; Wang, X.; Harper, J.F. Transcriptome Changes for Arabidopsis in Response to Salt, Osmotic, and Cold Stress. Plant Physiol. 2002, 130, 2129–2141. [Google Scholar] [CrossRef] [PubMed]
- Skirpan, A.L.; McCubbin, A.G.; Ishimizu, T.; Wang, X.; Hu, Y.; Dowd, P.E.; Ma, H.; Kao, T. Isolation and Characterization of Kinase Interacting Protein 1, a Pollen Protein That Interacts with the Kinase Domain of PRK1, a Receptor-Like Kinase of Petunia. Plant Physiol. 2001, 126, 1480–1492. [Google Scholar] [CrossRef]
- Ji, H.; Liu, J.; Chen, Y.; Yu, X.; Luo, C.; Sang, L.; Zhou, J.; Liao, H. Bioinformatic Analysis of Codon Usage Bias of HSP20 Genes in Four Cruciferous Species. Plants 2024, 13, 468. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Fu, Y.; Lee, Y.J.; Chern, M.; Li, M.; Cheng, M.; Dong, H.; Yuan, Z.; Gui, L.; Yin, J.; et al. The PGS1 Basic Helix-loop-helix Protein Regulates Fl3 to Impact Seed Growth and Grain Yield in Cereals. Plant Biotechnol. J. 2022, 20, 1311–1326. [Google Scholar] [CrossRef]
- Hong, J.K.; Je, J.; Song, C.; Hwang, J.E.; Lee, Y.-H.; Lim, C.O. Biochemical Analysis of a Chinese Cabbage Phytocystatin-1. Genes. Genom. 2012, 34, 13–18. [Google Scholar] [CrossRef]
- Yu, J.; Gao, L.; Liu, W.; Song, L.; Xiao, D.; Liu, T.; Hou, X.; Zhang, C. Transcription Coactivator ANGUSTIFOLIA3 (AN3) Regulates Leafy Head Formation in Chinese Cabbage. Front. Plant Sci. 2019, 10, 520. [Google Scholar] [CrossRef] [PubMed]
- Saha, G.; Park, J.-I.; Jung, H.-J.; Ahmed, N.U.; Kayum, M.A.; Chung, M.-Y.; Hur, Y.; Cho, Y.-G.; Watanabe, M.; Nou, I.-S. Genome-Wide Identification and Characterization of MADS-Box Family Genes Related to Organ Development and Stress Resistance in Brassica Rapa. BMC Genom. 2015, 16, 178. [Google Scholar] [CrossRef] [PubMed]
- Saha, G.; Park, J.-I.; Ahmed, N.U.; Kayum, M.A.; Kang, K.-K.; Nou, I.-S. Characterization and Expression Profiling of MYB Transcription Factors against Stresses and during Male Organ Development in Chinese Cabbage (Brassica rapa Ssp. Pekinensis). Plant Physiol. Biochem. 2016, 104, 200–215. [Google Scholar] [CrossRef] [PubMed]
- Ernst, M.; Robertson, J.L. The Role of the Membrane in Transporter Folding and Activity. J. Mol. Biol. 2021, 433, 167103. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Hua, Y.; Zhou, T.; Liu, Y.; Huang, J.; Yue, C. Global Landscapes of the Na+/H+ Antiporter (NHX) Family Members Uncover Their Potential Roles in Regulating the Rapeseed Resistance to Salt Stress. Int. J. Mol. Sci. 2020, 21, 3429. [Google Scholar] [CrossRef] [PubMed]
- Orovwode, H.; Ibukun, O.; Abubakar, J.A. A Machine Learning-Driven Web Application for Sign Language Learning. Front. Artif. Intell. 2024, 7, 1297347. [Google Scholar] [CrossRef]
- Cai, Z.; Tang, Q.; Song, P.; Tian, E.; Yang, J.; Jia, G. The m6A Reader ECT8 Is an Abiotic Stress Sensor That Accelerates mRNA Decay in Arabidopsis. Plant Cell 2024, 36, 2908–2926. [Google Scholar] [CrossRef]
- Murmu, S.; Sinha, D.; Chaurasia, H.; Sharma, S.; Das, R.; Jha, G.K.; Archak, S. A Review of Artificial Intelligence-Assisted Omics Techniques in Plant Defense: Current Trends and Future Directions. Front. Plant Sci. 2024, 15, 1292054. [Google Scholar] [CrossRef] [PubMed]
- Koh, E.; Sunil, R.S.; Lam, H.Y.I.; Mutwil, M. Confronting the Data Deluge: How Artificial Intelligence Can Be Used in the Study of Plant Stress. Comput. Struct. Biotechnol. J. 2024, 23, 3454–3466. [Google Scholar] [CrossRef]
- Zeng, H.; Zhuang, Y.; Yan, X.; He, X.; Qiu, Q.; Liu, W.; Zhang, Y. Machine Learning-Based Identification of Novel Hub Genes Associated with Oxidative Stress in Lupus Nephritis: Implications for Diagnosis and Therapeutic Targets. Lupus Sci. Med. 2024, 11, e001126. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Gao, Y.; Lu, Q.; Zhang, R.; Gui, J.; Liu, X.; Yue, Z. RiceSNP-BST: A Deep Learning Framework for Predicting Biotic Stress–Associated SNPs in Rice. Brief. Bioinform. 2024, 25, bbae599. [Google Scholar] [CrossRef] [PubMed]
- Monem, S.; Hassanien, A.E.; Abdel-Hamid, A.H. A Multi-View Feature Representation for Predicting Drugs Combination Synergy Based on Ensemble and Multi-Task Attention Models. J. Cheminform 2024, 16, 110. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Zhao, H.; Luo, Z.; Lin, Y.; Feng, W.; Li, Y.; Kong, F.; Li, X.; Fang, C.; Wang, X. SoyDNGP: A Web-Accessible Deep Learning Framework for Genomic Prediction in Soybean Breeding. Brief. Bioinform. 2023, 24, bbad349. [Google Scholar] [CrossRef] [PubMed]

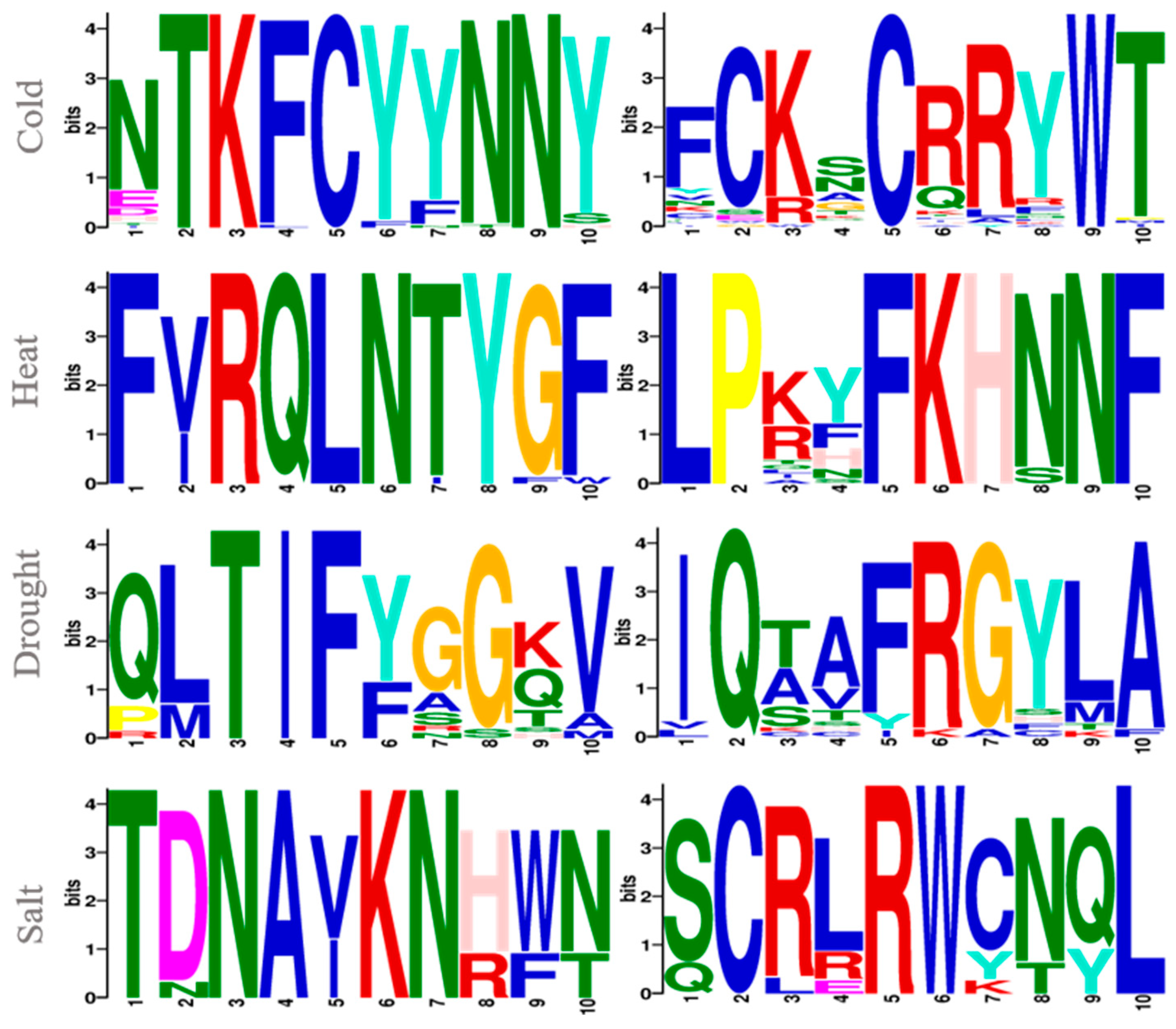


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Cold | Heat | Drought | Salt |
|---|---|---|---|---|
| Single stress | 728 | 663 | 449 | 328 |
| Positive set | 527 | 515 | 409 | 239 |
| Negative set | 1163 | 1175 | 1281 | 1451 |
| Unlabeled set | 40,291 | 40,356 | 40,570 | 40,691 |
| Stress | Feature | Model | Accuracy (%) | auROC (%) | auPRC (%) |
|---|---|---|---|---|---|
| Cold | CKSAAP | RF | 74.26 | 81.42 | 70.92 |
| DDE | BAG | 76.92 | 77.50 | 63.92 | |
| CKSAAP + DDE | RF | 73.67 | 80.86 | 71.22 | |
| Heat | CKSAAP | GBDT | 82.84 | 87.92 | 81.76 |
| DDE | RF | 73.08 | 79.59 | 65.73 | |
| CKSAAP + DDE | GBDT | 77.51 | 85.72 | 76.66 | |
| Drought | CKSAAP | XGB | 81.36 | 80.85 | 63.11 |
| DDE | SVM | 79.88 | 78.78 | 56.21 | |
| CKSAAP + DDE | RF | 75.74 | 80.62 | 62.54 | |
| Salt | CKSAAP | GBDT | 88.48 | 88.87 | 79.63 |
| DDE | BAG | 89.04 | 87.97 | 74.29 | |
| CKSAAP + DDE | RF | 83.15 | 88.15 | 76.79 |
| Stress | Feature | Model | Number of Features |
|---|---|---|---|
| Cold | CKSAAP | RF | 1045 |
| Heat | CKSAAP | GBDT | 1224 |
| Drought | CKSAAP | XGB | 1016 |
| Salt | CKSAAP | GBDT | 1018 |
| Rank | Cold | Likeliness | Heat | Likeliness | Drought | Likeliness | Salt | Likeliness |
|---|---|---|---|---|---|---|---|---|
| 1 | Bra017529 | 0.80 | Bra009415 | 0.99 | Bra020398 | 0.99 | Bra039970 | 0.99 |
| 2 | Bra023050 | 0.79 | Bra013731 | 0.99 | Bra002679 | 0.98 | Bra026062 | 0.99 |
| 3 | Bra025001 | 0.79 | Bra031896 | 0.99 | Bra031714 | 0.97 | Bra021958 | 0.99 |
| 4 | Bra010944 | 0.77 | Bra023806 | 0.99 | Bra036963 | 0.97 | Bra022658 | 0.99 |
| 5 | Bra023102 | 0.75 | Bra015720 | 0.99 | Bra008802 | 0.97 | Bra023592 | 0.99 |
| 6 | Bra022197 | 0.75 | Bra020597 | 0.99 | Bra040856 | 0.97 | Bra025086 | 0.99 |
| 7 | Bra013175 | 0.74 | Bra035105 | 0.99 | Bra016377 | 0.96 | Bra000458 | 0.99 |
| 8 | Bra002647 | 0.72 | Bra025461 | 0.99 | Bra019932 | 0.96 | Bra018594 | 0.99 |
| 9 | Bra023103 | 0.72 | Bra019045 | 0.99 | Bra007363 | 0.96 | Bra025850 | 0.98 |
| 10 | Bra032483 | 0.71 | Bra009416 | 0.99 | Bra034636 | 0.96 | Bra022945 | 0.98 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
You, X.; Shu, Y.; Ni, X.; Lv, H.; Luo, J.; Tao, J.; Bai, G.; Feng, S. MLAS: Machine Learning-Based Approach for Predicting Abiotic Stress-Responsive Genes in Chinese Cabbage. Horticulturae 2025, 11, 44. https://doi.org/10.3390/horticulturae11010044
You X, Shu Y, Ni X, Lv H, Luo J, Tao J, Bai G, Feng S. MLAS: Machine Learning-Based Approach for Predicting Abiotic Stress-Responsive Genes in Chinese Cabbage. Horticulturae. 2025; 11(1):44. https://doi.org/10.3390/horticulturae11010044
Chicago/Turabian StyleYou, Xiong, Yiting Shu, Xingcheng Ni, Hengmin Lv, Jian Luo, Jianping Tao, Guanghui Bai, and Shusu Feng. 2025. "MLAS: Machine Learning-Based Approach for Predicting Abiotic Stress-Responsive Genes in Chinese Cabbage" Horticulturae 11, no. 1: 44. https://doi.org/10.3390/horticulturae11010044
APA StyleYou, X., Shu, Y., Ni, X., Lv, H., Luo, J., Tao, J., Bai, G., & Feng, S. (2025). MLAS: Machine Learning-Based Approach for Predicting Abiotic Stress-Responsive Genes in Chinese Cabbage. Horticulturae, 11(1), 44. https://doi.org/10.3390/horticulturae11010044