
Bacterial and Fungal Communities of Table Grape Skins in Shanghai

 ,
, 
 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
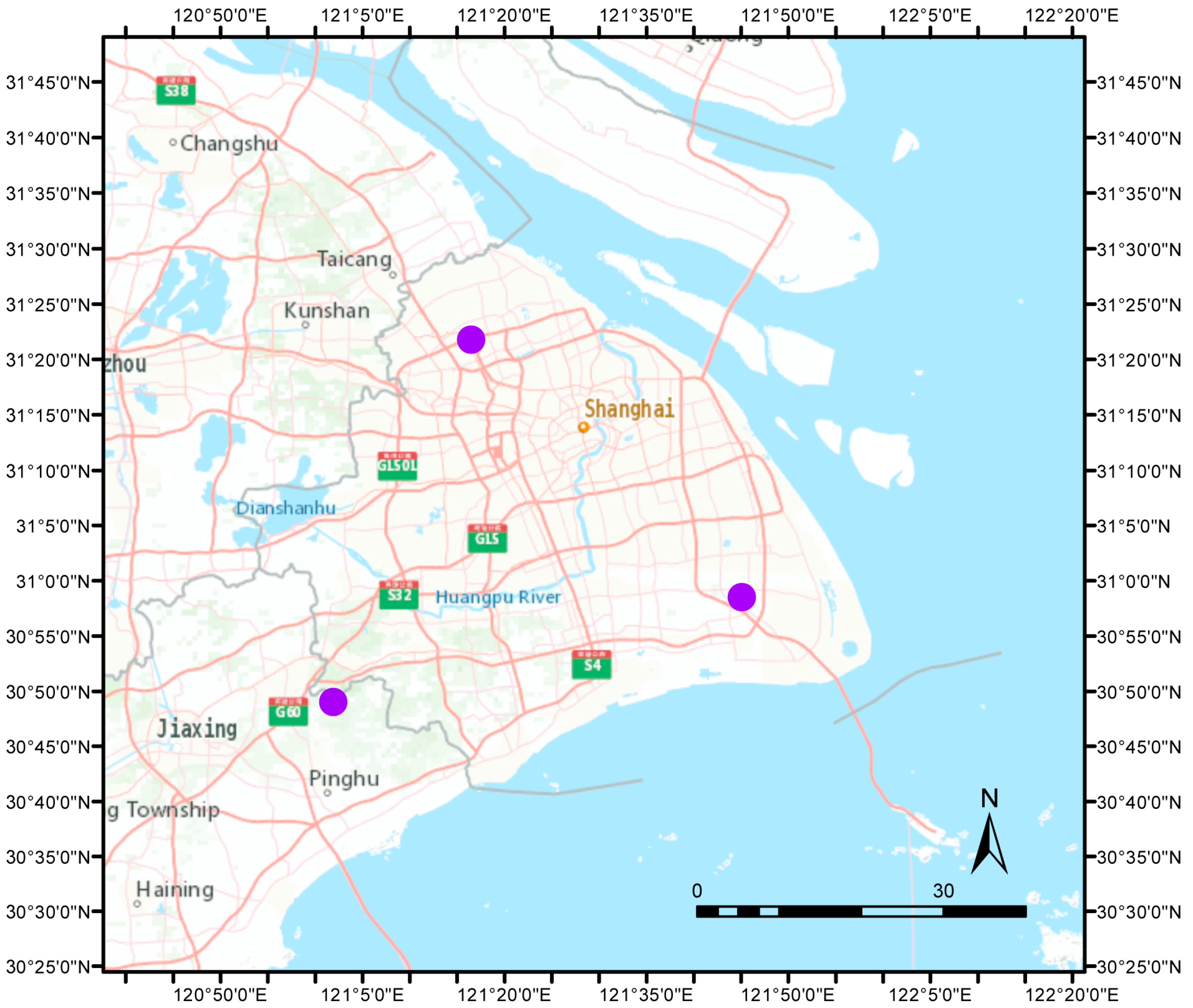
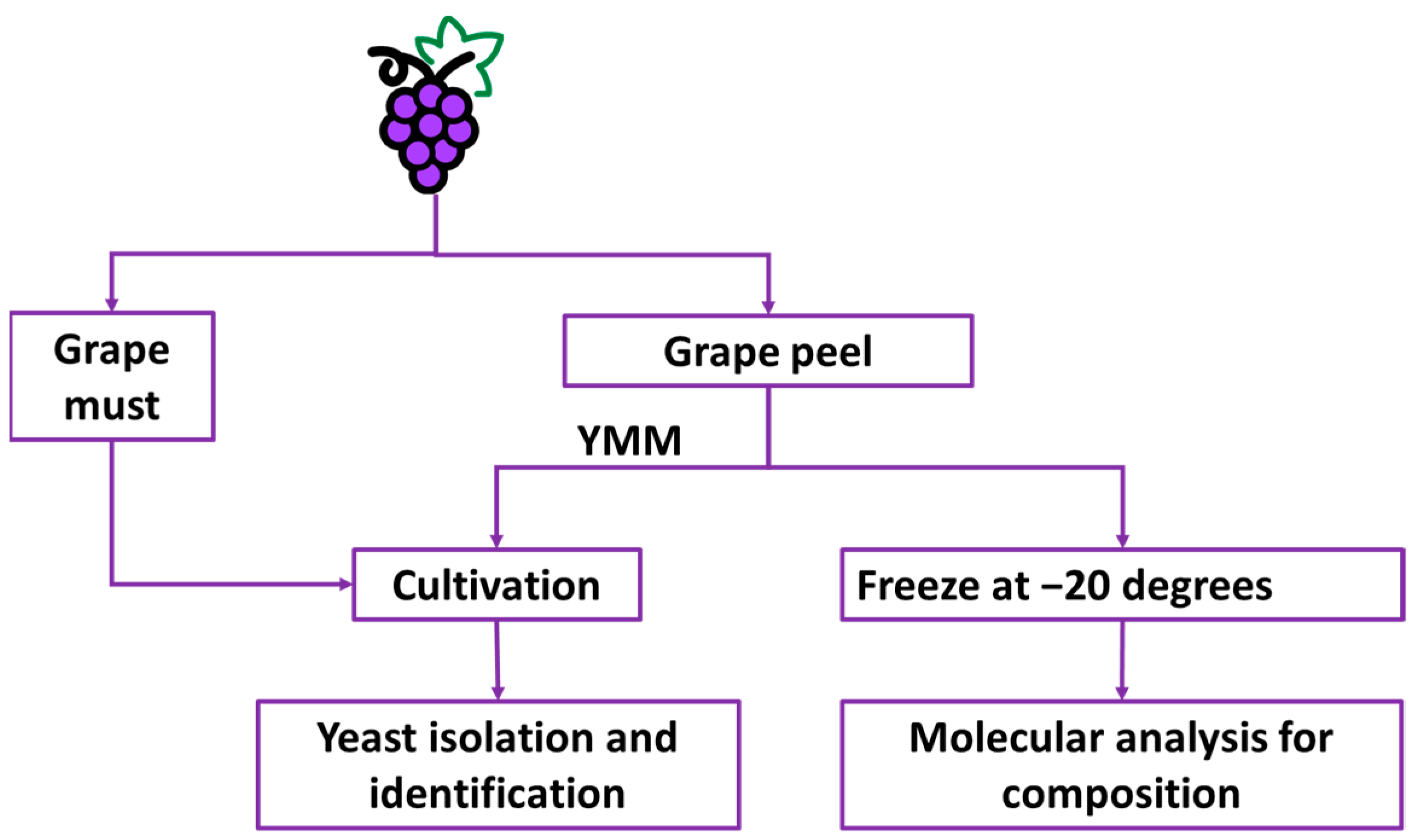
2.1. Study Area and Grape Sampling
2.2. Yeast Cultivation
2.3. Yeast Identification
2.4. DNA Extraction, Amplification, and Sequencing
2.5. Sequence Analysis and Bacterial Functional Prediction
2.6. Statistical Analysis
3. Results
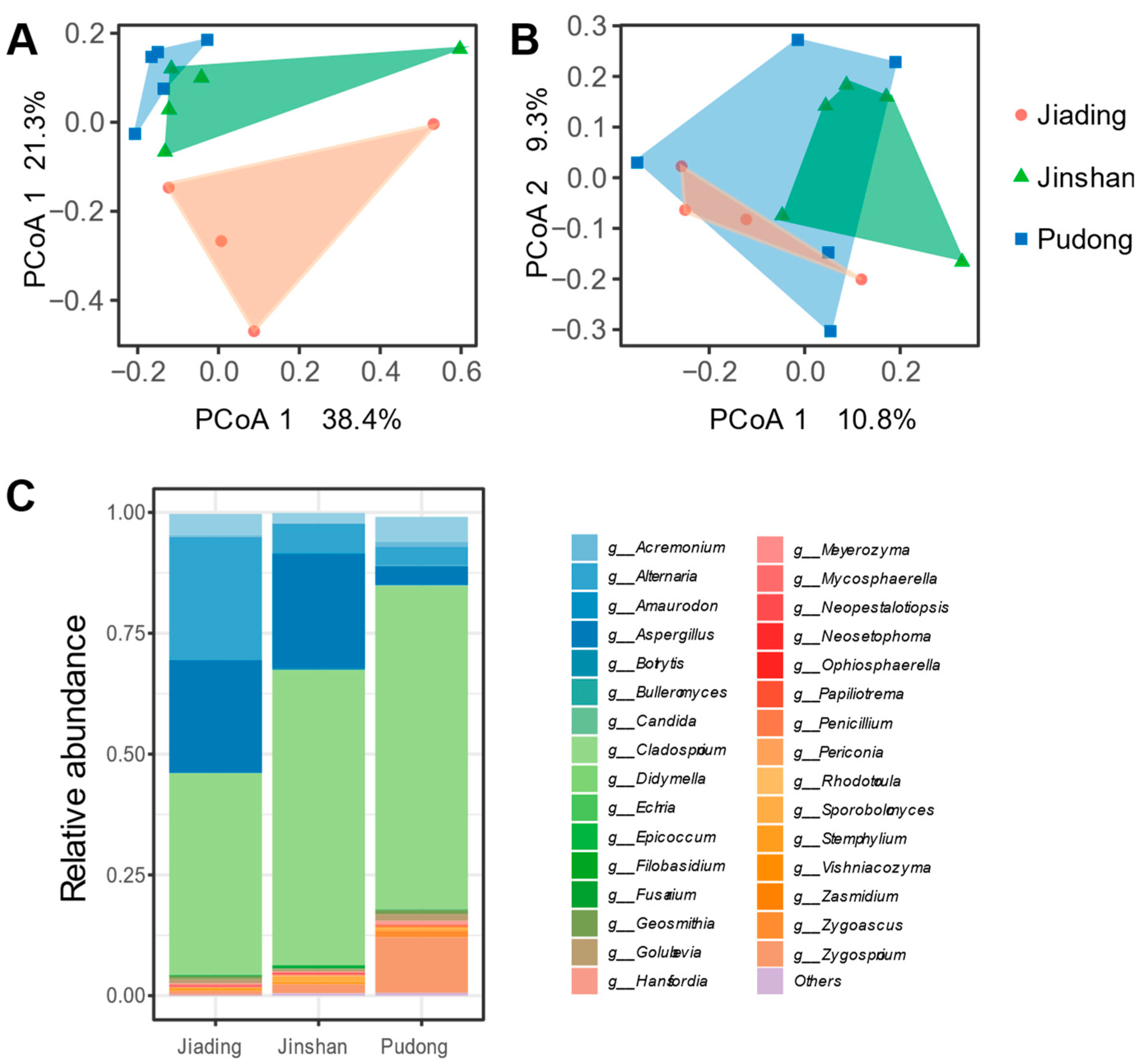
3.1. General Characteristics of Table Grape Microbiota
3.2. Potential Contributors to Grape Skin Mycobiota and Microbiota
3.3. Mycobiota and Microbiota of Different Table Grapes
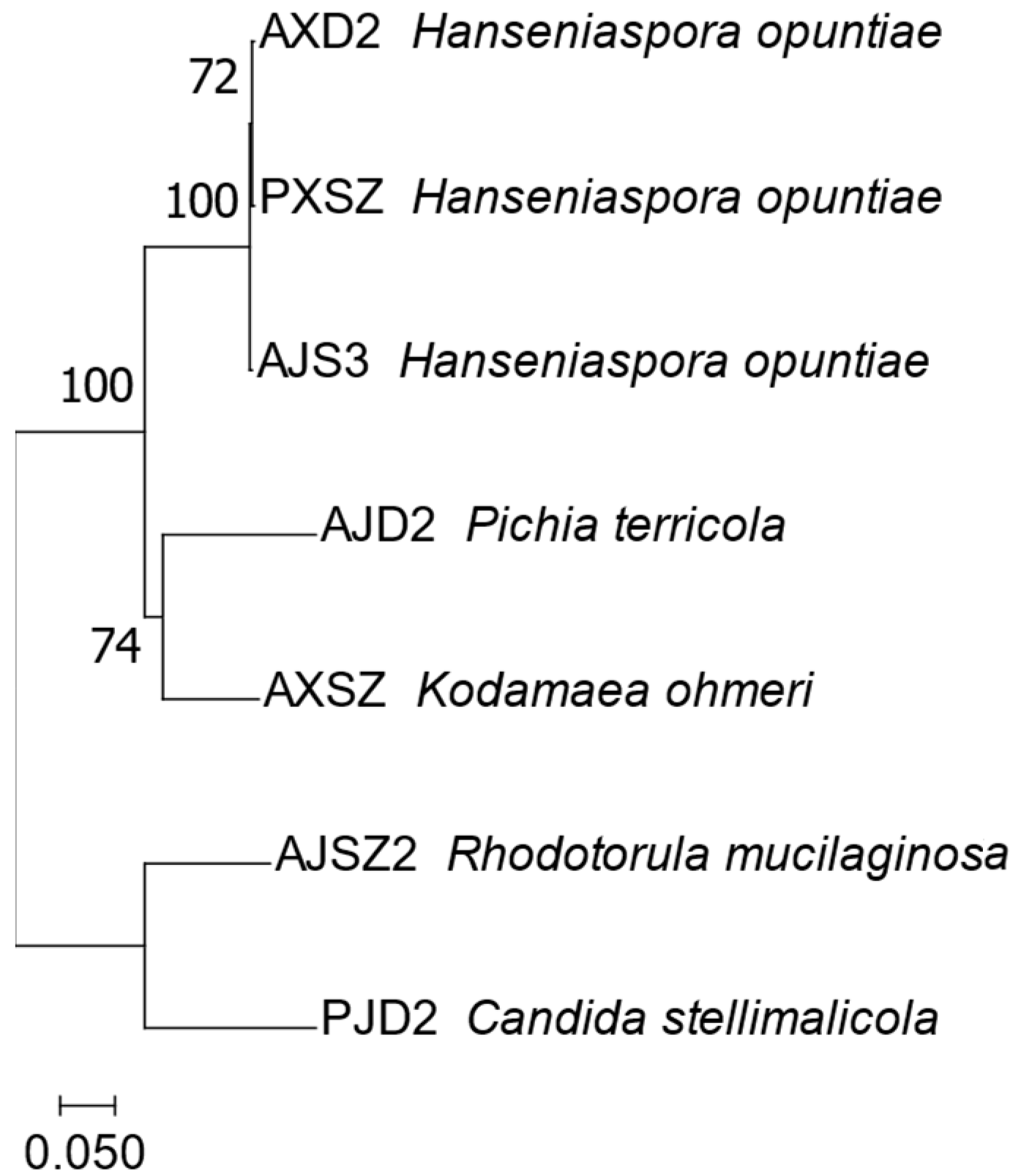
3.4. Cultivated Yeasts
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Creasy, G.L.; Creasy, L.L. Grapes (Crop Production Science in Horticulture), 2nd ed.; CABI: Wallingford, UK, 2018; Volume 27. [Google Scholar]
- Ozawa, T.; Amemiya, T.; Sato, T.; Furuya, J.; Furuya, K.; Miyake, M.; Saito, N.; Hirabayashi, T.; Mochiduki, T.; Kondo, M. Summer Black; A New Seedless Grape; Bulletin of the Yamanashi Fruit Tree Experiment Station; Yamanashi Fruit Tree Experiment Station: Yamanashi, Japan, 2000. [Google Scholar]
- Yamada, M.; Sato, A. Advances in table grape breeding in Japan. Breed. Sci. 2016, 66, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Sochorova, L.; Prusova, B.; Cebova, M.; Jurikova, T.; Mlcek, J.; Adamkova, A.; Nedomova, S.; Baron, M.; Sochor, J. Health effects of grape seed and skin extracts and their influence on biochemical markers. Molecules 2020, 25, 5311. [Google Scholar] [CrossRef] [PubMed]
- Barata, A.; Malfeito-Ferreira, M.; Loureiro, V. The microbial ecology of wine grape berries. Int. J. Food Microbiol. 2012, 153, 243–259. [Google Scholar] [CrossRef] [PubMed]
- Vitulo, N.; Lemos, W.J.F., Jr.; Calgaro, M.; Confalone, M.; Felis, G.E.; Zapparoli, G.; Nardi, T. Bark and grape microbiome of Vitis vinifera: Influence of geographic patterns and agronomic management on bacterial diversity. Front. Microbiol. 2019, 9, 3203. [Google Scholar] [CrossRef]
- Mezzasalma, V.; Sandionigi, A.; Guzzetti, L.; Galimberti, A.; Grando, M.S.; Tardaguila, J.; Labra, M. Geographical and cultivar features differentiate grape microbiota in Northern Italy and Spain vineyards. Front. Microbiol. 2018, 9, 946. [Google Scholar] [CrossRef] [PubMed]
- Bozoudi, D.; Tsaltas, D. Grape microbiome: Potential and opportunities as a source of starter cultures. In Grape and Wine Biotechnology; InTech: Houston, TX, USA, 2016. [Google Scholar]
- Morgan, H.H.; Toit, M.D.; Setati, M.E. The grapevine and wine microbiome: Insights from high-throughput amplicon sequencing. Front. Microbiol. 2017, 8, 820. [Google Scholar] [CrossRef] [PubMed]
- Begerow, D.; Nilsson, H.; Unterseher, M.; Maier, W. Current state and perspectives of fungal DNA barcoding and rapid identification procedures. Appl. Microbiol. Biotechnol. 2010, 87, 99–108. [Google Scholar] [CrossRef]
- Schoch, C.L.; Seifert, K.A.; Huhndorf, S.; Robert, V.; Spouge, J.L.; Levesque, C.A.; Chen, W.; Consortium, F.B.; List, F.B.C.A.; Bolchacova, E. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc. Natl. Acad. Sci. USA 2012, 109, 6241–6246. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Wei, R.; Wang, L.; Wang, W.; Wang, H.; Li, H. Exploring the ecological characteristics of natural microbial communities along the continuum from grape berries to winemaking. Food Res. Int. 2023, 167, 112718. [Google Scholar] [CrossRef]
- Gao, F.; Chen, J.; Xiao, J.; Cheng, W.; Zheng, X.; Wang, B.; Shi, X. Microbial community composition on grape surface controlled by geographical factors of different wine regions in Xinjiang, China. Food Res. Int. 2019, 122, 348–360. [Google Scholar] [CrossRef]
- González-Alonso, I.; Walker, M.E.; Vallejo-Pascual, M.-E.; Naharro-Carrasco, G.; Jiranek, V. Capturing yeast associated with grapes and spontaneous fermentations of the Negro Saurí minority variety from an experimental vineyard near León. Sci. Rep. 2021, 11, 3748. [Google Scholar] [CrossRef] [PubMed]
- Parada, A.E.; Needham, D.M.; Fuhrman, J.A. Every base matters: Assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ. Microbiol. 2016, 18, 1403–1414. [Google Scholar] [CrossRef] [PubMed]
- Apprill, A.; McNally, S.; Parsons, R.; Weber, L. Minor revision to V4 region SSU rRNA 806R gene primer greatly increases detection of SAR11 bacterioplankton. Aquat. Microb. Ecol. 2015, 75, 129–137. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.W.; Tsai, P.; Anfang, N.; Ross, H.A.; Goddard, M.R. Pyrosequencing reveals regional differences in fruit-associated fungal communities. Environ. Microbiol. 2014, 16, 2848–2858. [Google Scholar] [CrossRef] [PubMed]
- Bettenfeld, P.; Cadena, I.C.J.; Jacquens, L.; Fernandez, O.; Fontaine, F.; van Schaik, E.; Courty, P.E.; Trouvelot, S. The microbiota of the grapevine holobiont: A key component of plant health. J. Adv. Res. 2022, 40, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Bellí, N.; Marín, S.; Coronas, I.; Sanchis, V.; Ramos, A.J. Skin damage, high temperature and relative humidity as detrimental factors for Aspergillus carbonarius infection and ochratoxin A production in grapes. Food Control. 2007, 18, 1343–1349. [Google Scholar] [CrossRef]
- Kamilari, E.; Mina, M.; Karallis, C.; Tsaltas, D. Metataxonomic analysis of grape microbiota during wine fermentation reveals the distinction of xyprus regional terroirs. Front. Microbiol. 2021, 12, 726483. [Google Scholar] [CrossRef]
- Wirth, F.; Goldani, L.Z. Epidemiology of Rhodotorula: An emerging pathogen. Interdiscip. Perspect. Infect. Dis. 2012, 2012, 465717. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Li, Y.; Kudinha, T.; Xu, Y.; Liu, Z. Kodamaea ohmeri as an emerging human pathogen: A review and update. Front. Microbiol. 2021, 12, 736582. [Google Scholar] [CrossRef] [PubMed]
- Ioannou, P.; Papakitsou, I. Kodamaea ohmeri infections in humans: A systematic review. Mycoses 2020, 63, 636–643. [Google Scholar] [CrossRef] [PubMed]
- Kaur, R.; Wadhwa, A.; Agarwal, S.K. Rhodotorula mucilaginosa: An unusual cause of oral ulcers in AIDS patients. AIDS 2007, 21, 1068–1069. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Shin, J.H.; Kim, M.N.; Jung, S.I.; Park, K.H.; Cho, D.; Kee, S.J.; Shin, M.G.; Suh, S.P.; Ryang, D.W. Kodamaea ohmeri isolates from patients in a university hospital: Identification, antifungal susceptibility, and pulsed-field gel electrophoresis analysis. J. Clin. Microbiol. 2007, 45, 1005–1010. [Google Scholar] [CrossRef]
- Otag, F.; Kuyucu, N.; Erturan, Z.; Sen, S.; Emekdas, G.; Sugita, T. An outbreak of Pichia ohmeri infection in the paediatric intensive care unit: Case reports and review of the literature. Mycoses 2005, 48, 265–926. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
An, R.; Ma, Q.; Sun, S.; Zhang, H.; Lyu, C.; Wang, D.; Song, S. Bacterial and Fungal Communities of Table Grape Skins in Shanghai. Horticulturae 2024, 10, 560. https://doi.org/10.3390/horticulturae10060560
An R, Ma Q, Sun S, Zhang H, Lyu C, Wang D, Song S. Bacterial and Fungal Communities of Table Grape Skins in Shanghai. Horticulturae. 2024; 10(6):560. https://doi.org/10.3390/horticulturae10060560
Chicago/Turabian StyleAn, Ran, Qingchuan Ma, Sijie Sun, Hengcheng Zhang, Chenang Lyu, Dapeng Wang, and Shiren Song. 2024. "Bacterial and Fungal Communities of Table Grape Skins in Shanghai" Horticulturae 10, no. 6: 560. https://doi.org/10.3390/horticulturae10060560
APA StyleAn, R., Ma, Q., Sun, S., Zhang, H., Lyu, C., Wang, D., & Song, S. (2024). Bacterial and Fungal Communities of Table Grape Skins in Shanghai. Horticulturae, 10(6), 560. https://doi.org/10.3390/horticulturae10060560