
Assessment of Simple Sequence Repeat (SSR) Markers Derived from Whole-Genome Sequence (WGS) of Mungbean (Vigna radiata L. Wilczek): Cross-Species Transferability and Population Genetic Studies in Vigna Species

 ,
,  , , , ,
, , , ,
Abstract
:Simple Summary
Abstract
1. Introduction
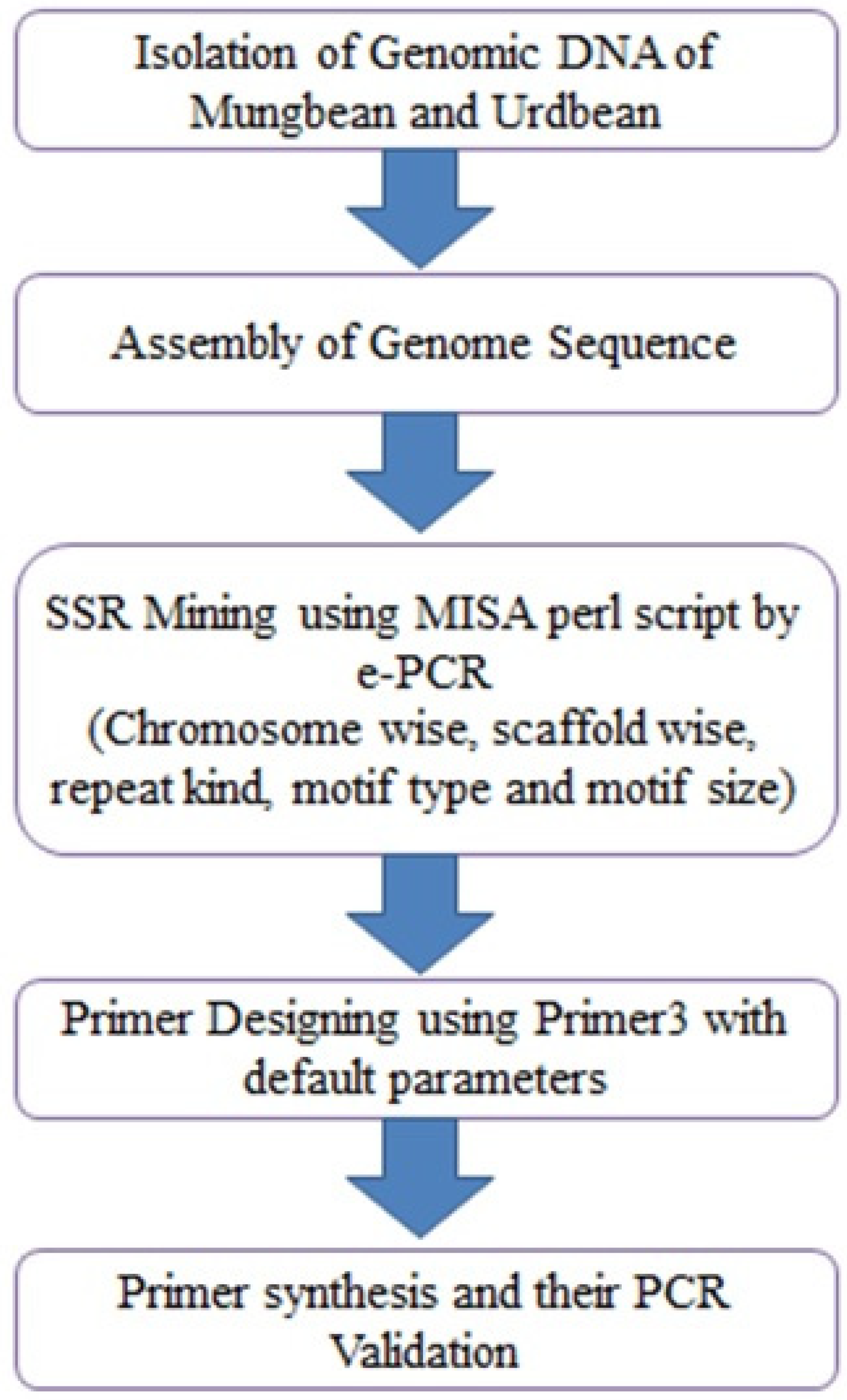
2. Materials and Methods
2.1. Plant Material
2.2. DNA Extraction and Quantification
2.3. SSR Marker Design
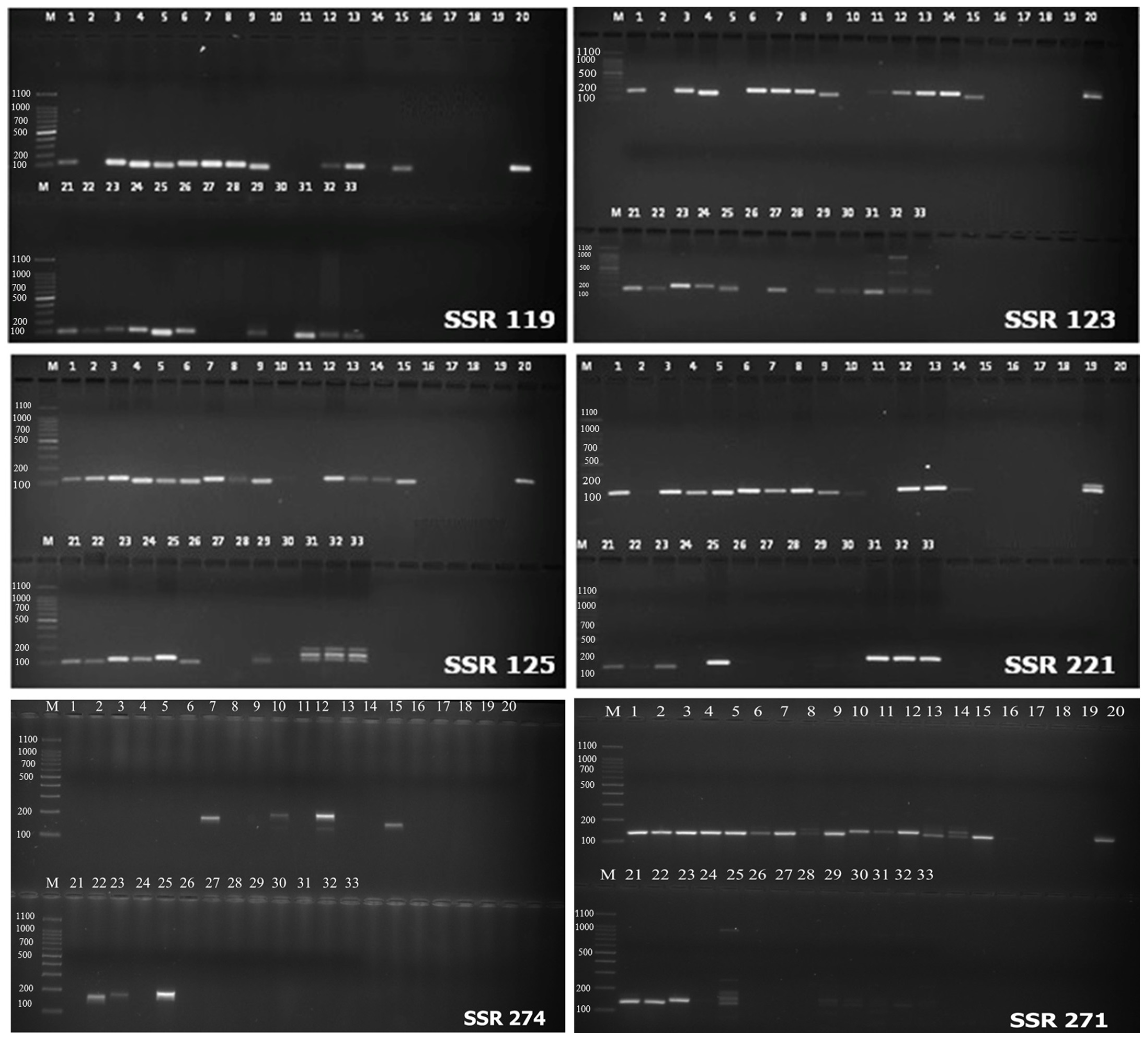
2.4. SSR Validation
2.5. Genetic Diversity, AMOVA, and PCoA in Vigna Species
2.6. Population Structure Analysis
2.7. Data Analysis
2.7.1. Polymorphic Information Content (PIC)
2.7.2. Effective Multiplex Ratio (EMR)
2.7.3. Marker Index (MI)
2.7.4. Resolving Power (RP)
3. Results
3.1. WGS-Based SSR Marker Development
3.2. Validation of SSR Markers in Vigna Species Accessions for Transferability Studies
3.3. SSR Marker Analysis
3.4. Genetic Diversity and Relationship among Different Vigna Species
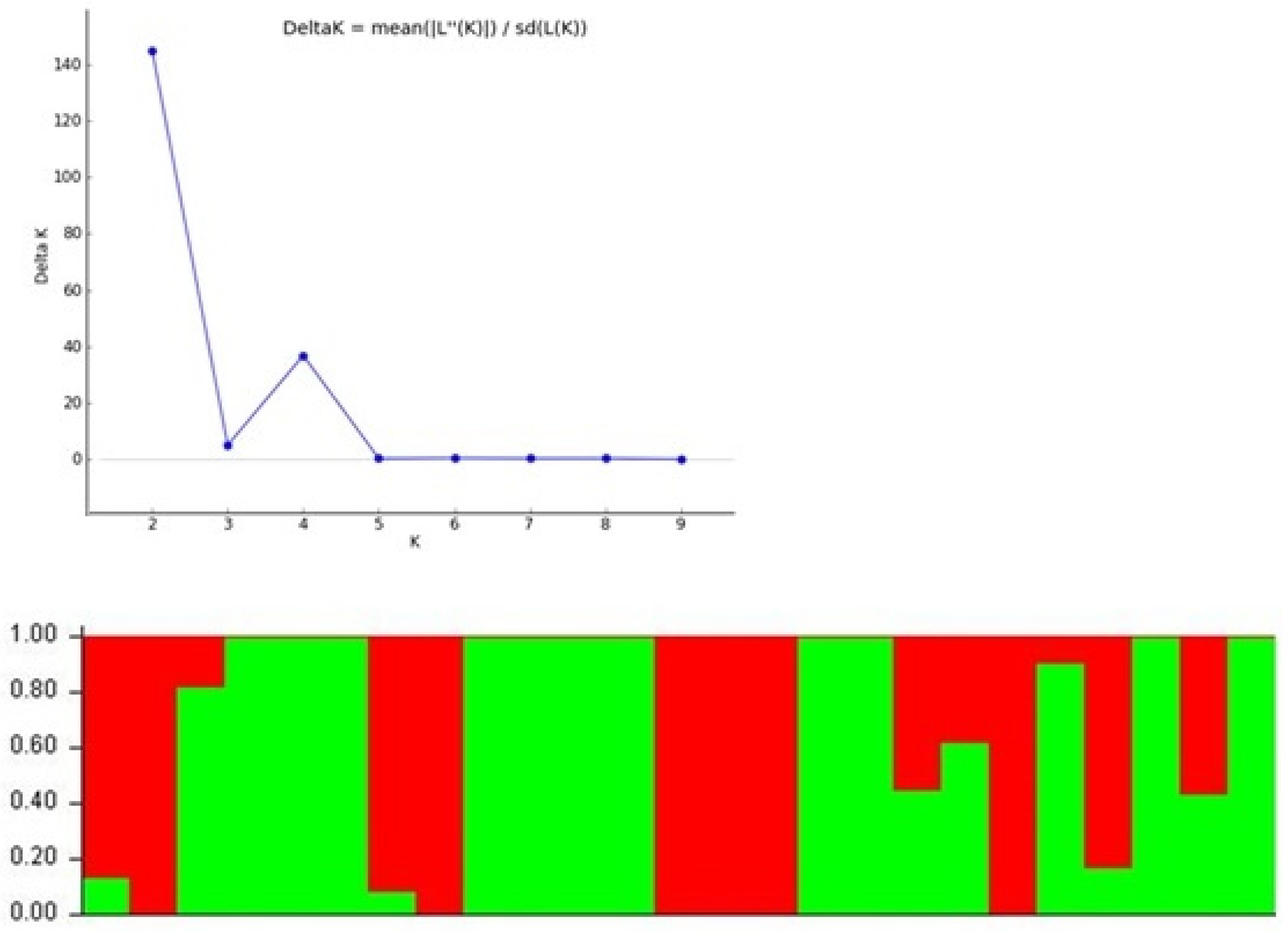
3.5. Population Structure Analysis
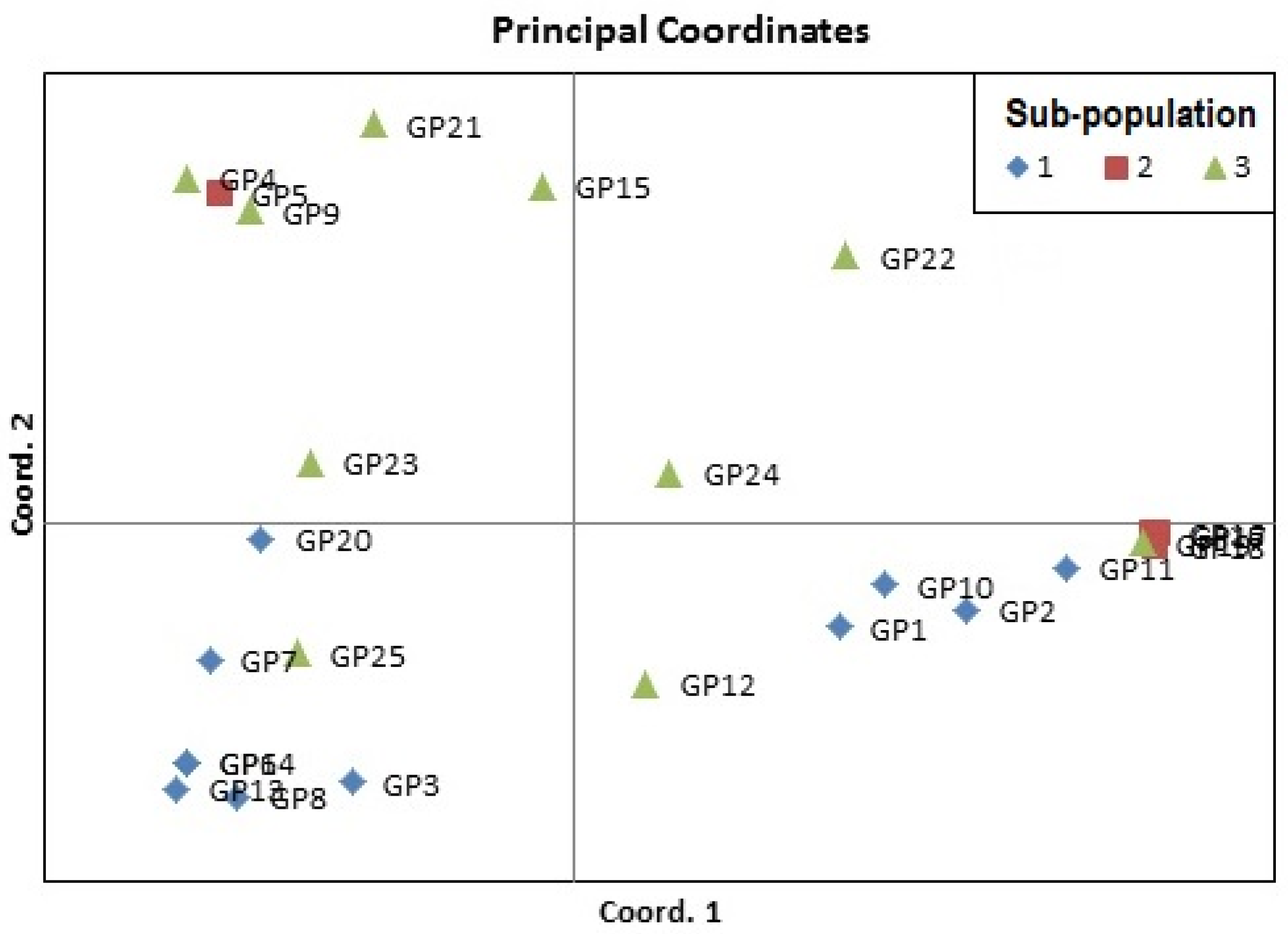
3.6. Analysis of Molecular Variance (AMOVA) and Principal Coordinate Analysis (PCoA)
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schrire, B.D. Tribe Phaseoleae. In Legumes of the World; Lewis, G., Schrire, B., Mackinder, B., Lock, M., Eds.; Royal Botanic Gardens: Kew, UK, 2005; pp. 393–431. [Google Scholar]
- Dikshit, H.K.; Singh, D.; Singh, A.; Jain, N.; Kumari, J.; Sharma, T.R. Utility of adzuki bean [Vigna angularis (Willd.) Ohwi & Ohashi] simple sequence repeat (SSR) markers in genetic analysis of mungbean and related Vigna spp. Afr. J. Biotechnol. 2012, 11, 13261–13268. [Google Scholar] [CrossRef]
- Akbar, W.; Akhtar, M.A.; Murtaza, G.; Hussain, A.; Javed, H.M.; Arshad, M.; Maqbool, M.A. Mungbean Yellow Mosaic Disease and its Management. J. Agric. Basic Sci. 2019, 4, 34–44. [Google Scholar]
- Kang, Y.J.; Kim, S.K.; Kim, M.Y.; Lestari, P.; Kim, K.H.; Ha, B.K.; Jun, T.H.; Hwang, W.J.; Lee, T.; Lee, J.; et al. Genome sequence of mungbean and insights into evolution within Vigna species. Nat. Commun. 2014, 5, 5443. [Google Scholar] [CrossRef] [PubMed]
- Rohilla, V.; Yadav, R.K.; Poonia, A.; Sheoran, R.; Kumari, G.; Shanmugavadivel, P.S.; Pratap, A. Association Mapping for Yield Attributing Traits and Yellow Mosaic Disease Resistance in Mung Bean [Vigna radiata (L.) Wilczek]. Front. Plant Sci. 2022, 12, 749439. [Google Scholar] [CrossRef] [PubMed]
- Hinz, R.; Sulser, T.B.; Hüfner, R.; Mason-D’Croz, D.; Dunston, S.; Nautiyal, S.; Ringler, C.; Schuengel, J.; Tikhile, P.; Wimmer, F.; et al. Agricultural development and land use change in India: A scenario analysis of trade-offs between UN sustainable development goals (SDGs). Earth’s Future 2020, 8, e2019EF001287. [Google Scholar] [CrossRef]
- Saini, P. Inheritance Studies and Mapping of Yellow Mosaic Disease Resistance in an Interspecific Cross of Mungbean (Vigna radiata (L.) Wilczek) and Urdbean (Vigna mungo (L.) Hepper). Ph.D. Dissertation, Punjab Agricultural University, Ludhiana, India, 2020; pp. 1–150. [Google Scholar]
- Somta, P.; Musch, W.; Kongsamai, B.; Chanprame, S.; Nakasathien, S.; Toojinda, T.; Sorajjapinun, W.; Seehalak, W.; Tragoonrung, S.; Srinives, P. New microsatellite markers isolated from mungbean (Vigna radiata (L.) Wilczek). Mol. Ecol. Resour. 2008, 8, 1155–1157. [Google Scholar] [CrossRef] [PubMed]
- Lestari, P.; Kim, S.K.; Reflinur; Kang, Y.J.; Dewi, N.; Lee, S.-H. Genetic diversity of mungbean (Vigna radiata L.) germplasm in Indonesia. Plant Genet. Resour. 2014, 12 (Suppl. S1), S91–S94. [Google Scholar] [CrossRef]
- Savithramma, D.L.; Ramakrishnan, C.K.D. Development and characterization of newly developed genomic SSR markers in Mung bean (Vigna radiata (L.) Wilczek). In Proceedings of the 2nd International Conference on Genetic & Protein Engineering, Atlanta, GA, USA, 14–16 November 2016; 43p. [Google Scholar]
- Somta, P.; Srinives, P. Genome research in mungbean [Vigna radiata (L.) Wilczek] and blackgram [V. mungo (L.) Hepper]. Sci. Asia 2007, 1, 69–74. [Google Scholar] [CrossRef]
- Chaitieng, B.; Kaga, A.; Tomooka, N.; Isemura, T.; Kuroda, Y.; Vaughan, D.A. Development of a black gram [Vigna mungo (L.) Hepper] linkage map and its comparison with an azuki bean [Vigna angularis (Willd.) Ohwi and Ohashi] linkage map. Theor. Appl. Genet. 2006, 113, 1261–1269. [Google Scholar] [CrossRef]
- Gupta, S.K.; Gopalakrishna, T. Development of unigene-derived SSR markers in cowpea (Vigna unguiculata) and their transferability to other Vigna species. Genome 2010, 53, 508–523. [Google Scholar] [CrossRef]
- Zhong, M.; Cheng, X.Z.; Wang, L.X.; Wang, S.H. Transferability of mungbean genomic-SSR markers in other Vigna species. Acta Agron Sin 2012, 38, 223–230. [Google Scholar] [CrossRef]
- Kaur, S.; Gill, R.K.; Bains, T.S.; Kaur, M.; Thakur, S. Comparative assessment of SSR markers derived from different sources in genetic diversity analysis of Vigna genotypes. Agric. Res. J. 2017, 54, 462–468. [Google Scholar] [CrossRef]
- Wang, L.X.; Cheng, X.Z.; Wang, S.H.; Liu, C.Y. Transferability of SSR markers for adzuki bean into mungbean. Acta Agron Sin 2009, 35, 816–820. [Google Scholar] [CrossRef]
- Choudhary, K.B.; Pratap, A.; Tomar, R. Cross genera marker transferability and genetic diversity analysis in elite cultivars of mungbean [Vigna radiata (L.) wilczek]. Legume Res.-Int. J. 2022, 45, 1065–1073. [Google Scholar] [CrossRef]
- Haddoudi, L.; Hdira, S.; Cheikh, N.B.; Mahjoub, A.; Abdelly, C.; Ludidi, N.; Badri, M. Assessment of genetic diversity in Tunisian populations of Medicago polymorpha based on SSR markers. Chil. J. Agric. Res. 2021, 81, 53–61. [Google Scholar] [CrossRef]
- Singh, B.; Das, A.; Parihar, A.K.; Bhagawati, B.; Singh, D.; Pathak, K.N.; Dwivedi, K.; Das, N.; Keshari, N.; Midha, R.L.; et al. Delineation of Genotype-by-Environment interactions for identification and validation of resistant genotypes in root-knot nematode (Meloidogyne incognita) using GGE biplot. Sci. Rep. 2020, 10, 4108. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.V.; Tan, S.G.; Quah, S.C.; Yusoff, K. Isolation of microsatellite markers in mungbean, Vigna radiata. Mol. Ecol. Notes 2002, 2, 96–98. [Google Scholar] [CrossRef]
- Kumar, S.V.; Tan, S.G.; Quah, S.C.; Yusoff, K. Isolation and characterization of seven tetranucleotide microsatellite loci in mungbean, Vigna radiata. Mol. Ecol. Notes 2002, 2, 293–295. [Google Scholar] [CrossRef]
- Miyagi, M.; Humphry, M.; Ma, Z.Y.; Lambrides, C.J.; Bateson, M.; Liu, C.J. Construction of bacterial artificial chromosome libraries and their application in developing PCR-based markers closely linked to a major locus conditioning bruchid resistance in Mungbean [Vigna radiata (L.) Wilczek]. Theor. Appl. Genet. 2004, 110, 151–156. [Google Scholar] [CrossRef]
- Gwag, J.G.; Chung, W.K.; Chung, H.K.; Lee, J.H.; Ma, K.H.; Dixit, A.; Park, Y.J.; Cho, E.G.; Kim, T.S.; Lee, S.H. Characterization of new microsatellite markers in mungbean. Mol. Ecol. Notes 2006, 6, 1132–1134. [Google Scholar] [CrossRef]
- Tangphatsornruang, S.; Somta, P.; Uthaipaisanwong, P.; Chanprasert, P.; Sangsrakru, D.; Seehalak, W.; Sommanas, W.; Tragoonrung, S.; Srinives, P. Characterization of microsatellites and gene contents from genome shotgun sequences of mungbean (Vigna radiata (L.) Wilczek). BMC Plant Biol. 2009, 9, 137. [Google Scholar] [CrossRef] [PubMed]
- Singh, N. Development of Cost Efficient and Rapid Method for Developing Homozygous SSR Markers in Mungbean Vigna radiata (L.) Wilczek and Its Validation in Vigna Species. Master’s Thesis, Tamil Nadu Agricultural University, Coimbatore, India, 2011. [Google Scholar]
- Singh, N.; Singh, H.; Nagarajan, P. Development of SSR markers in mung bean, Vigna radiata (L.) Wilczek using in silico methods. J. Crop Weed 2013, 9, 69–74. [Google Scholar]
- Shrivastava, D.; Verma, P.; Bhatia, S. Expanding the repertoire of microsatellite markers for polymorphism studies in Indian accessions of mungbean (Vigna radiata L. Wilczek). Mol. Biol. Rep. 2014, 41, 5669–5680. [Google Scholar] [CrossRef] [PubMed]
- Bangar, P.; Chaudhary, A.; Umdale, S.; Kumari, R.; Tiwari, B.; Kumar, S.; Gaikwad, A.B.; Bhat, K.V. Detection and characterization of polymorphic simple sequence repeats markers for the analysis of genetic diversity in Indian mungbean [Vigna radiata (L.) Wilczek]. Indian J. Genet. 2018, 78, 111–117. [Google Scholar] [CrossRef]
- Doyle, J.J.; Doyle, J.L. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar]
- Li, R.; Li, Y.; Kristiansen, K.; Wang, J. SOAP: Short oligonucleotide alignment program. Bioinformatics 2008, 24, 713–717. [Google Scholar] [CrossRef] [PubMed]
- Thiel, T.; Michalek, W.; Varshney, R.K.; Graner, A. Exploiting EST databases for the development of cDNA derived microsatellite markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 2003, 106, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Thakur, S. Whole Genome De Novo Assembly of Vigna mungo and Vigna radiata and In Silico Comparative Analysis for Marker Development. Ph.D. Dissertation, Punjab Agricultural University, Ludhiana, India, 2018; pp. 1–96. [Google Scholar]
- Shyu, C.; Foster, J.A.; Forney, L.J. Electronic polymerase chain reaction (ePCR) search algorithm. In Proceedings of the IEEE Computer Society Bioinformatics Conference, Stanford, CA, USA, 16 August 2002; p. 338. [Google Scholar]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research: An update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef]
- Earl, D.A. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Perrier, X.; Jacquemoud-Collet, J.P. DARwin Software, 5th ed.; Cirad: Montpellier, France, 2006. Available online: http://darwin.cirad.fr/darwin (accessed on 13 July 2023).
- Botstein, D.; White, R.L.; Skolnick, M.; Davis, R.W. Construction of a genetic linkage map in man using restriction fragment length polymorphism. Am. J. Hum. Genet. 1980, 32, 314–331. [Google Scholar] [PubMed]
- Powell, W.; Morgante, M.; Andre, C.; Hanafey, M.; Vogel, J.; Tingey, S.; Rafalski, A. The comparison of RFLP, RAPD, AFLP and SSR (microsatellite) markers for germplasm analysis. Mol. Breed. 1996, 2, 225–238. [Google Scholar] [CrossRef]
- Prevost, A.; Wilkinson, M.J. A new system of comparing PCR primers applied to ISSR fingerprinting of potato cultivars. Ther. Appl. Genet. 1999, 98, 107–112. [Google Scholar] [CrossRef]
- Nayak, S.N.; Song, J.; Villa, A.; Pathak, B.; Ayala-Silva, T.; Yang, X.; Todd, J.; Glynn, N.C.; Kuhn, D.N.; Glaz, B.; et al. Promoting utilization of Saccharum spp. genetic resources through genetic diversity analysis and core collection construction. PLoS ONE 2014, 9, 110856. [Google Scholar] [CrossRef] [PubMed]
- Nass, L.L.; Paterniani, E. Pre-breeding: A link between genetic resources and maize breeding. Sci. Agric. 2000, 57, 581–587. [Google Scholar] [CrossRef]
- Haussmann, B.I.G.; Parzies, H.K.; Presterl, T.; Susic, Z.; Mieddaner, T. Plant genetic resources in crop improvement. Plant Genet. Resour. 2004, 2, 3–21. [Google Scholar] [CrossRef]
- Acosta-Gallegos, J.; Kelly, J.D.; Gepts, P. Prebreeding in common bean and use of genetic diversity from wild germplasm. Crop Sci. 2007, 47, 44–59. [Google Scholar] [CrossRef]
- Shimelis, H.; Laing, M. Timelines in conventional crop improvement: Pre-breeding and breeding procedures. Aus. J. Crop Sci. 2012, 11, 1542–1549. [Google Scholar]
- Sharma, S.; Upadhyaya, H.D.; Varshney, R.K.; Gowda, C.L.L. Pre-breeding for diversification of primary gene pool and genetic enhancement of grain legume. Front. Plant Sci. 2013, 4, 309. [Google Scholar] [CrossRef]
- Kumar, V.; Shukla, Y.M. Pre-breeding: Its applications in crop improvement. Res. News U (RNFU) 2014, 16, 199–202. [Google Scholar]
- Meena, A.K.; Gurjar, D.; Kumhar, B.L. Pre-breeding is a bridge between wild species and improved genotypes—A review. Chem. Sci. Rev. Lett. 2017, 6, 1141–1151. [Google Scholar]
- Jain, S.K. Omprakash Pre-breeding: A Bridge between Genetic Resources and Crop Improvement. Int. J. Curr. Microbiol. App. Sci. 2019, 8, 1998–2007. [Google Scholar] [CrossRef]
- Singh, R.B.; Singh, B.; Singh, R.K. Evaluation of genetic diversity in Saccharum species clones and commercial varieties employing molecular (SSRs) and physiological markers. Indian J. Plant Genet. Resour. 2018, 31, 17–26. [Google Scholar] [CrossRef]
- Wang, M.L.; Barkley, N.A.; Gillaspie, G.A.; Pederson, G.A. Phylogenetic relationships and genetic diversity of the USDA Vigna germplasm collection revealed by gene-derived markers and sequencing. Genet. Res. 2008, 90, 467–480. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Somta, P.; Muto, C.; Iseki, K.; Naito, K.; Pandiyan, M.; Natesan, S.; Tomooka, N. Novel Genetic Resources in the Genus Vigna Unveiled from Gene Bank Accessions. PLoS ONE 2016, 11, e0147568. [Google Scholar] [CrossRef] [PubMed]
- Bisht, I.S.; Bhat, K.V.; Lakhanpaul, S.; Latha, M.; Jayan, P.K.; Biswas, B.K.; Singh, A.K. Diversity and genetic resources of wild Vigna species in India. Genet. Resour. Crop Evol. 2005, 52, 53–68. [Google Scholar] [CrossRef]
- Blair, M.W.; Hurtado, N.; Chavarro, C.M.; Munoz-Torres, M.C.; Pedraza, M.F.; Tomins, J.; Wing, R. Gene-based SSR markers for common bean (Phaseolus vulgaris L.) derived from root and leaf tissue ESTs: An integration of the BMC series. BMC Plant Biol. 2011, 11, 50. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.K.; Bansal, R.; Gopalakrishna, T. Development and characterization of genic SSR markers for mungbean (Vigna radiata (L.) Wilczek). Euphytica 2014, 195, 245–258. [Google Scholar] [CrossRef]
- Chen, H.; Qiao, L.; Wang, L.; Wang, S.; Blair, M.W.; Cheng, X. Assessment of genetic diversity and population structure of mungbean (Vigna radiata) germplasm using EST-based and genomic SSR markers. Gene 2015, 566, 175–183. [Google Scholar] [CrossRef]
- Gong, Y.M.; Xu, S.C.; Mao, W.H.; Hu, Q.Z.; Zhang, G.W.; Ding, J.; Li, Y.D. Developing new SSR markers from ESTs of pea (Pisum sativum L.). J. Zhejiang Univ. Sci. B 2010, 11, 702–707. [Google Scholar] [CrossRef]
- Choudhary, S.; Sethy, N.K.; Shokeen, B.; Bhatia, S. Development of chickpea EST-SSR markers and analysis of allelic variation across related species. Theor. Appl. Genet. 2009, 118, 591–608. [Google Scholar] [CrossRef] [PubMed]
- Blair, M.W.; Hurtado, N. EST-SSR markers from five sequenced cDNA libraries of common bean (Phaseolus vulgaris L.) comparing three bioinformatic algorithms. Mol. Ecol. Resour. 2013, 13, 688–695. [Google Scholar] [CrossRef]
- Bhardwaj, J.; Chauhan, R.; Swarnkar, M.K.; Chahota, R.K.; Singh, A.K.; Shankar, R.; Yadav, S.K. Comprehensive transcriptomic study on horse gram (Macrotyloma uniflorum): De novo assembly, functional characterization and comparative analysis in relation to drought stress. BMC Genom. 2013, 14, 647. [Google Scholar] [CrossRef] [PubMed]
- Jasrotia, R.S.; Yadav, P.K.; Iquebal, M.A.; Bhatt, S.B.; Arora, V.; Angadi, U.B.; Tomar, R.S.; Jaiswal, S.; Rai, A.; Kumar, D. VigSat DB: Genome-wide microsatellite DNA marker database of three species of Vigna for germplasm characterization and improvement. Database 2019, 2019, baz055. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Liu, L.; Wang, L.; Wang, S.; Somta, P.; Cheng, X. Development and validation of EST SSR markers from the transcriptome of adzuki bean (Vigna angularis). PLoS ONE 2015, 10, e0131939. [Google Scholar] [CrossRef]
- Luro, F.L.; Constantino, G.; Terol, J.; Argout, X.; Allario, T.; Wincker, P.; Talon, M.; Ollitrault, P.; Morillon, R. Transferability of the ESR-SSRs developed on Nules clementine (Citrus clementine Hort ex Tan) to other Citrus species and their effectiveness for genetic mapping. BMC Genom. 2008, 9, 287. [Google Scholar] [CrossRef] [PubMed]
- Mnejja, M.; Garcia-Mas, J.; Audergon, J.M.; Arús, P. Prunus microsatellite marker transferability across rosaceous crops. Tree Genet. Genomes 2010, 6, 689–700. [Google Scholar] [CrossRef]
- Somta, P.; Seehalak, W.; Srinives, P. Development, characterization and cross-species amplification of mungbean (Vigna radiata) genic microsatellite markers. Conserv. Genet. 2009, 10, 1939–1943. [Google Scholar] [CrossRef]
- Gupta, S.K.; Bansal, R.; Vaidya, U.J.; Gopalakrishna, T. Development of EST-derived microsatellite markers in mungbean [Vigna radiata (L.) Wilczek] and their transferability to other Vigna species. Indian J. Genet. 2012, 72, 468–471. [Google Scholar]
- Singh, A.; Dikshit, H.K.; Jain, N.; Singh, D.; Yadav, R.N. Efficiency of SSR, ISSR and RAPD markers in molecular characterization of mungbean and other Vigna species. Indian J. Biotechnol. 2014, 13, 81–88. [Google Scholar]
- Kaur, S.; Bains, T.S.; Sirari, A.; Kaur, S. Evaluation and molecular characterization of advanced interspecific lines for genetic improvement in mungbean [Vigna radiate (L.) Wilczek]. Legume Res. 2019, 42, 729–735. [Google Scholar] [CrossRef]
- Daware, A.; Das, S.; Srivastava, R.; Badoni, S.; Singh, A.K.; Agarwal, P.; Parida, S.K.; Tyagi, A.K. An Efficient Strategy Combining SSR Markers- and Advanced QTL-seq-driven QTL Mapping Unravels Candidate Genes Regulating Grain Weight in Rice. Front. Plant Sci. 2016, 7, 1535. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yang, S.; Chen, Y.; Zhang, S.; Zhao, Q.; Li, M.; Gao, Y.; Yang, L.; Bennetzen, J.L. Comparative genome-wide characterization leading to simple sequence repeat marker development for Nicotiana. BMC Genom. 2018, 19, 500. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhang, J.; Wang, Y.; Wang, H.; Wang, L.; Zhang, L.; Xiong, M.; He, W.; Yang, S.; Chen, Q.; et al. Development and Cross-Species Transferability of Novel Genomic-SSR Markers and Their Utility in Hybrid Identification and Trait Association Analysis in Chinese Cherry. Horticulturae 2022, 8, 222. [Google Scholar] [CrossRef]
- Pratap, A.; Gupta, S.; Malviya, N.; Tomar, R.; Maurya, R.; John, K.J.; Madhavan, L.; Singh, N.P. Genome scanning of Asiatic Vigna species for discerning population genetic structure based on microsatellite variation. Mol. Breed. 2015, 35, 178. [Google Scholar] [CrossRef]
- Kumari, G.; Roopa Lavanya, G.; Shanmugavadivel, P.S.; Singh, Y.; Singh, P.; Patidar, B.; Madhavan, L.; Gupta, S.; Singh, N.P.; Pratap, A. Genetic diversity and population genetic structure analysis of an extensive collection of wild and cultivated Vigna accessions. Mol. Genet. Genom. 2021, 296, 1337–1353. [Google Scholar] [CrossRef] [PubMed]
- Dachapak, S.; Somta, P.; Poonchaivilaisak, S.; Yimram, T.; Srinives, P. Genetic diversity and structure of the zombi pea (Vigna vexillata (L.) A. Rich) gene pool based on SSR marker analysis. Genetica 2017, 145, 189–200. [Google Scholar] [CrossRef]
- Sarr, A.; Bodian, A.; Gbedevi, K.M.; Ndir, K.N.; Ajewole, O.O.; Gueye, B.; Foncéka, D.; Diop, E.A.; Diop, B.M.; Cissé, N.; et al. Genetic Diversity and population structure analyses of wild relatives and cultivated cowpea (Vigna unguiculata (L.) Walp.) from senegal using simple sequence repeat markers. Plant Mol. Biol. Rep. 2020, 39, 112–124. [Google Scholar] [CrossRef]
- Singh, R.B.; Mahenderakar, M.D.; Jugran, A.K.; Singh, R.K.; Srivastava, R.K. Assessing genetic diversity and population structure of sugarcane cultivars, progenitor species and genera using microsatellite (SSR) markers. Gene 2020, 753, 144800. [Google Scholar] [CrossRef]
- Paliwal, R.; Singh, R.; Choudhury, D.R.; Tiwari, G.; Kumar, A.; Bhat, K.C.; Singh, R. Molecular Characterization of Tinospora cordifolia (Willd.) Miers Using Novel g-SSR Markers and Their Comparison with EST-SSR and SCoT Markers for Genetic Diversity Study. Genes 2022, 13, 2042. [Google Scholar] [CrossRef]
- Noble, T.J.; Tao, Y.; Mace, E.S.; Williams, B.; Jordan, D.R.; Douglas, C.A.; Mundree, S.G. Characterization of linkage disequilibrium and population structure in a mungbean diversity panel. Front. Plant Sci. 2018, 8, 2102. [Google Scholar] [CrossRef] [PubMed]
- Xiong, H.; Shi, A.; Mou, B.; Qin, J.; Motes, D.; Lu, W.; Ma, J.; Weng, Y.; Yang, W.; Wu, D. Genetic diversity and population structure of cowpea (Vigna unguiculata L. Walp). PLoS ONE 2016, 11, e0160941. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Chen, H.; Hu, L.; Wang, L.; Wang, S.; Wang, M.L.; Cheng, X. Genetic diversity and a population structure analysis of accessions in the Chinese cowpea [Vigna unguiculata (L.) Walp.] germplasm collection. Crop J. 2017, 5, 363–372. [Google Scholar] [CrossRef]
- Luo, Z.; Brock, J.; Dyer, J.M.; Kutchan, T.; Schachtman, D.; Augustin, M.; Ge, Y.; Fahlgren, N.; Abdel-Haleem, H. Genetic Diversity and Population Structure of a Camelina sativa Spring Panel. Front. Plant Sci. 2019, 10, 184. [Google Scholar] [CrossRef]
- Saxena, S.; Kole, P.R.; Bhat, K.V.; Pandey, S.P. Species diversity and relationship among Vigna radiata (L.) wilczek and its close wild relatives. Bangladesh J. Botany 2016, 45, 567–573. [Google Scholar]
- Pandiyan, M.; Senthil, N.; Ramamoorthi, N.; Muthiah, A.R.; Tomooka, N.; Duncan, V.; Tomooka, N.; Jayaraj, T. Interspecifc hybridization of Vigna radiata × 13 wild Vigna species for developing MYMV donor. Electron. J. Plant Breed. 2010, 1, 600–610. [Google Scholar]
- Kumar, S.; Gupta, S.; Chandra, S.; Singh, B.B. How wide is the genetic base of pulse crops. In Pulses in New Perspective; Ali, M., Singh, B.B., Kumar, S., Dhar, V., Eds.; Indian Society of Pulses Research and Development: Kanpur, India, 2004; pp. 211–221. [Google Scholar]
- Yan, H.; Qi, H.; Li, Y.; Wu, Y.; Wang, Y.; Chen, J.; Yu, J. Assessment of the Genetic Relationship and Population Structure in Oil-Tea Camellia Species Using Simple Sequence Repeat (SSR) Markers. Genes 2022, 13, 2162. [Google Scholar] [CrossRef]
- Frankham, R.; Ballou, J.D.; Briscoe, D.A. Introduction to Conservation Genetics; Cambridge University Press: Cambridge, UK, 2002. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Designation | Accessions | Designation | Accessions |
|---|---|---|---|
| GP1 | V. umbellata (Cultivated) | GP14 | V. stipulacea |
| GP2 | V. umbellata (Cultivated) | GP15 | V. radiata var. radiata |
| GP3 | V. umbellata | GP16 | V. radiata var. mungo |
| GP4 | V. sublobata | GP17 | V. radiata var. mungo |
| GP5 | V. sublobata | GP18 | V. radiata var. mungo |
| GP6 | V. trilobata | GP19 | V. slyestris |
| GP7 | V. trilobata | GP20 | V. glabrescence |
| GP8 | V. trilobata | GP21 | V. radiata var. satulosa |
| GP9 | V. trilobata | GP22 | V. vexillata |
| GP10 | V. aconitifolia | GP23 | V. hainiana |
| GP11 | V. aconitifolia | GP24 | V. dalzelliana |
| GP12 | V. aconitifolia (TMV-1) | GP25 | V. unguiculata |
| GP13 | V. stipulacea | ||
| Parameters | Number of SSRs | |
|---|---|---|
| V. radiata cv. ML267 | V. mungo cv. Mash114 | |
| SSR Mining | ||
| SSR sequences examined | 444,059 | 471,725 |
| SSRs identified | 225,359 | 218,508 |
| SSR-containing sequences | 130,125 | 126,749 |
| Sequences containing more than 1 SSR | 50,760 | 46,626 |
| SSRs present in compound formation | 16,201 | 15,565 |
| Repeat Types | ||
| Mononucleotide | 173,536 (77%) | 170,071 (77.83%) |
| Dinucleotide | 29,559 (13.12%) | 27,625 (12.64%) |
| Trinucleotide | 19,732 (8.76%) | 18,490 (8.46%) |
| Tetranucleotide | 1939 (0.86%) | 1794 (0.82%) |
| Pentanucleotide | 410 (0.18%) | 369 (0.17%) |
| Hexanucleotide | 183 (0.08%) | 159 (0.08%) |
| Dinucleotide Repeat | N | Number | Percentage |
|---|---|---|---|
| (AT)n | 6, 7, 8, 9, 10, 11, 12, 13, 14, 17 | 69 | 27.6 |
| (AG)n | 6, 7, 8, 9, 10, 13, 14, 16, 17, 22 | 30 | 12.0 |
| (AC)n | 6, 7, 8, 9 | 16 | 6.40 |
| (TA)n | 6, 7, 8, 9, 10, 11, 12, 13, 20 | 62 | 24.8 |
| (TC)n | 6, 7. 8, 9, 10, 11, 13, 14, 16, 17 | 31 | 12.4 |
| (TG)n | 6, 7, 9 | 11 | 4.40 |
| (CT)n | 6, 7, 8, 9, 12, 14 | 13 | 5.20 |
| (CA)n | 6, 7 | 02 | 0.80 |
| (GA)n | 6, 7, 8, 10, 12, 19, 20 | 10 | 4.00 |
| (GT)n | 6, 7, 14 | 06 | 2.40 |
| Cluster 1 | Cluster 2 | Cluster 3 | |||
|---|---|---|---|---|---|
| Sub-Cluster 1a | Sub-Cluster 1b | Sub-Cluster 2a | Sub-Cluster 2b | Sub-Cluster 3a | Sub-Cluster 3b |
| GP5 (V. sublobata) | GP24 (V. dalzelliana) | GP17 (V. radiata var. mungo) | GP12 (V. aconitifolia TMV-1) | GP8 (V. trilobata) | GP3 (V. umbellata) |
| GP4 (V. sublobata) | GP23 (V. hainiana) | GP16 (V. radiata var. mungo) | GP6 (V. trilobata) | ||
| GP9 (V. trilobata) | GP25 (V. unguiculata) | GP18 (V. radiata var. mungo) | GP7 (V. trilobata) | ||
| GP15 (V. radiata var. radiata) | GP19 (V. slyestris) | GP14 (V. stipulacea) | |||
| GP22 (V. vexillata) | GP11 (V. aconitifolia) | GP13 (V. stipulacea) | |||
| GP21 (V. radiata var. setulosa) | GP10 (V. aconitifolia) | ||||
| GP20 (V. glabrescence) | GP2 (V. umbellata cultivated) | ||||
| GP1 (V. umbellata cultivated) | |||||
| Pop ID | Pop 1 | Pop 2 | Pop 3 |
|---|---|---|---|
| Pop 2 | 0.374 | - | - |
| Pop 3 | 0.208 | 0.442 | - |
| Pop 4 | 0.250 | 0.458 | 0.189 |
| Composition of Sub-Population 1 | Composition of Sub-Population 2 |
|---|---|
| GP1 (V. umbellata cultivated) | GP3 (V. umbellata) |
| GP2 (V. umbellata cultivated) | GP4 (V. sublobata) |
| GP10 (V. aconitifolia) | GP5 (V. sublobata) |
| GP11 (V. aconitifolia) | GP6 (V. trilobata) |
| GP12 (V. aconitifolia TMV-1) | GP7 (V. trilobata) |
| GP16 (V. radiata var. mungo) | GP8 (V. trilobata) |
| GP17 (V. radiata var. mungo) | GP9 (V. trilobata) |
| GP18 (V. radiata var. mungo) | GP13 (V. stipulacea) |
| GP20 (V. glabrescence) | GP14 (V. stipulacea) |
| GP22 (V. vexillata) | GP15 (V. radiata var. radiata) |
| GP24 (V. dalzelliana) | GP19 (V. slyestris) |
| GP21 (V. radiata var. setulosa) | |
| GP23 (V. hainiana) | |
| GP25 (V. unguiculata) |
| Source of Variation | Df | Sum of Squares | Mean Sum of Squares | Estimated Variance | % Variance | F Statistics |
|---|---|---|---|---|---|---|
| Among populations | 2 | 470.62 | 235.31 | 7.24 | 10 | 0.105 |
| Among individuals | 22 | 2706.18 | 123.00 | 61.15 | 89 | 0.989 |
| Within individuals | 25 | 17.50 | 0.700 | 0.70 | 1 | 0.990 |
| Total | 49 | 3194.30 | 69.09 | 100 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saini, P.; Sirari, A.; Gnanesh, B.N.; Mandahal, K.S.; Ludhar, N.K.; Nagpal, S.; Patel, S.A.H.; Akhatar, J.; Saini, P.; Pratap, A.; et al. Assessment of Simple Sequence Repeat (SSR) Markers Derived from Whole-Genome Sequence (WGS) of Mungbean (Vigna radiata L. Wilczek): Cross-Species Transferability and Population Genetic Studies in Vigna Species. Horticulturae 2024, 10, 34. https://doi.org/10.3390/horticulturae10010034
Saini P, Sirari A, Gnanesh BN, Mandahal KS, Ludhar NK, Nagpal S, Patel SAH, Akhatar J, Saini P, Pratap A, et al. Assessment of Simple Sequence Repeat (SSR) Markers Derived from Whole-Genome Sequence (WGS) of Mungbean (Vigna radiata L. Wilczek): Cross-Species Transferability and Population Genetic Studies in Vigna Species. Horticulturae. 2024; 10(1):34. https://doi.org/10.3390/horticulturae10010034
Chicago/Turabian StyleSaini, Pawan, Asmita Sirari, Belaghihalli N. Gnanesh, Kamalpreet Singh Mandahal, Navkiran Kaur Ludhar, Sharon Nagpal, S. A. H. Patel, Javed Akhatar, Pooja Saini, Aditya Pratap, and et al. 2024. "Assessment of Simple Sequence Repeat (SSR) Markers Derived from Whole-Genome Sequence (WGS) of Mungbean (Vigna radiata L. Wilczek): Cross-Species Transferability and Population Genetic Studies in Vigna Species" Horticulturae 10, no. 1: 34. https://doi.org/10.3390/horticulturae10010034
APA StyleSaini, P., Sirari, A., Gnanesh, B. N., Mandahal, K. S., Ludhar, N. K., Nagpal, S., Patel, S. A. H., Akhatar, J., Saini, P., Pratap, A., Bains, T. S., & Yadav, I. S. (2024). Assessment of Simple Sequence Repeat (SSR) Markers Derived from Whole-Genome Sequence (WGS) of Mungbean (Vigna radiata L. Wilczek): Cross-Species Transferability and Population Genetic Studies in Vigna Species. Horticulturae, 10(1), 34. https://doi.org/10.3390/horticulturae10010034