Abstract
The maltose α-amylase AmyM from Bacillus stearothermophilus can be used for flour modification, baked goods preservation, and maltose production. Here, we optimized the recombinant expression of AmyM in Bacillus subtilis WB800 via several strategies. By screening the optimal promoter, a double promoter combination (P43 and PamyL) could improve the expression level of AmyM by 61.25%, compared with the strong promoter P43. Then, we optimized the secretion efficiency of recombinant AmyM by over-expressing the molecular chaperone prsA gene. SDS-PAGE results suggested that over-expression of the prsA could improve the secretion efficiency of AmyM to the extracellular environment. The extracellular enzyme activity of AmyM was increased by 101.58% compared to the control strain. To further improve the expression of AmyM, we introduced the hemoglobin gene of Vitreoscilla (vgb) into the AmyM recombinant strain. The results revealed that the introduction of vgb could promote the transcription and translation of AmyM in B. subtilis. This may be due to the increasing level of intracellular NADPH and NADP+ caused by the expression of vgb. By this strategy, the expression level of AmyM was increased by 204.08%. Finally, we found the recombinant AmyM showed an optimal temperature of 65 °C and an optimal pH of 5.5. Our present results provided an effective strategy for increasing the heterologous expression level of AmyM in B. subtilis.
1. Introduction
Maltogenic amylase is a member of glycoside hydrolase family 13. The product of the sustained action of maltogenic amylase on starch is maltose, hence its name maltose α-amylase. The hydrolysis product of maltogenic amylase is in the α conformation, and it can also hydrolyze the polysaccharide chains and cyclic dextrins that are modified at the reducing end, thus indicating that it is an endonuclease [1]. In 1999, Dauter et al. [2] characterized the complexes that are formed by the maltogenic amylase AmyM from Bacillus stearothermophilus with maltose and acarbose and, therefore, determined the three-dimensional structure of AmyM. Unlike other α-amylases, maltose amylase AmyM contains five structural domains, which are very similar to CGTases. Maltogenic amylase could enhance slow digestion starch and release more bioactive compound content [3]. Exogenous maltose α-amylase of bacterial origin is commonly used in wheat bread production to slow down the aging of the crumb [4]. In addition, maltose amylase has some potential applications in the pharmaceutical field. Sahar Roozbehi et al. developed an enzyme-triggered system based on β-cyclodextrin (β-CD) for the controlled release of hydrophobic drugs in the presence of maltose amylase [5].
B. subtilis was discovered in 1958 as a host that could be used for transformation, and since then, the genetic manipulation system of B. subtilis has been further developed [6]. In 1997, the whole genome of B. subtilis 168 was sequenced. With the invention of DNA recombination techniques, especially the discovery of the resistance plasmid in Staphylococcus aureus by Gryczan and Ehrlich in 1977–1978 as vectors for B. subtilis, genetic engineering work in B. subtilis was further developed rapidly [7]. B. subtilis is commonly used as a host for the production of amylases, lipases, glycosidases, and so on because of its safety, ease of handling, and high secretion capacity. The amylase gene of B. subtilis WB800 was knocked out in our laboratory, so the strain was chosen as the expression host to avoid the effect of B. subtilis amylase on the experimental results.
The promoters and signal peptide as important components are often the primary target for the optimization of gene expression. Rémi Dulermo et al. [8] found that the fitness of the promoter and the gene was more important than the promoter strength alone. Ying Wang et al. [9] developed the hybrid model for promoter identification, which could successfully identify and screen on both eukaryotic and prokaryotic datasets. Yu Zhao et al. [10] found that the hybrid promoter strength could be fine-tuned to upstream activation sequences (UAS), TATA box, and core promoter. Through the screening of promoters and signal peptides, Hui Wang et al. [11] found that the amylase gene promoter PamyQ from Bacillus amylolyticus and the signal peptide SPaprE from the alkaline protease gene of Bacillus clausii (PamyQ-SPaprE) was successfully used in the production of maltogenic α-amylase Gs-MAase. Kang Zhang et al. replaced the native signal peptide of pullulanase with that encoded by ywtF (a signal peptide in the Sec secretion pathway of B. subtilis) and increased extracellular pullulanase activity by an additional 12% [12]. Lingqia Su et al. screened the best signal peptide for the expression of α/β-CGTase from 173 signal peptides of B. subtilis WS11 [13]. Haiquan Yang et al. increased the enzymatic activity of alkaline amylase in the fermenter by 250.6-fold compared to the control strain after a series of modifications on the promoter and signal peptide [14].
After the work of promoter screening and signal peptide screening by Yutong Li et al. to express the amyM gene in Bacillus subtilis, the shake flask fermentation enzyme activity was only 250.7 U/mL [15]. Since screening promoters and signal peptides is a massive effort, and often with little success, we need to find other ways to effectively increase the heterologous expression level of AmyM. In this study, we first screened the most suitable promoter for the expression of AmyM. Although this is a conventional strategy, the promoter is a crucial element in the initiation of transcriptional translation, so it was necessary to select the most suitable promoter. AmyM is secreted outside of the cell via the Sec pathway, and prsA is an important molecular chaperone protein in the Sec pathway that promotes the secretion of intracellular proteins and, thus, reduces inclusion body formation. Our second strategy was to optimize the secretory pathway of AmyM by over-expressing prsA, thereby reducing the formation of inclusion bodies and increasing extracellular expression. Because it was not possible to determine whether the expression of prsA would be counterproductive, we selected two promoters with different expression strengths to express prsA and analyzed its role on AmyM expression. As B. subtilis is an aerobic micro-organism, the growth of the bacterium is severely affected in a low-oxygen environment, resulting in low cell product production. Therefore, our third research strategy was to introduce a vgb gene into B. subtilis. This gene increases the intracellular content of NADP+ and NADPH [16], which might enhance cellular metabolism and increase the expression level of AmyM.
2. Materials and Methods
2.1. Strains and Plasmids
E. coli mach1-T1 was used for plasmid construction. The strain B. subtilis WB800 (ΔamyE), which was modified in the laboratory in previous experiments, was used for gene expression. Shuttle plasmid PBE-SamyQ was used to clone and express the amyM gene (EC 3.2.1.133). The basic shuttle plasmid PWSCas9n was used for knockout and knock-in on the genome of WB800. Plasmid PMD-amyM-18T in E. coli Mach1 T1 was the template of the amyM gene. All of the above strains and plasmids were preserved by this laboratory.
2.2. Chemical Reagents and Enzymes
Primer STAR Max DNA Polymerase was purchased from TaKaRa Biotech (Dalian, China) Co., Ltd. Green Taq™ Mix was purchased from Vazyme Bio (Nanjing, China) Co., Ltd. Seamless Assembly Cloning kit was purchased from Clone Smarter (Beijing, China) Co., Ltd. All restriction enzymes were purchased from Thermo Fisher Scientific (Shanghai, China) Co., Ltd. HiPure Plasmid Micro Kit, HiPure DNA Pure Micro Kits, HiPure Gel Pure DNA Mini Kit, and the bacterial total RNA extraction kit were purchased from Magen Bio (Shanghai, China) Co., Ltd. Evo M-MLV Reverse Transcription Kit II was purchased from Accurate Bio (Changsha, China) Co., Ltd. The Coenzyme II NADP(H) Content Assay Kit was purchased from Boxbio (Beijing, China) Co., Ltd. Other chemicals were purchased from Newprobe Bio (Beijing, China) Co., Ltd.
2.3. Construction of AmyM Expression Cassette with Different Promoters
In this study, plasmids containing seven different promoter combinations were constructed. The B. subtilis genome was used as a template to amplify the P43 promoter and the PamyE promoter with primer pairs F1/R1 and F3/R3, respectively. The Bacillus amyloliquefaciens genome was used as a template to amplify the Pcd promoter and PamyS promoter with primer pairs F2/R2 and F4/R4, respectively. The Bacillus licheniformis genome was used as a template to amplify the PamyL promoter with primer pair F5/R5 and then overlapped the amplified single promoter to obtain different promoter combinations. The amplified fragments were detected by 1.1% agarose gel electrophoresis and then recovered by either a HiPure DNA Pure Micro Kit or HiPure Gel Pure DNA Mini Kit. The PBE-SamyQ shuttle plasmid was double-digested with EcorI and BamHI and recovered with a HiPure Gel Pure DNA Mini Kit. The recovered amplified fragments and linearized vector were ligated with Seamless Assembly Cloning kit at a molar ratio of 1:3 and transformed into E. coli mach1-T1. The plasmids containing different promoters were obtained after DNA sequencing. All primer sequences used are shown in Table 1.

Table 1.
Primers used for plasmid construction.
2.4. Construction of a Knockout Plasmid(pWSCas9n-ΔsrfAC) and Three Knock-in Plasmids(pWSCas9n-PamyX-prsA, pWSCas9n-P43-prsA, and pWSCas9n-P43-vgb)
CRISPR Cas9 technology was used for gene knockout and knock-in, and the basic plasmid was the pWSCas9n shuttle plasmid that was constructed in this laboratory. To construct the knockout plasmid pWSCas9n-ΔsrfAC, the P43 promoter without RBS was amplified by primers P43-F1/P43-R1; the 20 bp sgRNA sequences were included in primer P43-R1 and gRNA-F1 as a homologous arm in which P43 and gRNA overlapped; the gRNA was amplified by primers gRNA-F1/gRNA-R1; the homologous upstream sequences were amplified by primers UP-F1/UP-R1; the homologous downstream sequences were amplified by primers Down-F1/Down-R1. These four amplified DNA fragments had overlapped each other and ligated with the linearized plasmid pWSCas9n by In-Fusion cloning. To construct the knockout plasmid pWSCas9n-PamyX-prsA, the P43-sgRNA-gRNA-UP was amplified by primers P43-F1/UP-R1; the PamyX promoter was amplified by primers PamyX-F/PamyX-R; the prsA was amplified by primers prsA-F/prsA-R; the homologous downstream sequences were amplified by primers Down-F1/Down-R1. Then, these four DNA fragments were overlapped and ligated with the linearized plasmid pWSCas9n via In-Fusion cloning. When the P43 promoter without RBS was replaced with the PnprE promoter without RBS and the PamyX promoter was replaced with the P43 promoter with RBS, the plasmid named pWSCas9n-P43-prsA was constructed. Similarly, the vgb sequences were amplified by primers vgb-F/vgb-R and then replaced with the prsA sequences, which can construct the plasmid named pWSCas9n-P43-vgb. All plasmids were transformed into E. coli mach1-T1 to achieve cloning and identified by DNA sequencing. All primer sequences used are shown in Table 1.
2.5. Knockout of srfAC Gene, Knock-in of prsA Gene, and Vgb Gene
The identification of the successful role of the editing plasmid was based on the method of Qinglong Xin [17], with slight improvements. The correct knockout and knock-in plasmids were transformed into B. subtilis WB800 (ΔamyE) according to the electro-transformation method of Guoqiang Cao [18], then spread on an LB plate containing 50 ug/mL of kanamycin and cultured at 30 °C for about 14 h. After the transformants on LB plate were verified via colony PCR, the transformants with the correct plasmids were picked into liquid LB medium containing 50 µg/mL kanamycin and 100 µM/mL IPTG for induction at 30 °C for 24 h at 220 rpm. Then, the induction culture was diluted to the appropriate multiple and spread on the LB plate for 14 h at 30 °C. Finally, four single colonies were randomly selected for colony PCR and DNA sequencing to determine if the editing was successful. Successfully edited transformants were incubated for 24 h at 37 °C in liquid LB medium without antibiotics to lose intracellular plasmids for traceless gene knockout and gene knock-in.
2.6. Expression and Purification of Recombinant AmyM
In order to compare the effect of seven promoters, plasmids containing seven different promoters were transformed into B. subtilis WB800. After the transformants were verified via colony PCR, and the transformants with the correct plasmids were picked into liquid LB medium containing 50 µg/mL kanamycin and incubated at 37 °C for 10 h to obtain the seed solution, which was inoculated into 50 mL of TB medium at 2% inoculum for fermentation at 33 °C and 220 rpm. A sample of 1 mL was taken at 24 h, 48 h, and 72 h of fermentation, and the supernatant obtained after centrifugation was the crude enzyme solution. The fermentation broth of the seven strains containing different promoters was verified via SDS-PAGE.
The plasmid named pBE-P43-SamyQ-amyM was transformed into WB800-WT, WB800-ΔsrfAC, WB800-PamyX-prsA, and WB800-P43-prsA for fermentation, and the AmyM protein content in the cells was observed by breaking the bacteria.
To verify whether the hemoglobin VHB could enhance the oxygen uptake capacity of WB800, pBE-P43-SamyQ-amyM was transformed into WB800-WT and WB800-P43-vgb for fermentation. The fermentation system was 100 mL, and the rest of the conditions were the same as before. The crude enzyme broth was purified in three steps. In the first step, the fermentation broth was transferred to a 50 mL centrifuge tube and centrifuged at 10,000× g for 15 min. The supernatant was collected and held at 60 °C for 10 min to remove the poorly heat-stable heteroproteins. In the second step, the enzyme was further purified by using the AKTA Protein Purification System by loading the enzyme solution onto a HisTrp HP column that used buffer A (500 mM NaCl, 20 mM Tris-HCl, pH 8.0) and eluting the enzyme with buffer B (500 mM NaCl, 20 mM Tris-HCl, 500 mM imidazole, pH 8.0). In the third step, the eluted target protein was desalted by using a dialysis bag, and then protein purity was checked via SDS-PAGE.
2.7. RT-PCR Verificaiton of the Expression of amyM, prsA, and vgb
WB800-WT-amyM, WB800-ΔsrfAC-amyM, WB800-PamyX-prsA-amyM, and WB800-P43-prsA-amyM were inoculated simultaneously for fermentation, and 1.5 mL of fermentation broth was taken from each at 48 h. Then, the fermentative organisms obtained after centrifugation were used for RNA extraction. RNA extraction was performed with a bacterial total RNA extraction kit (Magen Bio, Shanghai, China). According to the instructions of Evo M-MLV Reverse Transcription Kit II (Accurate Bio, Changsha, China), the extracted RNA was first removed from the gRNA and then reverse-transcribed. cDNA after reverse transcription was diluted to the same concentration for RT-PCR. The transcript amounts of the internal reference gene gapA and prsA gene were semi-quantified via RT-PCR results.
The WB800-WT-amyM and WB800-P43-vgb-amyM were inoculated simultaneously for fermentation, and the subsequent extraction of RNA and verification of the transcript levels of the vgb and amyM genes were performed as before.
2.8. Detection of NADP+ and NADPH Content in the Cytoplasm
WB800-WT-amyM and WB800-P43-vgb-amyM were inoculated and fermented simultaneously. Fermentation broth was taken at 18 h, 24 h, 36 h, 48 h, and 60 h of fermentation. Then, NADP+ and NADPH in the bacterium were extracted, and concentrations were determined according to the instructions of the Coenzyme II NADP(H) Content Assay Kit, and all samples were replicated three times.
2.9. Enzyme Activity Assay
The enzyme activity assay was determined via the modified DNS method: 0.2 M phosphate buffer (pH 6.0) and 400 μL 1% soluble starch solution preheated at 60 °C for 10 min. Then, 200 μL of the diluted enzyme solution was added and placed in a water bath at 60 °C for another 10 min, and the reaction was stopped by adding 800 μL of 0.4 M NaOH solution. After the reaction, 200 μL of reaction solution was taken from each reaction tube; then, 300 μL of DNS solution was added and boiled for 7 min for color development. After that, 200 μL of reaction mixture was added into a 96-well plate to measure the absorbance value at 540 nm by using an Infinite M200 multifunctional ELISA. One unit of enzyme activity was defined as the amount of maltogenic amylase required to produce 1 μmoL of maltose per minute under the above conditions.
2.10. Determination of Optimum Temperature and Thermostability of the Recombinant AmyM
The optimum temperature of the enzyme was determined by measuring the relative enzymatic activity in phosphate buffer (pH 6.0) under different temperatures (30 °C, 35 °C, 40 °C, 45 °C, 50 °C, 55 °C, 60 °C, 65 °C, 70 °C, 75 °C, 80 °C). The enzyme thermostability was determined by measuring the residual enzymatic activity after incubation in phosphate buffer (pH 5.5) at different temperatures (30–80 °C) for 20 min. The enzyme activity of the untreated sample was defined as 100%.
2.11. Determination of Optimum pH and pH Stability of the Recombinant AmyM
The optimum pH of the enzyme was determined by measuring the relative enzymatic activity at 65 °C under different pH (3.0, 3.5, 4.0, 4.5, 5.0, 5.5, 6.0, 6.5, 7.0, 7.5, 8.0). The enzyme pH stability was determined by measuring the residual enzymatic activity after incubation at 65 °C in different pH (3.0–8.0) for 20 min. Enzyme activity of the untreated sample defined as 100%. The reaction buffers of different pH are prepared as follows: 50 mM citrate buffer was used to prepare reaction buffers at pH 3.0, 3.5, 4.0, 4.5, and 5.0, 5.5; 50 mM phosphate buffer was used to prepare reaction buffers at pH 6.0, 6.5, and 7.0; 50 mM Tris-HCl buffer was used to prepare reaction buffers at pH 7.5 and 8.0.
3. Results
3.1. Selection of Suitable Promoters for the Recombinant Expression of Maltogenic Amylase AmyM
The promoter is an important transcriptional progenitor, and a suitable strong promoter can significantly increase the transcription level of the target gene. In this study, promoter P43, Pcd, PamyE, PamyS, and PamyL were used to express amyM (Figure 1a). SDS-PAGE results illustrated that the band of AmyM protein appeared at approximately 75 kDa in all seven recombinant strains with the target gene amyM. Strains P43-amyM and Pcd-P43-amyM owned the similar target protein bands (Figure 1b). Strains PamyL-P43-amyM and PamyS-P43-amyM also owned the similar target protein bands (Figure 1b), while the content of amyM protein in strains PamyE-PamyL-amyM, PamyS-PamyL-amyM, and PamyE-PamyS-PamyL-amyM was relatively low. Protein purification also suggested that the target protein amyM was expressed in the recombinant strain.
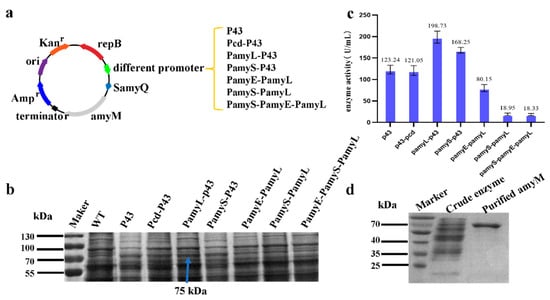
Figure 1.
Selection of suitable promoters for the expression of amyM. (a) Construction of seven amyM expression cassettes containing different promoters. (b) SDS-PAGE analysis of amyM recombinant strains. WT—wild-type strain without amyM expression cassette. (c) Enzyme activity assay. (d) Protein purification: the amount of protein sampled was 0.07 mg for the crude enzyme solution and 0.02 mg for the purified protein. The arrows in the figure point to the location of the target protein bands, which only need to be labeled as shown at 75 kDa.
The enzyme activity assay showed that the enzyme activities of PamyL-P43-amyM, PamyS-P43-amyM, P43-amyM, and Pcd-P43-amyM were 198.73 U/mL, 168.25 U/mL, 123.24 U/mL, and 121.05 U/mL, respectively (Figure 1c). Unbelievably, the enzyme activities of the rest recombinant strains were low, especially PamyS-PamyL-amyM and PamyE-PamyS-PamyL-amyM (less than 20 U/mL).
SDS-PAGE and enzyme activity assay showed that the strong promoter P43 was the most suitable promoter for the expression of amyM, especially in the recombination form of P43 and an amylase promoter derived from B. licheniformis (PamyL). The highest enzyme activity of 198.73 U/mL was achieved in 50 mL of TB medium at 72 h of fermentation (Figure 1b).
3.2. Knockout of the Surfactin Synthetase srfAC Gene and Over-Expression of the Molecular Chaperone prsA Gene Enhances the Extracellular Expression of AmyM
The srfAC gene encodes a surfactin synthetase in B. subtilis, but it is a non-essential gene in the growth of B. subtilis. Conversely, in B. subtilis fermentation, the presence of this gene can lead to increased foaming, resulting in a detrimental effect on fermentation. Molecular chaperone prsA plays a key role in the Sec secretion pathway of intracellular proteins in B. subtilis. Therefore, we attempted to optimize the AmyM secretion pathway by knocking out the srfAC gene and expressing the prsA gene at the srfAC locus.
Plasmid pWSCas9n-ΔsrfAC was used to knock out the srfAC gene (Figure 2a,b). Two plasmids, namely, pWSCas9n-PamyX-prsA and pWSCas9n-P43-prsA, were constructed for the expression of the prsA gene (Figure 2c–f) by using the promoter PamyX derived from the amylase gene of Bacillus amyloliquefaciens and the promoter P43 derived from B. subtilis as the promoters, respectively. These two plasmids were used to integrate the prsA gene with different promoters at the srfAC locus.
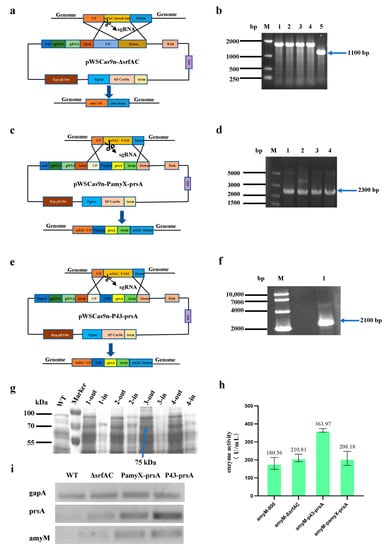
Figure 2.
The effect of srfAC and prsA on the secretion and expression of AmyM. (a) Construction of pWSCas9n-ΔsrfAC plasmid; (b) identification of srfAC knockout; (c) construction of pWSCas9n-PamyX-prsA plasmid; (d) identification of PamyX-PrsA knock-in; (e) construction of pWSCas9n-P43-prsA plasmid; (f) identification of P43-prsA knock-in; (g) SDS-PAGE analysis of amyM expression: WT—wild-type strain without amyM expression cassette, 1-in—WB800-WT-amyM fermentation supernatant, 1-out—WB800-WT-amyM fermentation cell extract, 2-in—WB800-ΔsrfAC-amyM fermentation supernatant, 2-out—WB800-ΔsrfAC-amyM fermentation cell extract, 3-in—WB800-P43-prsA fermentation supernatant, 3-out—WB800-P43-prsA fermentation cell extract, 4-in—WB800-PamyX-prsA fermentation supernatant, 4-out—WB800-PamyX-prsA fermentation cell extract; (h) enzyme activity assay; (i) validation of the transcription of prsA and amyM by semi-quantitative RT-PCR (23 PCR cycles): gapA—internal reference gene, prsA—target gene, amyM—target gene; WT—WB800-WT strain with amyM expression cassette, ΔsrfAC—WB800-ΔsrfAC strain with amyM expression cassette, PamyX-prsA—WB800-PamyX-prsA strain with amyM expression cassette, P43-prsA—WB800-P43-prsA strain with amyM expression cassette.
Finally, amyM was recombinantly expressed in these three hosts: WB800-ΔsrfAC, WB800-PamyX-prsA, and WB800-P43-prsA. The SDS-PAGE result showed that the fermentation supernatants of WB800-WT-amyM, WB800-ΔsrfAC-amyM, WB800-PamyX-prsA-amyM, and WB800-P43-prsA-amyM all contained the target protein AmyM with the molecular mass of 75 kDa (Figure 2g). Importantly, by expressing the prsA gene under the control of the P43 promoter, there was no AmyM docked in cells, suggesting that the over-expression of prsA contributed to the effective secretion of AmyM. Semi-quantitative RT-PCR results revealed that the transcription level of prsA under the control of the P43 promoter was higher than that under the control of the PamyX promoter (Figure 2i). This may be the reason that the secretion of AmyM was more effective when prsA was expressed with the P43 promoter instead of the PamyX promoter (Figure 2g). We also found it interesting that the transcription levels of the amyM gene were similarly significantly increased in strains that over-expressed prsA. Furthermore, the expression level of AmyM was also enhanced to 363.97 U/mL with the help of prsA (P43 promoter, Figure 2h). Taken together, the deletion of srfAC and the over-expression of prsA not only increased the secretion efficiency of AmyM but also improved the transcription level of amyM.
3.3. Expression of vgb Gene Enhances Cellular Metabolic Viability, Thereby Increasing the Expression of AmyM
The vgb gene is derived from the bacterium Trichoderma hyopneumoniae and it could enhance cellular oxygen uptake under low oxygen conditions, promoting cell growth and the accumulation of metabolites. Since B. subtilis is an aerobic bacterium, in this study, a vgb gene was integrated in the srfAC locus of the B. subtilis genome and verified whether it could enhance the expression of the exogenous target protein AmyM.
The vgb gene was successfully integrated into the srfAC locus by using CRISPR/Cas9 technology, obtaining recombinant strain WB800-P43-vgb (Figure 3a,b). Then, AmyM was expressed in the WB800-P43-vgb strain. The SDS-PAGE result revealed that the protein concentration of AmyM in the WB800-P43-vgb strain was higher than the strain without vgb (Figure 3c). The enzyme activity of WB800-P43-vgb-AmyM was much higher than that of WB800-WT-AmyM, reaching 554.43 U/mL (Figure 3d), which was 3.04 times higher than that of WB800-WT-AmyM. Semi-quantitative RT-PCR also showed that vgb was successfully expressed in the WB800-P43-vgb-AmyM strain and that AmyM was highly transcribed (Figure 3e). The above results suggested that vgb gene could significantly improve the expression level of AmyM.
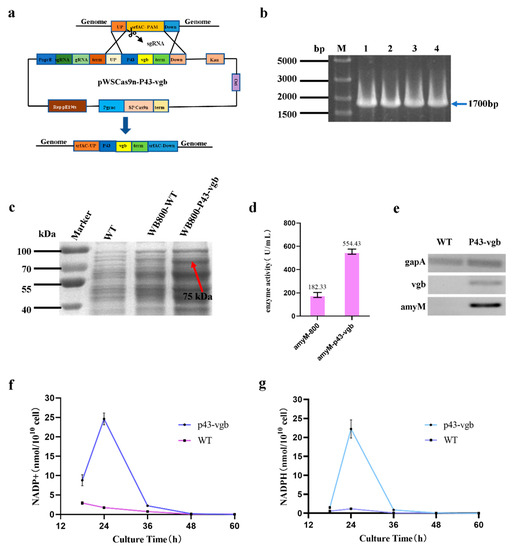
Figure 3.
The effect of vgb on the expression of AmyM. (a) Construction of pWSCas9n-P43-vgb plasmid; (b) identification of P43-vgb knock-in; (c) SDS-PAGE analysis: WT—wild-type strain without AmyM expression cassette, WB800-WT—WB800-WT-AmyM fermentation supernatant, WB800-P43-vgb—WB800-P43-vgb-AmyM fermentation supernatant; (d) enzyme activity assay; (e) validation of the transcription of vgb and AmyM via semi-quantitative RT-PCR (23 PCR cycles): gapA—internal reference gene, vgb—target gene, AmyM—target gene; WT—WB800-WT strain with AmyM expression cassette, P43-vgb—WB800-P43-vgb strain with AmyM expression cassette; (f) NADP+ content determination; (g) NADPH content determination.
Furthermore, we measured the concentration of intracellular NADP+ and NADPH contents (Figure 3f,g). The results showed that, in strain WB800-WT, the concentrations of both NADP+ and NADPH were very low, especially that of NADPH. By introducing vgb in the recombinant strain WB800-p43-vgb, the levels of both substances were higher than in WB800-WT, especially after 24 h of incubation, when the difference between the two strains reached its maximum. We supposed that the expression of vgb optimized the energy status of the recombinant strain and then improved the expression level of the target gene AmyM.
3.4. The Characteristics of the Recombinant AmyM
In a preliminary investigation of the enzymatic properties of purified AmyM, we found that the optimum temperature of AmyM was 65 °C (Figure 4a) and the optimum pH was 5.5 (Figure 4c). We also investigated the stability of AmyM at different temperatures and pH. The results revealed that AmyM could keep good stability in a wide range of temperatures and pH (Figure 4b,d), which may give it potential applications in baking.
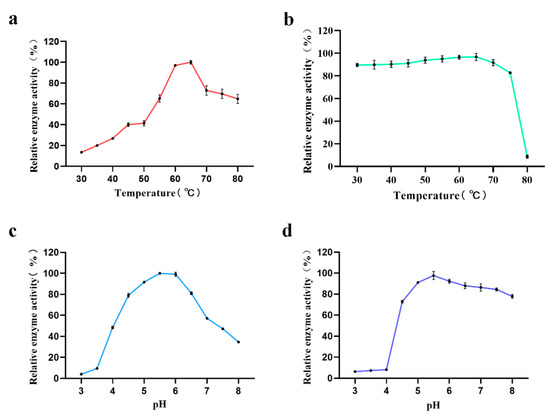
Figure 4.
Characteristics of the recombinant AmyM. (a) Determination of optimum temperature (incubate in pH 6.0 buffer for 10 min). (b) Determination of temperature stability (hold in pH 5.5 buffer and corresponding temperature for 20 min, then incubate at 65 °C, pH 5.5 buffer for 10 min). (c) Determination of optimum pH (incubate at 65 °C for 10 min). (d) Determination of pH stability (hold at 65 °C and corresponding pH for 20 min, then incubate at 65 °C, pH 5.5 buffer for 10 min).
4. Discussion
Maltogenic amylase is a novel amylase, with maltose as the main hydrolysate, and it has important applications in industry, medicine, and baking. Researchers have studied this enzyme from simple cloning and heterologous expression to exploring strategies to improve its expression level, commonly used tools such as promoter screening, signal peptide optimization, and terminator selection. While conventional research tools have been effective in increasing enzyme expression, in most cases, their effect is limited. Therefore, we should think about how to improve the heterologous expression level of the enzyme from other aspects. In this study, we considered three aspects of the strategies to increase the heterologous expression of AmyM: improving the transcription level of the target gene, optimizing the target protein secretion pathway, and improving the cellular metabolic energy. The results of this study show that all three strategies have good positive effects.
The promoter plays a key role in regulating gene transcription, and dual promoters have attracted the attention of researchers because of their high efficiency and continuity [19]. Yi Rao et al. expressed α-amylase with a double promoter p43-pykzA, which increased the enzymatic activity of α-amylase by 1.85-fold compared to single promoter p43 [19]. He Li et al. expressed α-amylase AmyZ1 with the double promoter PspoVG-PspoVG142, and its activity was 3.11-fold higher than that using the single promoter PgroE [20]. B. subtilis has been widely used as a host to express proteins for a variety of industrial applications. Thus, the effective expression of recombinant proteins relies heavily on the efficient and powerful promoters in B. subtilis [21]. For the selection of promoters, we first selected the strong promoter P43 from B. subtilis and the promoter Pcd from B. amyloliquefaciens, which belongs to the same type as P43. In addition, since AmyM belongs to a class of amylases, we selected the amylase promoter PamyE from B. subtilis, the amylase promoter PamyS from B. amylolyticus, and the amylase promoter PamyL from B. licheniformis. In this study, seven different promoters were constructed and screened, from which the most effective double promoter, P43-PamyL, was selected. P43-PamyL increased the expression level of AmyM by 61.25% compared to the traditional strong promoter P43 in B. subtilis. Although the use of a B. subtilis-derived strong promoter in tandem with an amylase promoter was beneficial in increasing the transcriptional level of AmyM, the enzymatic activity data showed that there is still much room to improve the expression of AmyM.
The Sec secretion pathway is the predominant protein secretion pathway in B. subtilis, with over 90% of extracellular proteins being secreted through this pathway [22]. The prsA protein is a membrane-anchored peptidyl propyl cis-trans isomerase in B. subtilis and most other Gram-positive bacteria [23]. It catalyzes the post-translocation folding of the exported protein and is essential in the Sec secretion pathway of B. subtilis. It was found that prsA promotes the secretion of intracellular proteins into the extracellular compartment, thereby increasing the amount of protein in the extracellular supernatant. In addition, it is essential for the normal growth of B. subtilis. Liyu Xu et al. expressed prsA lipoprotein in tandem with the β-mannanase gene in B. subtilis 168, in which β-mannanase secretion was increased by 15.4% compared to the control strain [24]. Ane Quesada-Ganuza et al. [25] increased the final production of five extracellular α-amylases by up to 2.5-fold by co-expressing heterologous prsA. Considering that the high expression of prsA has the potential to adversely affect the host, we did not select the plasmid to express prsA but integrated the prsA gene into the srfAC locus on the genome of B. subtilis WB800. To ensure the rigor of the study, we first knocked out 500 bp sequence of the srfAC gene alone and examined whether knocking out the gene alone could affect the expression level of AmyM. Then, we integrated two different strength promoters for the over-expression of the prsA gene and investigated whether the two strains showed different effect. The results revealed that the expression level of AmyM is slightly higher in WB800-ΔsrfAC than in WB800-WT, with an increase in enzyme activity of approximately 16.75% (Figure 2h), while the enzyme activity level of AmyM in WB800-pamyX-prsA does not seem to be increased. Encouragingly, the strain over-expressing prsA by using the p43 promoter showed a significant positive effect, with the enzymatic activity of AmyM in this strain increasing by 101.58% compared to that in the wild-type strain.
To analyze the effect of prsA on the secretion of AmyM, we compared intracellular AmyM concentration between strain with prsA and strain without prsA. The SDS-PAGE results revealed that there was no intracellular AmyM band in the sample of WB800-p43-prsA, while clear bands of the target protein were seen in WB800-WT, WB800-ΔsrfAC, and WB800-pamyX-prsA (Figure 2g). This result demonstrates that prsA acts as a molecular chaperone protein that effectively prevents the target protein AmyM from forming inclusion bodies and promotes its secretion. This strategy significantly increases its extracellular expression level of AmyM. The role of prsA depends on its own expression controlled by different promoters, and promoter P43 acts as the best promoter. To further verify the accuracy of the results, we performed semi-quantitative RT-PCR validation on four strains, WB800-WT, WB800-ΔsrfAC, WB800-pamyX-prsA, and WB800-p43-prsA. The results showed that the transcription level of prsA in WB800-p43-prsA was the highest (Figure 2i). The amount of prsA transcripts was also higher in WB800-pamyX-prsA than in WB800-WT and WB800-ΔsrfAC. In addition, it was also uncanny to us that in strains WB800-P43-prsA and WB800-PamyX-prsA, AmyM transcription was significantly higher than in strains WB800-WT and WB800-ΔsrfAC. Regrettably, the amount of intracellular AmyM protein was not significantly reduced, and the extracellular supernatant enzyme activity of AmyM was not improved in the WB800-PamyX-prsA strain. After performing time-series RNA-seq experiments, researchers found that the over-expression of prsA can affect ATP biosynthetic activity, amino acid metabolism, and cell wall stability [26]. Therefore, we speculate that the expression of prsA may need to reach a certain level in order to be effective, and that small amounts of over-expression cannot lead to a significant effect. Based on these results, we recommend that a strong promoter should be selected when prsA is expressed via genomic integration. This will achieve the aim of optimizing the secretory pathway of the target protein and, thus, increase its extracellular expression.
The third strategy in this study was to integrate a vgb gene derived from Vitreoscilla into the WB800 genome. The expression product of vgb is the hemoglobin VHB. VHB was the first bacterial hemoglobin to be discovered and is a soluble hemoglobin with a high rate of oxygen dissociation that promotes cell growth, product synthesis, and cellular resilience [27]. We integrated P43-vgb at the srfAC locus, and the enzyme activity of the strain integrated with the vgb gene was increased by 204.08%, compared with the wild-type strain. RT-PCR results showed that the vgb gene was transcriptionally expressed in WB800-p43-vgb, and there was a substantial increase in the transcription level of the AmyM gene in this strain (Figure 3e). This result also confirms that VHB can indeed increase the level of cell product accumulation in B. subtilis.
Huaiyuan Zhang et al. introduced the vgb gene into Mucor circinelloides and found that it could alleviate oxygen limitation, thereby promoting cell growth and fatty acid synthesis [28]. Jun-feng Li et al. transformed the vgb gene into S. bombicola O-13-1 and found that expression of the gene increased the efficiency of intracellular oxygen utilization under oxygen-enriched or oxygen-limited conditions [29]. A study by Karim E. Jaén et al. found that expression of the vgb gene reduced the accumulation of fermentation by-products in the strain and that the cells showed higher oxidative activity [30].
By measuring the levels of NADP+ and NADPH in WB800-WT and WB800-P43-vgb, we found that in WB800-WT, NADP+ was gradually decreasing with increasing incubation time, while NADPH first increased briefly at 24 h and then began to decrease. In WB800-P43-vgb, on the other hand, both NADP+ and NADPH first showed an increasing trend and reached a maximum content after 24 h of incubation, and then the content started to decrease with a longer incubation time. It is undeniable that the levels of NADP+ and NADPH in WB800-P43-vgb were higher than in WB800-WT during the first 48 h incubation. The difference was most distinct at 24 h of incubation, when the levels of NADP+ and NADPH in WB800-P43-vgb were 14 and 18.9 times higher than in WB800-WT, respectively. NADPH is considered a key cofactor in antioxidant defense and reductive biosynthesis [31], not only in the biosynthesis of key metabolites but also in the regeneration of reduced forms of glutathione, thioredoxin, and peroxisomal proteins [32]. Xiumin Ding et al. [33] constructed two NADPH regeneration systems and, therefore, improved the adaptability between substance and energy metabolism in the MK-7 synthesis pathway in B. subtilis. Hui-Jun Du et al. [34] increased the amount of NADPH in E. coli cells by 134.4%, resulting in a 3.7-fold increase in the yield of the target product. Yuzheng Wu [33] et al. found that elevated intracellular NADPH levels in B. subtilis increased the conversion of inositol. This phenomenon also demonstrated that the intracellular oxygen utilization efficiency was significantly enhanced in WB800-P43-vgb, and the intracellular respiratory metabolism was strong, which facilitated cell growth and metabolite accumulation, thus increasing the expression of AmyM.
In a preliminary investigation of the enzymatic properties of AmyM, we found that the relative enzyme activity of the target protein was low at reaction temperatures below 50 °C (Figure 4a). When the temperature was greater than 50 °C, the relative enzyme activity tended to rise linearly, eventually reaching a maximum at 65 °C before starting to decline again. In the previous report, the optimum temperature of AmyM was 60 °C [15,35]. In our study, the relative enzyme activity of AmyM reached a maximum at 65 °C, but the difference was smaller than that at 60 °C. Therefore, we believe that the difference of 5 °C between the optimum temperature that we determined and that determined in the previous study is due to experimental error. This indicates that the enzyme is suitable for catalysis at medium-to-high temperatures and is a medium-to-high temperature amylase. Yingqi Ruan et al. screened a mutant via error-prone PCR with an optimum temperature of 65 °C, which proved to be superior to the wild type with an optimum temperature of only 60 °C in a bread-baking application [36]. Combined with the temperature stability measurement, the residual activity of AmyM was above 90% after 20 min of treatment between 40 °C and 70 °C (Figure 4b), indicating that the enzyme has good temperature stability and a wide range of action temperature. Interestingly, after 20 min of treatment at 80 °C, the enzyme activity dropped sharply to a residual activity of less than 10%, and it is assumed that if the enzyme is continuously heated, it will lose its activity completely. This is an important characteristic in baking applications. In baking, the core temperature can eventually reach above 95 °C, which is sufficient to inactivate the enzyme to prevent the enzyme from over-reacting and making the dough sticky, ultimately destroying the fluffiness of the bread.
As for the optimum pH of AmyM, it can be seen that the relative enzyme activity of AmyM was below 10% at pH values less than 4 (Figure 4c). The relative enzyme activity remained above 80% between 4.5 and 6.5, with the highest enzyme activity at pH 5.5 and a rapid decrease in enzyme activity when the pH was greater than 7.5. The optimum pH of AmyM is consistent with that in previous reports [15,35]. When the pH stability of AmyM was measured, it was seen that after treating the enzyme solution with a buffer of a pH value less than 4.0 for 20 min, the residual enzyme activity was less than 10% (Figure 4d). In contrast, when pH was greater than 4.5, the enzyme had good stability with a wide pH range.
5. Conclusions
In this study, we significantly increased the extracellular expression of maltose amylase AmyM by increasing the transcriptional level of the target gene, optimizing the protein secretion pathway, and improving cellular metabolic activity. As for promoter screening, we found that the strong promoter p43 combined with an amylase promoter PamyL gave the best results, with a 61.25% increase in extracellular enzyme activity compared to the control strain. As for the protein secretion pathway, we over-expressed the prsA gene via various promoters for the first time and showed that the preferred strong promoter gives good results, with extracellular AmyM activity increasing by 101.58% compared to the control strain. Although many studies have shown that cellular metabolic activity can be improved by integrating the vgb gene, we attempted to introduce the gene to promote the expression of AmyM in B. subtilis. The results were surprising, with the introduction of vgb resulting in a 204.08% increase in AmyM activity, suggesting an effective strategy that has important implications in other organisms, particularly aerobic ones. In addition, the enzymatic properties of AmyM have been determined to have excellent temperature and pH stability, and its optimum reaction temperature and pH are mild, which renders it a promising candidate for industrial applications. Our future research will focus on increasing the specific enzymatic activity of AmyM through rational or semi-rational design, aiming to further broaden its action temperature and pH in order to increase its utilization value.
Author Contributions
Conceptualization, Y.C. and B.W.; methodology, Y.C., Q.X., L.P. and B.W.; validation, Y.C. and B.W.; formal analysis, Y.C. and B.W.; investigation, Y.C., L.P. and B.W.; resources, Y.C., Q.X., L.P., and B.W.; writing—original draft preparation, Y.C. and B.W.; visualization, Y.C. and B.W.; funding acquisition, Y.C., L.P. and B.W. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by R&D project in key areas of Guangdong Province (grant number 2020B020226007),the National Key Research and Development Program of China (Project No.2021YFC2100200), the Natural Science Foundation of Guangdong Province (grant number 2022A1515010291), and R&D projects in key areas of Guangdong Province (grant number 2022B0202010001).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All relevant data generated or analyzed during this study are included in this article.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bijttebier, A.; Goesaert, H.; Delcour, J.A. Amylase action pattern on starch polymers. Biologia 2008, 63, 989–999. [Google Scholar] [CrossRef]
- Dauter, Z.; Dauter, M.; Brzozowski, A.M.; Christensen, S.; Borchert, T.V.; Beier, L.; Wilson, K.S.; Davies, G.J. X-ray structure of Novamyl, the five-domain ‘maltogenic’ alpha-amylase from Bacillus stearothermophilus: Maltose and Acarbose Complexes at 1.7 Å Resolution. Biochemistry 1999, 38, 8385–8392. [Google Scholar] [CrossRef]
- Nguyen, N.T.L.; Nguyen, B.D.T.; Dai, T.T.X.; Co, S.H.; Do, T.T.; Tong Thi, A.N.; Oladapo, I.J.; Nguyen Cong, H. Influence of germinated brown rice-based flour modified by MAse on type 2 diabetic mice and HepG2 cell cytotoxic capacity. Food Sci. Nutr. 2021, 9, 781–793. [Google Scholar] [CrossRef] [PubMed]
- Rebholz, G.F.; Sebald, K.; Dirndorfer, S.; Dawid, C.; Hofmann, T.; Scherf, K.A. Impact of exogenous maltogenic α-amylase and maltotetraogenic amylase on sugar release in wheat bread. Eur. Food Res. Technol. 2021, 247, 1425–1436. [Google Scholar] [CrossRef]
- Roozbehi, S.; Dadashzadeh, S.; Sajedi, R.H. An enzyme-mediated controlled release system for curcumin based on cyclodextrin/cyclodextrin degrading enzyme. Enzym. Microb. Technol. 2021, 144, 109727. [Google Scholar] [CrossRef] [PubMed]
- Kunst, F.; Ogasawara, N.; Moszer, I.; Albertini, A.M.; Danchin, A.J.N. The complete genome sequence of the Gram-positive bacterium Bacillus subtilis. Nature 1997, 390, 249–256. [Google Scholar] [CrossRef]
- Ehrlich, S.D.; Bierne, H.; d’Alençon, E.; Vilette, D.; Petranovic, M.; Noirot, P.; Michel, B. Mechanisms of illegitimate recombination. Gene 1993, 135, 161–166. [Google Scholar] [CrossRef]
- Dulermo, R.; Brunel, F.; Dulermo, T.; Ledesma-Amaro, R.; Vion, J.; Trassaert, M.; Thomas, S.; Nicaud, J.M.; Leplat, C. Using a vector pool containing variable-strength promoters to optimize protein production in Yarrowia lipolytica. Microb. Cell Fact. 2017, 16, 31. [Google Scholar] [CrossRef]
- Wang, Y.; Peng, Q.; Mou, X.; Wang, X.; Li, H.; Han, T.; Sun, Z.; Wang, X. A successful hybrid deep learning model aiming at promoter identification. BMC Bioinform. 2022, 23, 206. [Google Scholar] [CrossRef]
- Zhao, Y.; Liu, S.; Lu, Z.; Zhao, B.; Wang, S.; Zhang, C.; Xiao, D.; Foo, J.L.; Yu, A. Hybrid promoter engineering strategies in Yarrowia lipolytica: Isoamyl alcohol production as a test study. Biotechnol. Biofuels 2021, 14, 149. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, X.; Qiu, J.; Wang, K.; Meng, K.; Luo, H.; Su, X.; Ma, R.; Huang, H.; Yao, B. Development of Bacillus amyloliquefaciens as a high-level recombinant protein expression system. J. Ind. Microbiol. Biotechnol. 2019, 46, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Su, L.; Wu, J. Enhancing Extracellular Pullulanase Production in Bacillus subtilis Through dltB Disruption and Signal Peptide Optimization. Appl. Biochem. Biotechnol. 2022, 194, 1206–1220. [Google Scholar] [CrossRef]
- Su, L.; Li, Y.; Wu, J. Efficient secretory expression of Bacillus stearothermophilus alpha/beta-cyclodextrin glycosyltransferase in Bacillus subtilis. J. Biotechnol. 2021, 331, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Ma, Y.; Zhao, Y.; Shen, W.; Chen, X. Systematic engineering of transport and transcription to boost alkaline alpha-amylase production in Bacillus subtilis. Appl. Microbiol. Biotechnol. 2020, 104, 2973–2985. [Google Scholar] [CrossRef]
- Li, Y.; Su, Y.; Wu, J.; Wu, D. Recombinant Expression and Fermentation Optimization of B. stearothermophilu Maltogenic Amylases in Bacillus subtilis. J. Food Sci. Biotechnol. 2020, 39, 9. [Google Scholar]
- Tsai, P.S.; Hatzimanikatis, V.; Bailey, J.E.J.B. Bioengineering. Effect of Vitreoscilla hemoglobin dosage on microaerobic Escherichia coli carbon and energy metabolism. Biotechnol. Bioeng. 1996, 49, 139–150. [Google Scholar] [CrossRef]
- Xin, Q.; Chen, Y.; Chen, Q.; Wang, B.; Pan, L. Development and application of a fast and efficient CRISPR-based genetic toolkit in Bacillus amyloliquefaciens LB1ba02. Microb. Cell Fact. 2022, 21, 99. [Google Scholar] [CrossRef]
- Cao, G.; Zhang, X.; Zhong, L.; Lu, Z. A modified electro-transformation method for Bacillus subtilis and its application in the production of antimicrobial lipopeptides. Biotechnol. Lett. 2011, 33, 1047–1051. [Google Scholar] [CrossRef]
- Rao, Y.; Cai, D.; Wang, H.; Xu, Y.; Xiong, S.; Gao, L.; Xiong, M.; Wang, Z.; Chen, S.; Ma, X. Construction and application of a dual promoter system for efficient protein production and metabolic pathway enhancement in Bacillus licheniformis. J. Biotechnol. 2020, 312, 1–10. [Google Scholar] [CrossRef]
- Li, H.; Yao, D.; Pan, Y.; Chen, X.; Fang, Z.; Xiao, Y. Enhanced extracellular raw starch-degrading alpha-amylase production in Bacillus subtilis by promoter engineering and translation initiation efficiency optimization. Microb. Cell Fact. 2022, 21, 127. [Google Scholar] [CrossRef]
- Liu, Y.; Shi, C.; Li, D.; Chen, X.; Li, J.; Zhang, Y.; Yuan, H.; Li, Y.; Lu, F. Engineering a highly efficient expression system to produce BcaPRO protease in Bacillus subtilis by an optimized promoter and signal peptide. Int. J. Biol. Macromol. 2019, 138, 903–911. [Google Scholar] [CrossRef] [PubMed]
- Tjalsma, H.; Antelmann, H.; Jongbloed, J.D.; Braun, P.G.; Darmon, E.; Dorenbos, R.; Dubois, J.Y.; Westers, H.; Zanen, G.; Quax, W.J.; et al. Proteomics of protein secretion by Bacillus subtilis: Separating the “secrets” of the secretome. Microbiol. Mol. Biol. Rev. 2004, 68, 207–233. [Google Scholar] [CrossRef]
- Hyyrylainen, H.L.; Marciniak, B.C.; Dahncke, K.; Pietiainen, M.; Courtin, P.; Vitikainen, M.; Seppala, R.; Otto, A.; Becher, D.; Chapot-Chartier, M.P.; et al. Penicillin-binding protein folding is dependent on the PrsA peptidyl-prolyl cis-trans isomerase in Bacillus subtilis. Mol. Microbiol. 2010, 77, 108–127. [Google Scholar] [CrossRef]
- Xu, L.; Zhang, Y.; Dong, Y.; Qin, G.; Zhao, X.; Shen, Y. Enhanced extracellular beta-mannanase production by overexpressing PrsA lipoprotein in Bacillus subtilis and optimizing culture conditions. J. Basic Microbiol. 2022, 62, 815–823. [Google Scholar] [CrossRef] [PubMed]
- Quesada-Ganuza, A.; Antelo-Varela, M.; Mouritzen, J.C.; Bartel, J.; Becher, D.; Gjermansen, M.; Hallin, P.F.; Appel, K.F.; Kilstrup, M.; Rasmussen, M.D.; et al. Identification and optimization of PrsA in Bacillus subtilis for improved yield of amylase. Microb. Cell Fact. 2019, 18, 158. [Google Scholar] [CrossRef]
- Geissler, A.S.; Poulsen, L.D.; Doncheva, N.T.; Anthon, C.; Seemann, S.E.; Gonzalez-Tortuero, E.; Breuner, A.; Jensen, L.J.; Hjort, C.; Vinther, J.; et al. The impact of PrsA over-expression on the Bacillus subtilis transcriptome during fed-batch fermentation of alpha-amylase production. Front. Microbiol. 2022, 13, 909493. [Google Scholar] [CrossRef]
- Yu, F.; Zhao, X.; Wang, Z.; Liu, L.; Yi, L.; Zhou, J.; Li, J.; Chen, J.; Du, G. Recent Advances in the Physicochemical Properties and Biotechnological Application of Vitreoscilla Hemoglobin. Microorganisms 2021, 9, 1455. [Google Scholar] [CrossRef]
- Zhang, H.; Kang, X.; Wang, R.; Xin, F.; Chang, Y.; Zhang, Y.; Song, Y. Overexpression of Shinorhizobium meliloti flavohemoglobin improves cell growth and fatty acid biosynthesis in oleaginous fungus Mucor circinelloides. Biotechnol. Lett. 2022, 44, 595–604. [Google Scholar] [CrossRef]
- Li, J.F.; Li, H.F.; Yao, S.M.; Zhao, M.J.; Dong, W.X.; Liang, S.K.; Xu, X.Y. Vitreoscilla Hemoglobin Improves Sophorolipid Production in Starmerella Bombicola O-13-1 Under Oxygen Limited Conditions. Front. Bioeng. Biotechnol. 2021, 9, 773104. [Google Scholar] [CrossRef]
- Jaen, K.E.; Velazquez, D.; Delvigne, F.; Sigala, J.C.; Lara, A.R. Engineering E. coli for improved microaerobic pDNA production. Bioprocess Biosyst. Eng. 2019, 42, 1457–1466. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Kou, J.; Qin, J.; Li, L.; Zhang, Z.; Pan, Y.; Xue, Y.; Du, W. NADPH levels affect cellular epigenetic state by inhibiting HDAC3-Ncor complex. Nat. Metab. 2021, 3, 75–89. [Google Scholar] [CrossRef]
- Yadav, S.; Mody, T.A.; Sharma, A.; Bachhawat, A.K. A Genetic Screen To Identify Genes Influencing the Secondary Redox Couple NADPH/NADP(+) in the Yeast Saccharomyces cerevisiae. G3 2020, 10, 371–378. [Google Scholar] [CrossRef]
- Wu, Y.; Kawabata, H.; Kita, K.; Ishikawa, S.; Tanaka, K.; Yoshida, K.I. Constitutive glucose dehydrogenase elevates intracellular NADPH levels and luciferase luminescence in Bacillus subtilis. Microb. Cell Fact. 2022, 21, 266. [Google Scholar] [CrossRef]
- Du, H.-J.; Luo, W.; Appiah, B.; Zou, Z.-C.; Yang, Z.-H.; Zeng, R.; Luo, L. Promotion of the Asymmetric Reduction of Prochiral Ketone with Recombinant E. coli Through Strengthening Intracellular NADPH Supply by Modifying EMP and Introducing NAD Kinase. Catal. Lett. 2021, 151, 2527–2536. [Google Scholar] [CrossRef]
- Sun, Y.; Duan, X.; Wang, L.; Wu, J. Enhanced maltose production through mutagenesis of acceptor binding subsite +2 in Bacillus stearothermophilus maltogenic amylase. J. Biotechnol. 2016, 217, 53–61. [Google Scholar] [CrossRef]
- Ruan, Y.; Zhang, R.; Xu, Y. Directed evolution of maltogenic amylase from Bacillus licheniformis R-53: Enhancing activity and thermostability improves bread quality and extends shelf life. Food Chem. 2022, 381, 132222. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).