

Engineering the C3N Pathway as a Short Detour for De Novo NAD+ Biosynthesis in Saccharomyces cerevisiae

Abstract
:1. Introduction
2. Materials and Methods
2.1. Strains, Plasmids, and Growth Conditions
2.2. General DNA Manipulations and Sequence Analyses
2.3. Construction of S. cerevisiae BY01
2.4. Expression of phzD, phzE, and dhbX Individually in S. cerevisiae
2.5. Construction of S. cerevisiae BY00-Con, BY00-C3N, BY01-Con, and BY01-C3N
2.6. Growth of S. cerevisiae Strains on YMMN Plates
2.7. Measurement of Intracellular NAD(H) Concentrations
2.8. Spectroscopic Analysis
2.9. Sequence Data
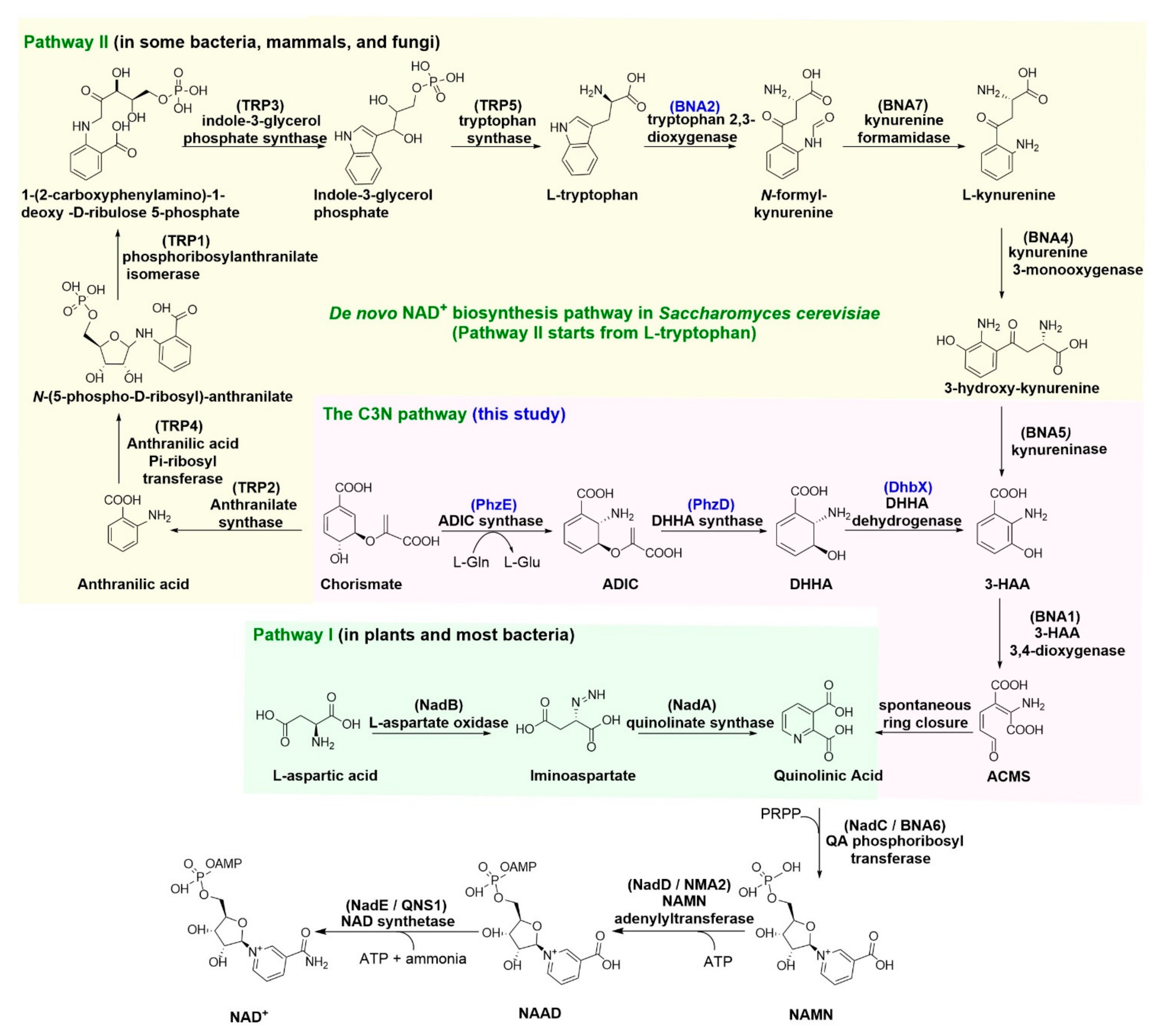
3. Results
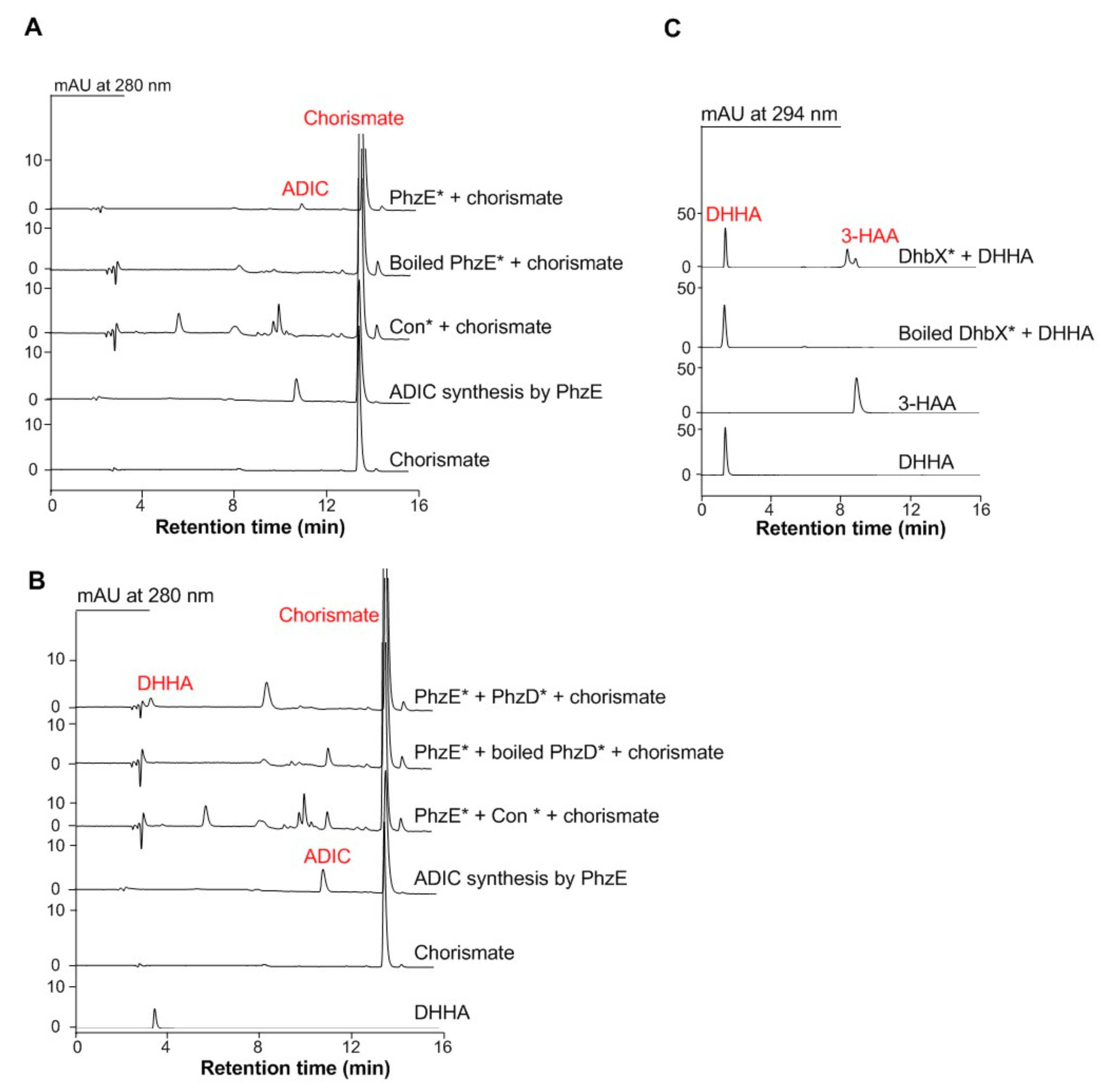
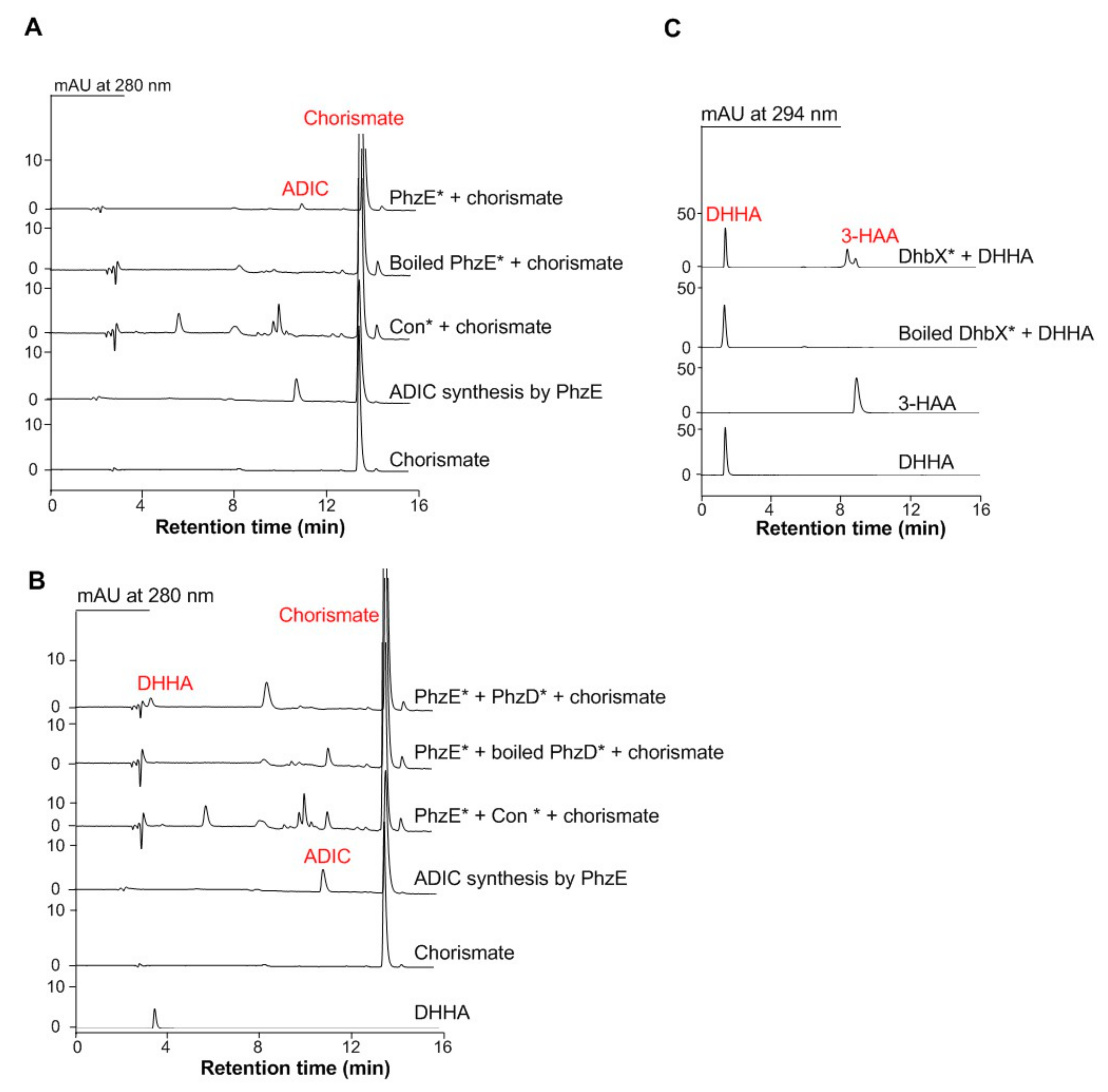
3.1. Functional Expression of the C3N Pathway Genes in S. cerevisiae
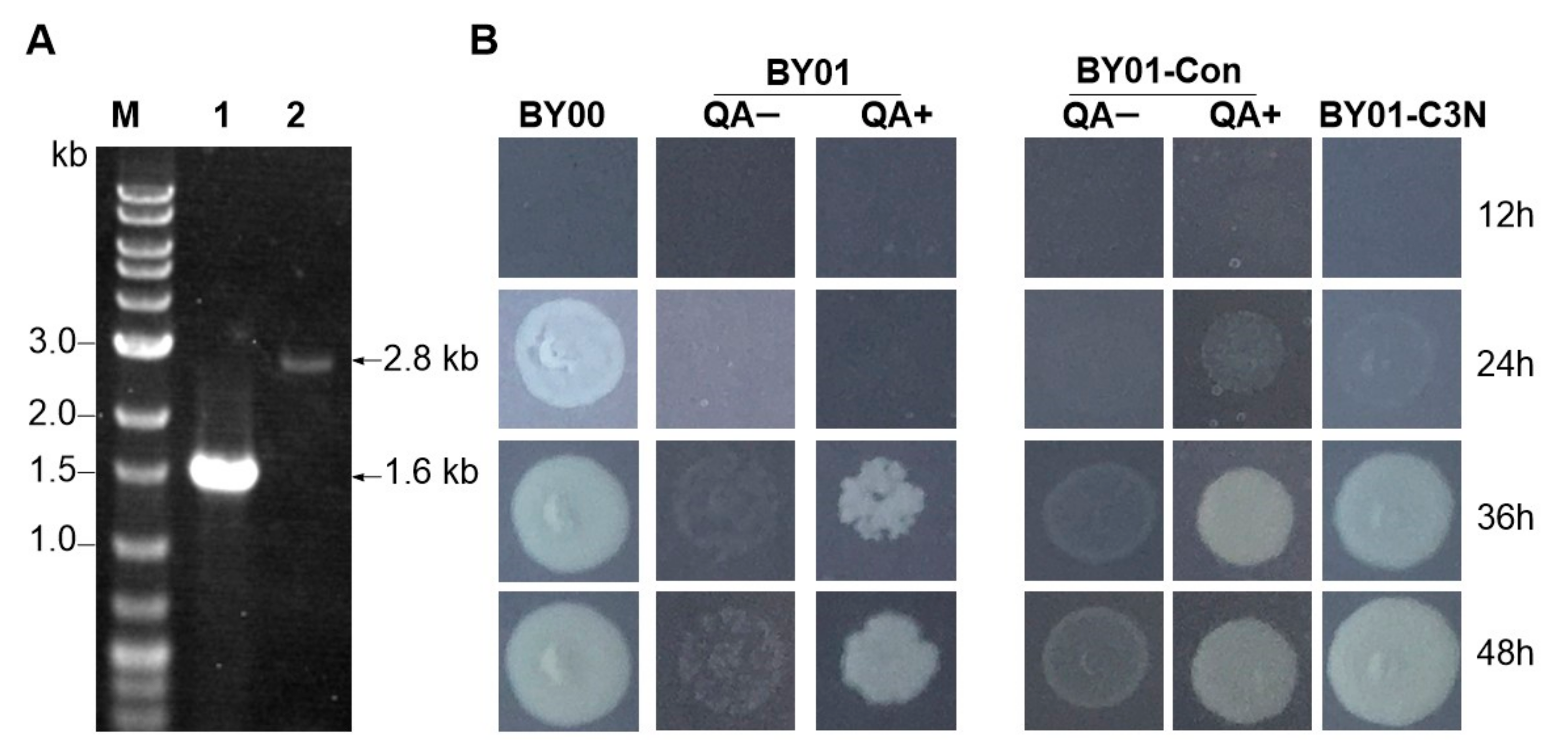
3.2. Construction of the C3N Pathway in S. cerevisiae
3.3. Expanding the NAD(H) Pool of S. cerevisiae by the C3N Pathway
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Selles, V.L.; Kelly, C.L.; Mordaka, P.M.; Heap, J.T. Review of NAD(P)H-dependent oxidoreductases: Properties, engineering and application. Biochim. Et Biophys. Acta (BBA) Proteins Proteom. 2018, 1866, 327–347. [Google Scholar] [CrossRef] [PubMed]
- Covarrubias, A.J.; Perrone, R.; Grozio, A.; Verdin, E. NAD+ metabolism and its roles in cellular processes during ageing. Nat. Rev. Mol. Cell Biol. 2021, 22, 119–141. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Chen, B.; Fang, Y.; Tan, T. Cofactor engineering for more efficient production of chemicals and biofuels. Biotechnol. Adv. 2017, 35, 1032–1039. [Google Scholar] [CrossRef] [PubMed]
- Ying, W. NAD+/NADH and NADP+/NADPH in cellular functions and cell death: Regulation and biological consequences. Antioxid. Redox Signal. 2018, 10, 179–206. [Google Scholar] [CrossRef] [PubMed]
- Katsyuba, E.; Auwerx, J. Modulating NAD+ metabolism, from bench to bedside. EMBO J. 2017, 36, 2670–2683. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.J.; Yang, W.; Wang, L.; Zhu, Z.; Zhang, S.; Zhao, Z.K. Engineering NAD+ availability for Escherichia coli whole-cell biocatalysis: A case study for dihydroxyacetone production. Microb. Cell Factories 2013, 12, 103. [Google Scholar] [CrossRef]
- Li, F.; Li, Y.; Cao, Y.; Wang, L.; Liu, C.; Shi, L.; Song, H. Modular engineering to increase intracellular NAD(H/+) promotes rate of extracellular electron transfer of Shewanella oneidensis. Nat. Commun. 2018, 9, 3637. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, H.; Zhao, G.; Caiyin, Q.; Qiao, J. Redox cofactor engineering in industrial microorganisms: Strategies, recent applications and future directions. J. Ind. Microbiol. Biotechnol. 2018, 45, 313–327. [Google Scholar] [CrossRef]
- Sharma, S.; Hsieh, Y.-C.; Dietze, J.; Bockwoldt, M.; Strømland, Ø.; Ziegler, M.; Heiland, I. Early Evolutionary Selection of NAD Biosynthesis Pathway in Bacteria. Metabolites 2022, 12, 569. [Google Scholar] [CrossRef]
- Heuser, F.; Schroer, K.; Lütz, S.; Bringer-Meyer, S.; Sahm, H. Enhancement of the NAD(P)H pool in Escherichia coli for biotransformation. Eng. Life Sci. 2007, 7, 343–353. [Google Scholar] [CrossRef]
- Katoh, A.; Uenohara, K.; Akita, M.; Hashimoto, T. Early steps in the biosynthesis of NAD+ in Arabidopsis start with aspartate and occur in the plastid. Plant Physiol. 2006, 141, 851–857. [Google Scholar] [CrossRef] [PubMed]
- Kurnasov, O.; Goral, V.; Colabroy, K.; Gerdes, S.; Anantha, S.; Osterman, A.; Begley, T.P. NAD biosynthesis: Identification of the tryptophan to quinolinate pathway in bacteria. Chem. Biol. 2003, 10, 1195–1204. [Google Scholar] [CrossRef] [PubMed]
- Rodionov, D.A.; De Ingeniis, J.; Mancini, C.; Cimadamore, F.; Zhang, H.; Osterman, A.L.; Raffaelli, N. Transcriptional regulation of NAD+ metabolism in bacteria: NrtR family of Nudix-related regulators. Nucleic Acids Res. 2008, 36, 2047–2059. [Google Scholar] [CrossRef] [PubMed]
- Malkowski, S.N.; Spencer, T.C.J.; Breaker, R.R. Evidence that the nadA motif is a bacterial riboswitch for the ubiquitous enzyme cofactor NAD+. RNA 2019, 25, 1616–1627. [Google Scholar] [CrossRef] [PubMed]
- Croft, T.; Venkatakrishnan, P.; Raj, C.J.T.; Groth, B.; Cater, T.; Salemi, M.R.; Phinney, B.; Lin, S.J. N-terminal protein acetylation by NatB modulates the levels of Nmnats, the NAD+ biosynthetic enzymes in Saccharomyces cerevisiae. J. Biol. Chem. 2020, 295, 7362–7375. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Li, X.L.; Horsman, G.P.; Li, P.W.; Wang, M.; Li, J.E.; Zhang, Z.; Liu, W.; Wu, B.; Tao, Y.; et al. Construction of an alternative NAD+ de novo biosynthesis pathway. Adv. Sci. 2021, 8, 2004632. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, K.; Yang, Y.; Davis, I.; Liu, A. Observing 3-hydroxyanthranilate-3,4-dioxygenase in action through a crystalline lens. Proc. Natl. Acad. Sci. USA 2020, 117, 19720–19730. [Google Scholar] [CrossRef]
- Hammer, S.K.; Avalos, J.L. Harnessing yeast organelles for metabolic engineering. Nat. Chem. Biol. 2017, 13, 823–832. [Google Scholar] [CrossRef]
- Chen, R.E.; Thorner, J. Function and regulation in MAPK signaling pathways: Lessons learned from the yeast Saccharomyces cerevisiae. Biochim. Biophys. Acta 2007, 1773, 1311–1340. [Google Scholar] [CrossRef]
- Gershon, H. Gershon, D. The budding yeast, Saccharomyces cerevisiae, as a model for aging research: A critical review. Mech. Ageing Dev. 2000, 120, 1–22. [Google Scholar] [CrossRef]
- Raj, C.J.T.; Lin, S.J. Cross-talk in NAD+ metabolism: Insights from Saccharomyces cerevisiae. Curr. Genet. 2019, 65, 1113–1119. [Google Scholar]
- Oliveira, A.P.; Ludwig, C.; Zampieri, M.; Weisser, H.; Aebersold, R.; Sauer, U. Dynamic phosphoproteomics reveals TORC1-dependent regulation of yeast nucleotide and amino acid biosynthesis. Sci. Signal. 2015, 8, rs4. [Google Scholar] [CrossRef] [PubMed]
- Gietz, R.D.; Schiestl, R.H. High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat. Protoc. 2017, 2, 31–34. [Google Scholar] [CrossRef] [PubMed]
- Inoue, H.; Nojima, H.; Okayama, H. High efficiency transformation of Escherichia coli with plasmids. Gene 1990, 96, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Hao, T.; Xie, Z.; Wang, M.; Liu, L.; Zhang, Y.; Wang, W.; Zhang, Z.; Zhao, X.; Li, P.; Guo, Z.; et al. An anaerobic bacterium host system for heterologous expression of natural product biosynthetic gene clusters. Nat. Commun. 2019, 10, 3665. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Chen, M.; Tang, W.; Guo, Z.; Zhang, Y.; Wang, M.; Horsman, G.P.; Zhong, J.; Lu, Z.; Chen, Y. Initiating polyketide biosynthesis by on-line methyl esterification. Nat. Commun. 2021, 12, 4499. [Google Scholar] [CrossRef]
- Kern, S.E.; Price-Whelan, A.; Newman, D.K. Extraction and measurement of NAD(P)+ and NAD(P)H. In Pseudomonas Methods and Protocols; Filloux, A., Ramos, J.L., Eds.; Humana Press: New York, NY, USA, 2014; pp. 311–323. [Google Scholar]
- Day, A.; Schneider, C.; Schneider, B.L. Yeast Cell Synchronization. In Cell Cycle Checkpoint Control Protocols; Lieberman, H., Ed.; Humana Press: New York, NY, USA, 2004; pp. 55–76. [Google Scholar]
- Sporty, J.; Lin, S.J.; Kato, M.; Ognibene, T.; Stewart, B.; Turteltaub, K.; Bench, G. Quantitation of NAD+ biosynthesis from the salvage pathway in Saccharomyces cerevisiae. Yeast 2009, 26, 363–369. [Google Scholar] [CrossRef]
- Tang, W.; Guo, Z.; Cao, Z.; Wang, M.; Li, P.; Meng, X.; Zhao, X.; Xie, Z.; Wang, W.; Zhou, A.; et al. d-Sedoheptulose-7-phosphate is a common precursor for the heptoses of septacidin and hygromycin B. Proc. Natl. Acad. Sci. USA 2018, 115, 2818–2823. [Google Scholar] [CrossRef]
- Culbertson, J.E.; Toney, M.D. Expression and characterization of PhzE from P. aeruginosa PAO1: Aminodeoxyisochorismate synthase involved in pyocyanin and phenazine-1-carboxylate production. Biochim. Biophys. Acta 2013, 183, 240–246. [Google Scholar] [CrossRef]
- Parsons, J.F.; Calabrese, K.; Eisenstein, E.; Ladner, J.E. Structure and mechanism of Pseudomonas aeruginosa PhzD, an isochorismatase from the phenazine biosynthetic pathway. Biochemistry 2003, 42, 5684–5693. [Google Scholar] [CrossRef]
- Iwamoto, Y.; Lee, I.S.; Tsubaki, M.; Kido, R. Tryptophan 2,3-dioxygenase in Saccharomyces cerevisiae. Can. J. Microbiol. 1995, 41, 19–26. [Google Scholar] [CrossRef]
- Almeida, A.M.; Marchiosi, R.; Abrahão, J.; Constantin, R.P.; dos Santos, W.D.; Ferrarese-Filho, O. Revisiting the shikimate pathway and highlighting their enzyme inhibitors. Phytochem. Rev. 2023, 1–37. [Google Scholar] [CrossRef]
- Lynch, J.H.; Dudareva, N. Aromatic amino acids: A complex network ripe for future exploration. Trends Plant Sci. 2020, 25, 670–681. [Google Scholar] [CrossRef]
- Hubrich, F.; Müller, M.; Andexer, J.N. Chorismate- and isochorismate converting enzymes: Versatile catalysts acting on an important metabolic node. Chem. Commun. 2021, 57, 2441–2463. [Google Scholar] [CrossRef]
- Wu, S.; Chen, W.; Lu, S.; Zhang, H.; Yin, L. Metabolic engineering of shikimic acid biosynthesis pathway for the production of shikimic acid and its branched products in microorganisms: Advances and Prospects. Molecules 2022, 27, 4779. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strains | NAD(H) [mM] | NAD+ [mM] |
|---|---|---|
| BY00 | 3.30 ± 0.45 | 1.90 ± 0.24 |
| BY00-Con | 2.92 ± 0.20 | 1.40 ± 0.19 |
| BY01-C3N | 3.13 ± 0.06 | 1.65 ± 0.09 |
| BY00-C3N | 4.59 ± 0.37 | 2.30 ± 0.25 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, X.; Tang, Y.; Ding, Y.; Li, P.; Chen, Y. Engineering the C3N Pathway as a Short Detour for De Novo NAD+ Biosynthesis in Saccharomyces cerevisiae. Fermentation 2023, 9, 886. https://doi.org/10.3390/fermentation9100886
Li X, Tang Y, Ding Y, Li P, Chen Y. Engineering the C3N Pathway as a Short Detour for De Novo NAD+ Biosynthesis in Saccharomyces cerevisiae. Fermentation. 2023; 9(10):886. https://doi.org/10.3390/fermentation9100886
Chicago/Turabian StyleLi, Xinli, Yue Tang, Yong Ding, Pengwei Li, and Yihua Chen. 2023. "Engineering the C3N Pathway as a Short Detour for De Novo NAD+ Biosynthesis in Saccharomyces cerevisiae" Fermentation 9, no. 10: 886. https://doi.org/10.3390/fermentation9100886
APA StyleLi, X., Tang, Y., Ding, Y., Li, P., & Chen, Y. (2023). Engineering the C3N Pathway as a Short Detour for De Novo NAD+ Biosynthesis in Saccharomyces cerevisiae. Fermentation, 9(10), 886. https://doi.org/10.3390/fermentation9100886