
Changes and Driving Mechanism of Microbial Community Structure during Paocai Fermentation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Paocai Production and Sampling
2.2. Determination of Physicochemical Indexes in Paocai
2.3. Metagenomic DNA Extraction
2.4. PCR Amplification and MiSeq Highthroughput Sequencing
2.5. Sequencing Data Processing and Bioinformatics Analysis
3. Results
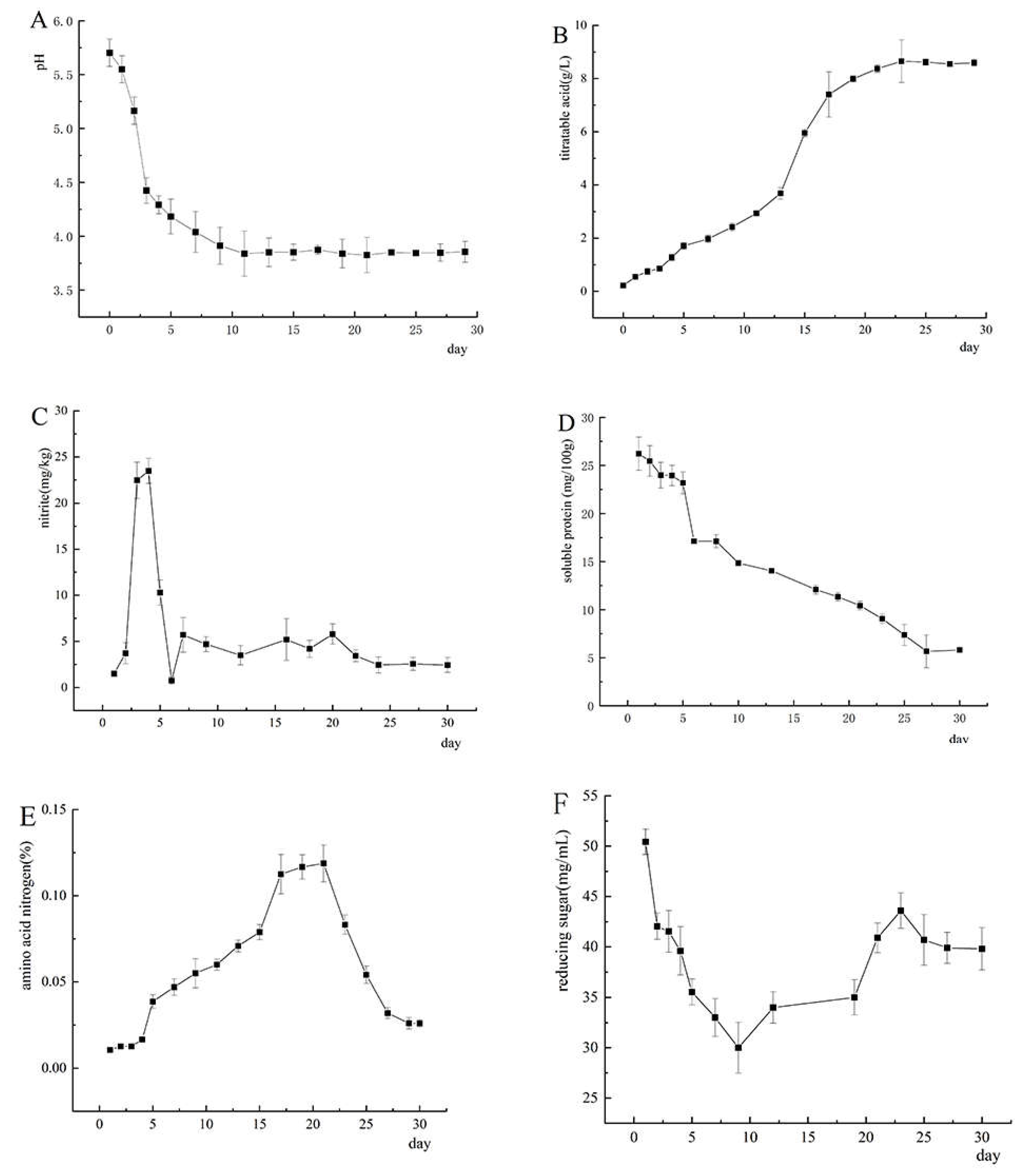
3.1. The Trend of Physicochemical Indexes in Paocai
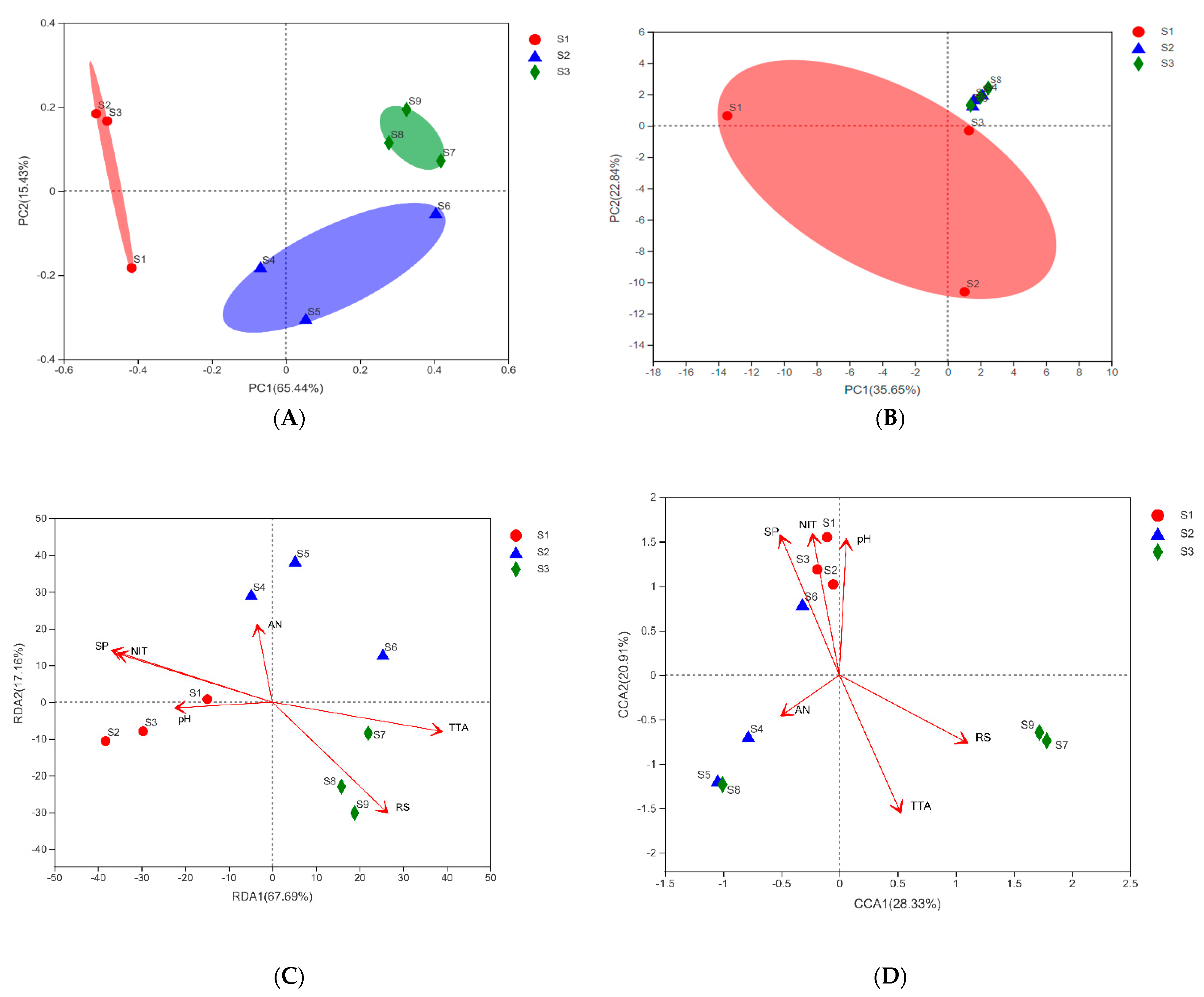
3.2. Sample Grouping Analysis
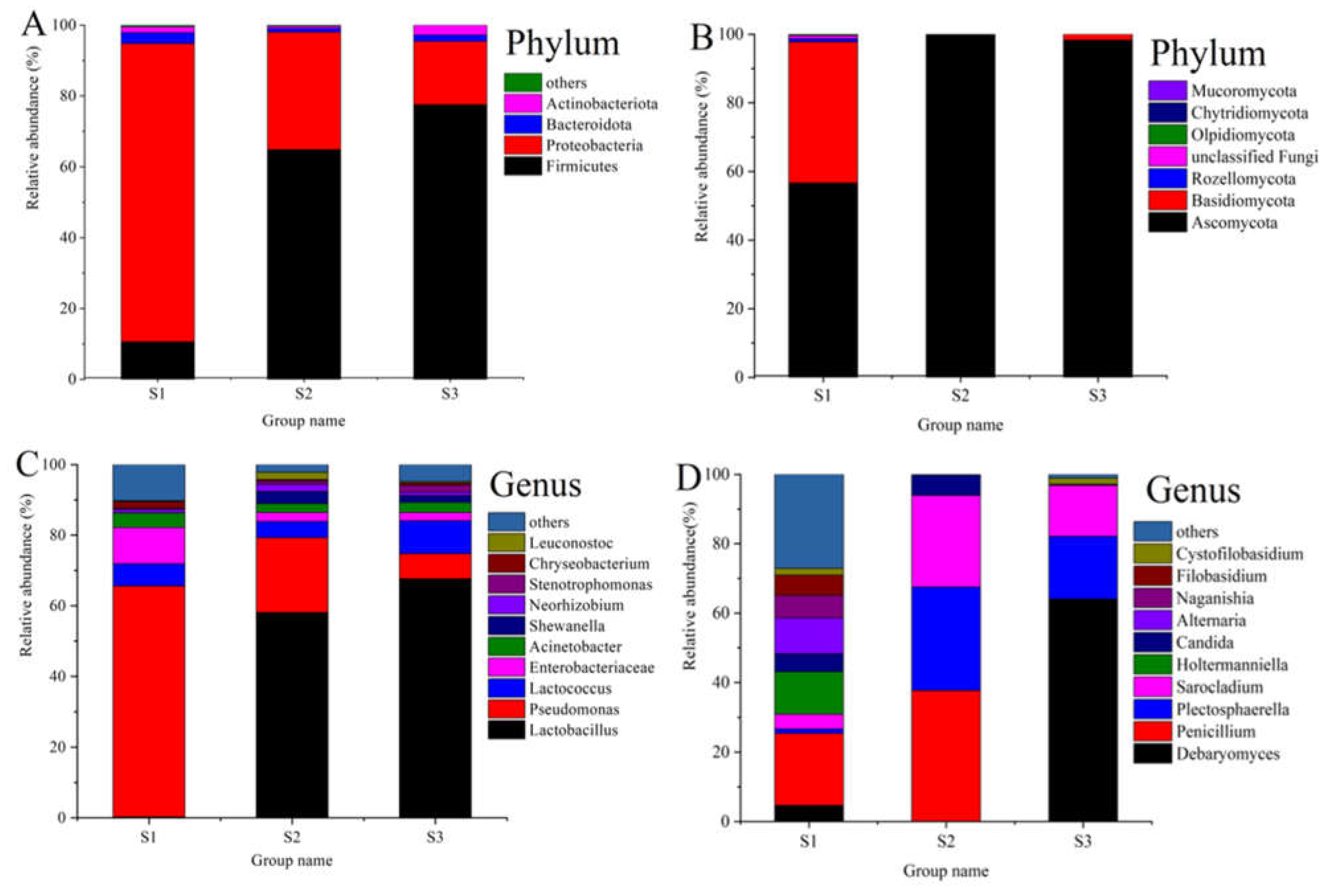
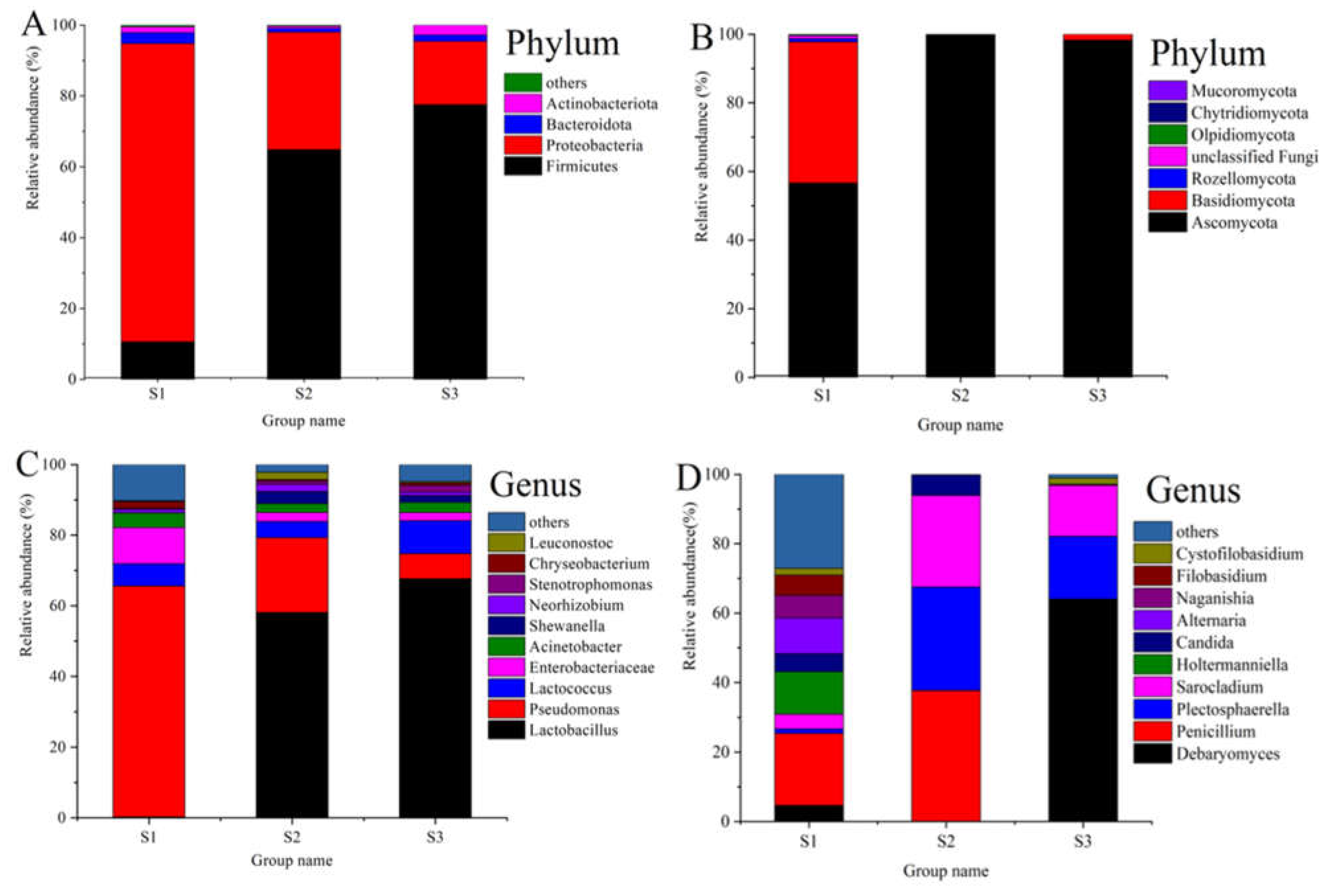
3.3. Composition of Bacterial and Fungal Communities in Paocai Samples
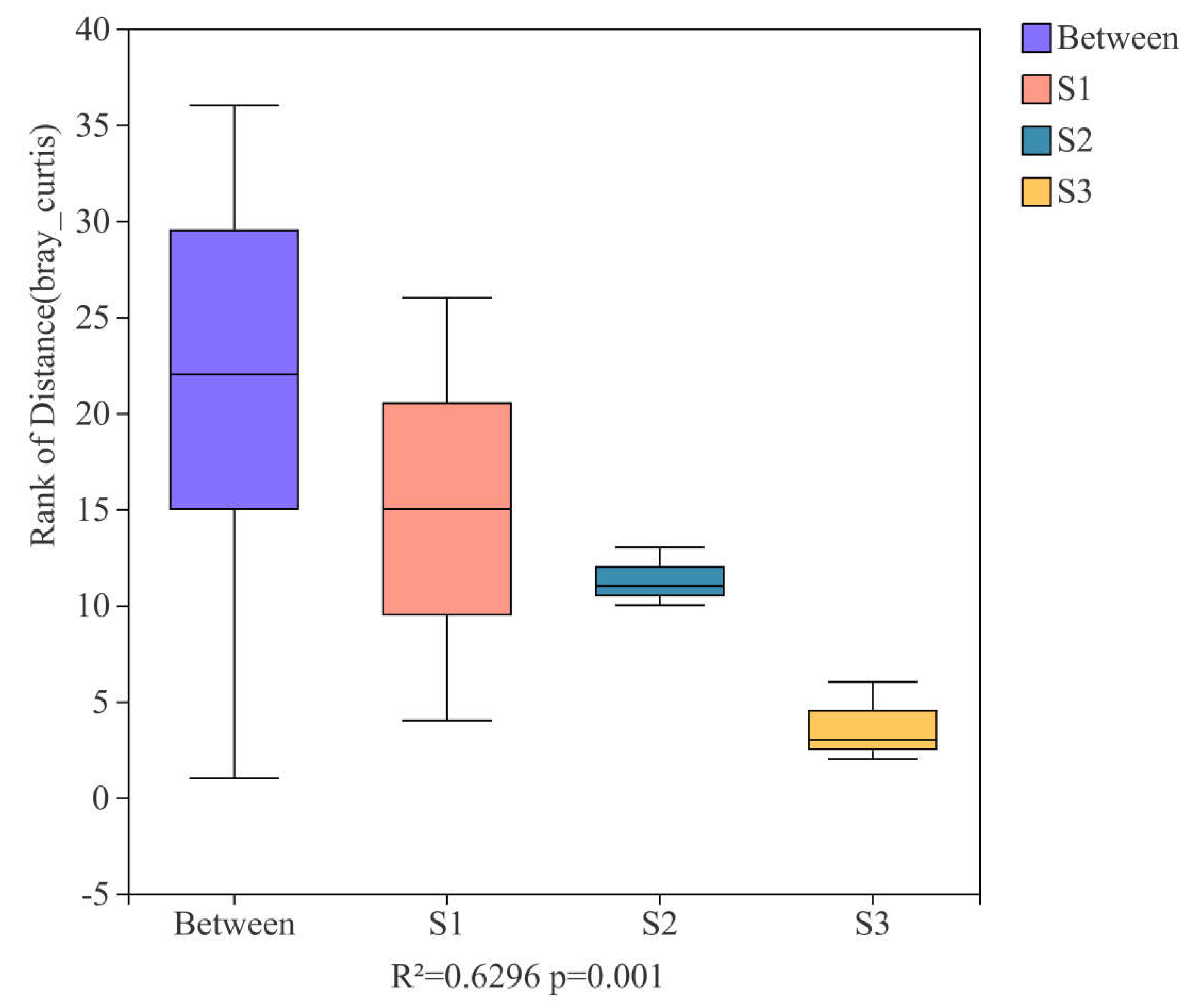
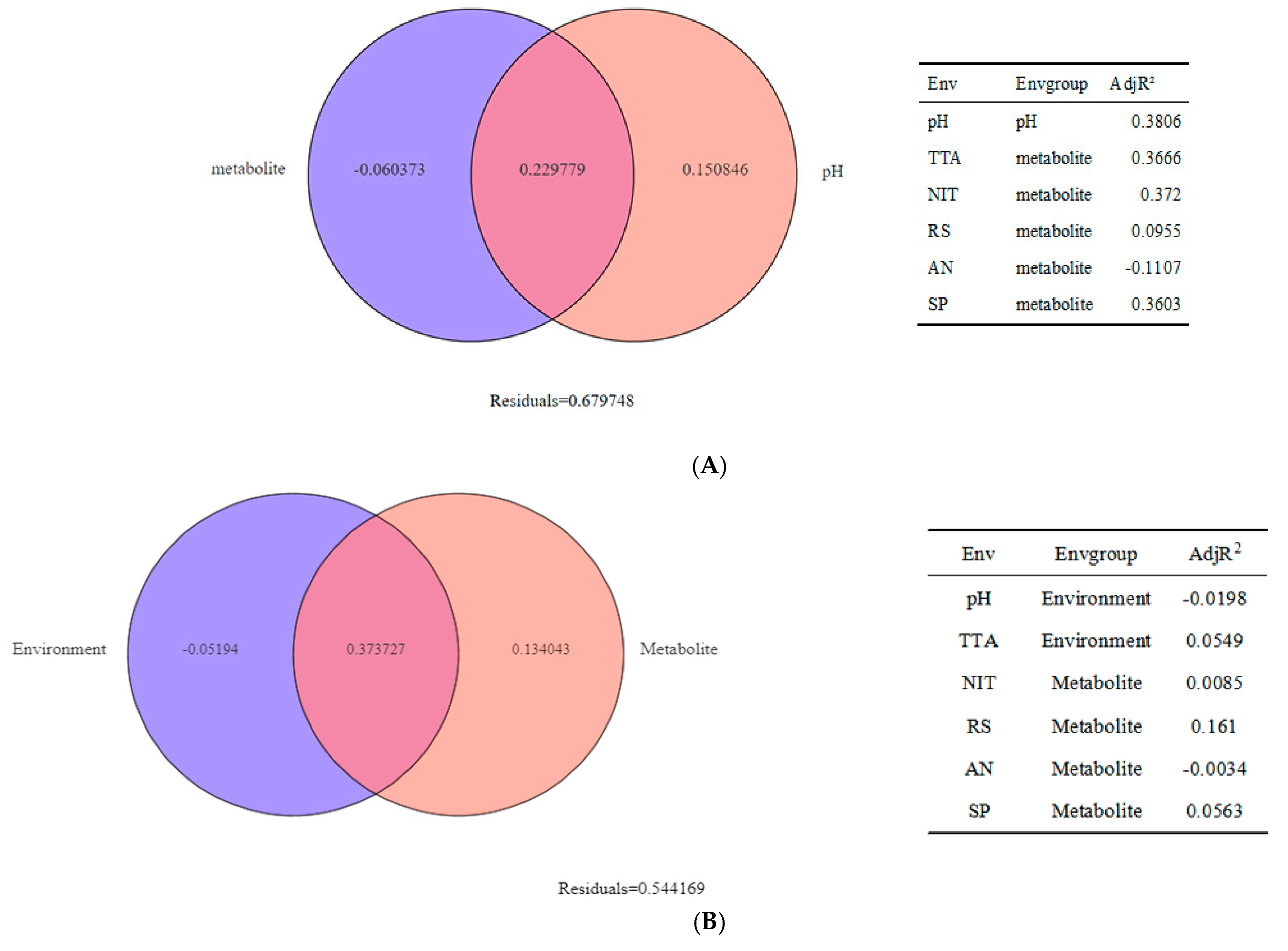
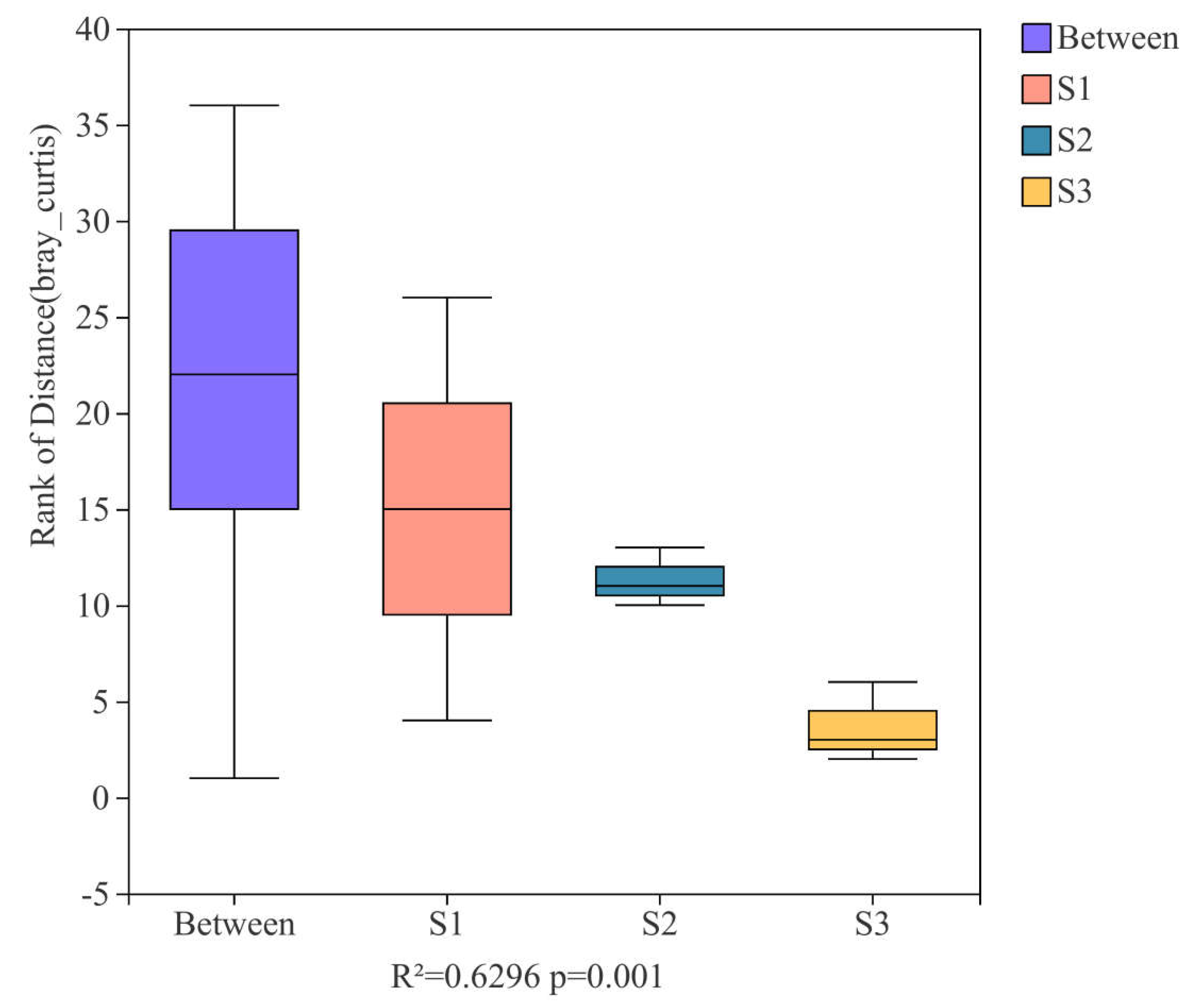
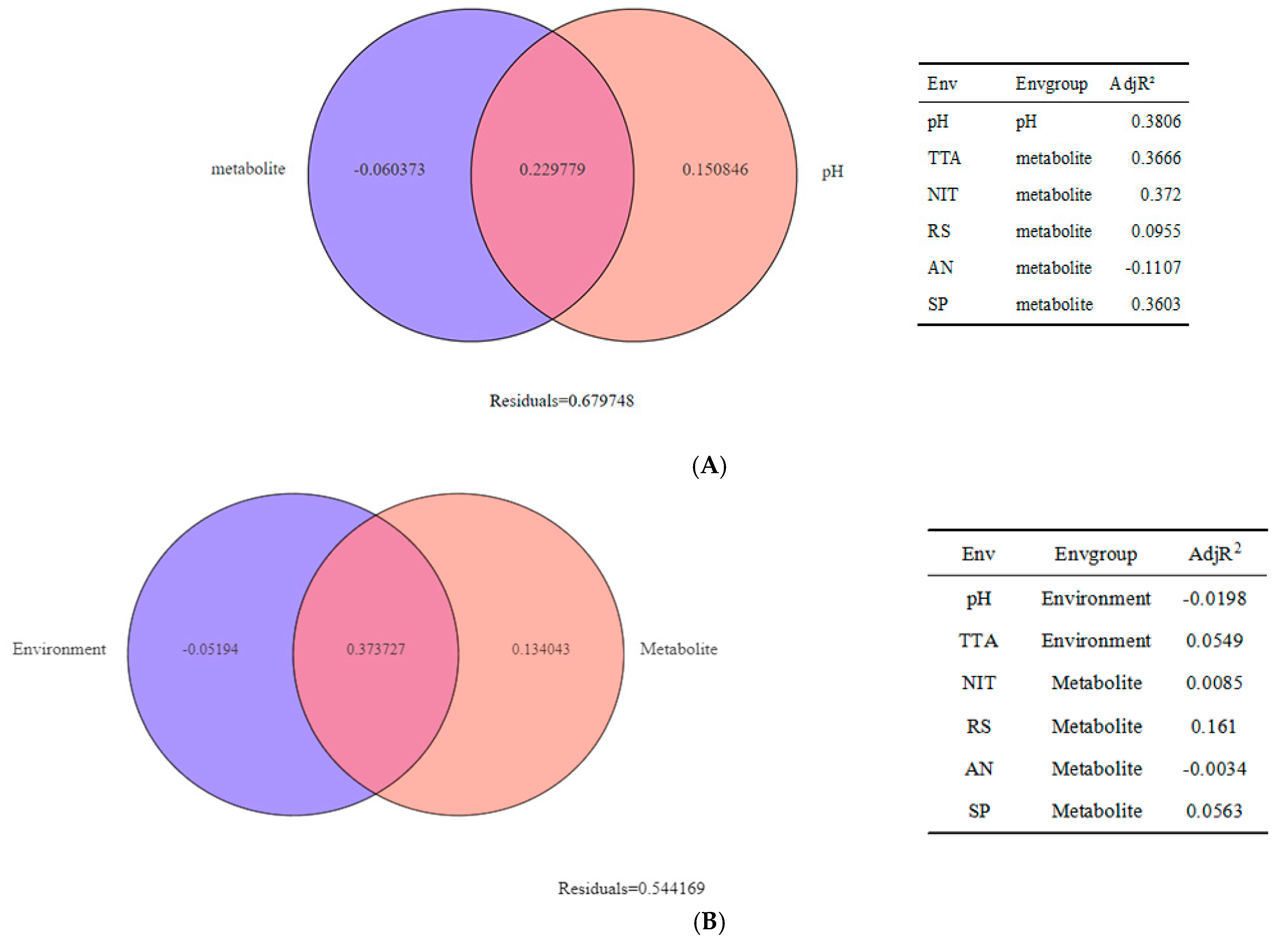
3.4. Research on Beta Diversity and Driving Factors of Samples
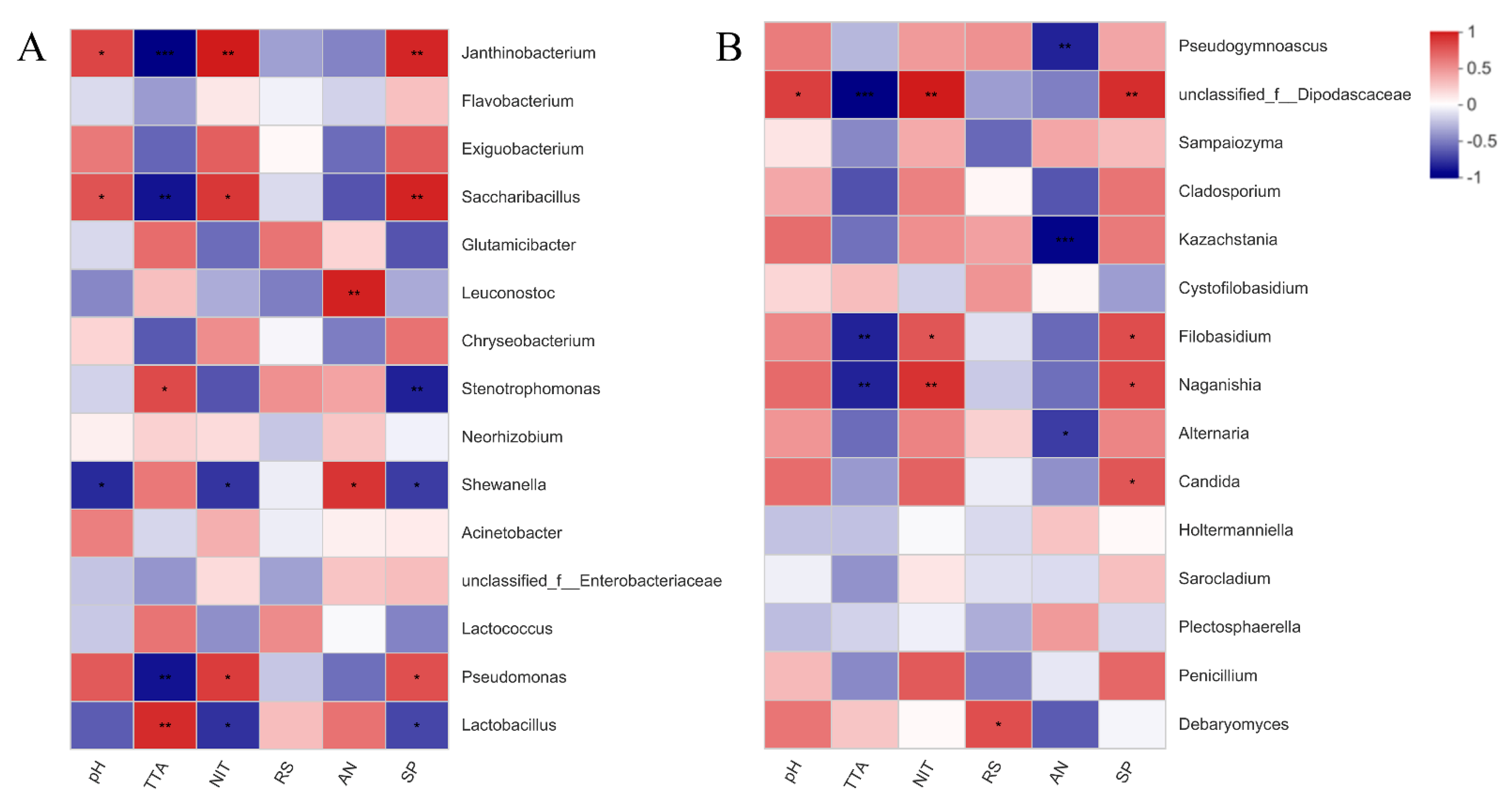
3.5. Correlation of Bacterial and Fungal Communities in Fermented Paocai
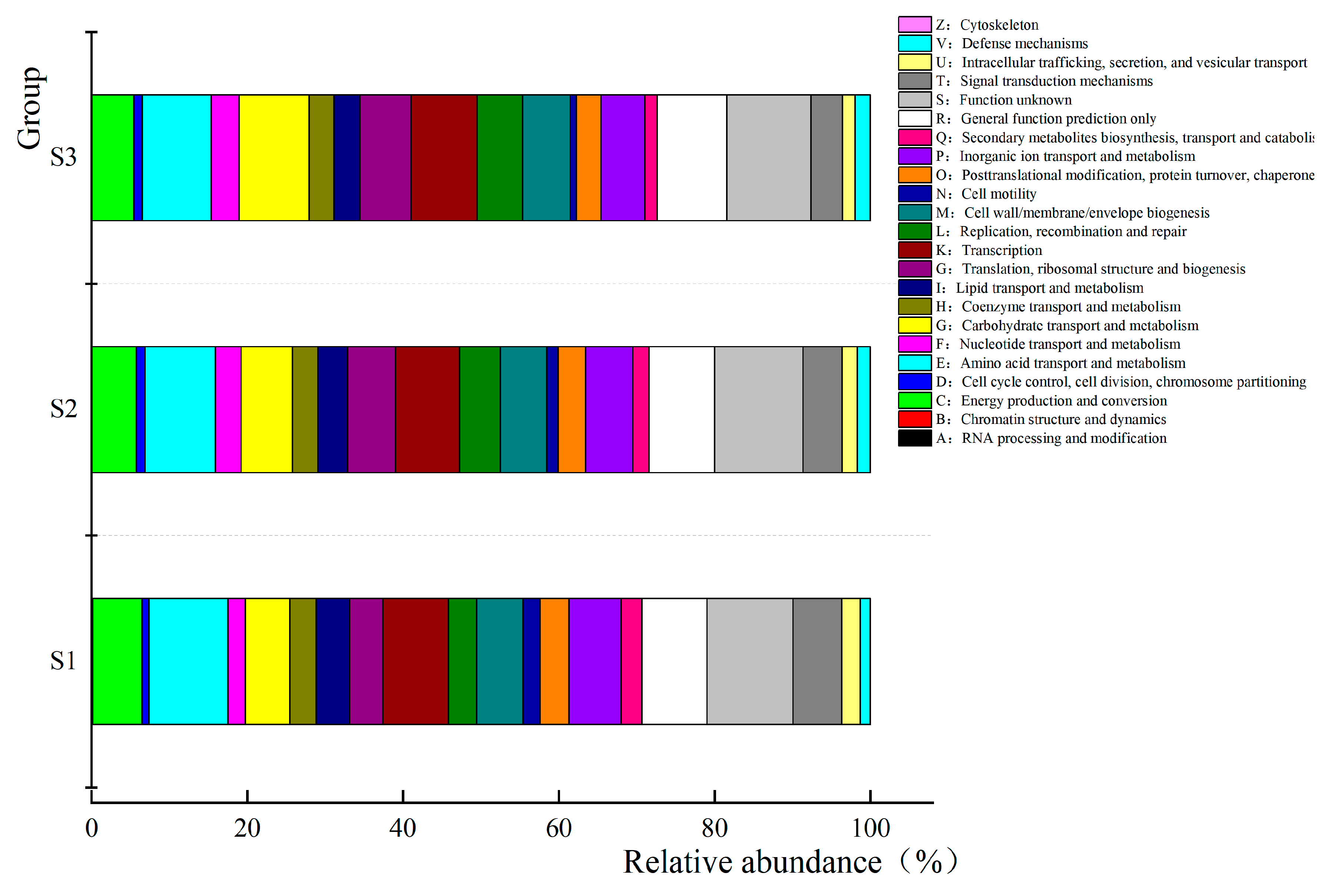
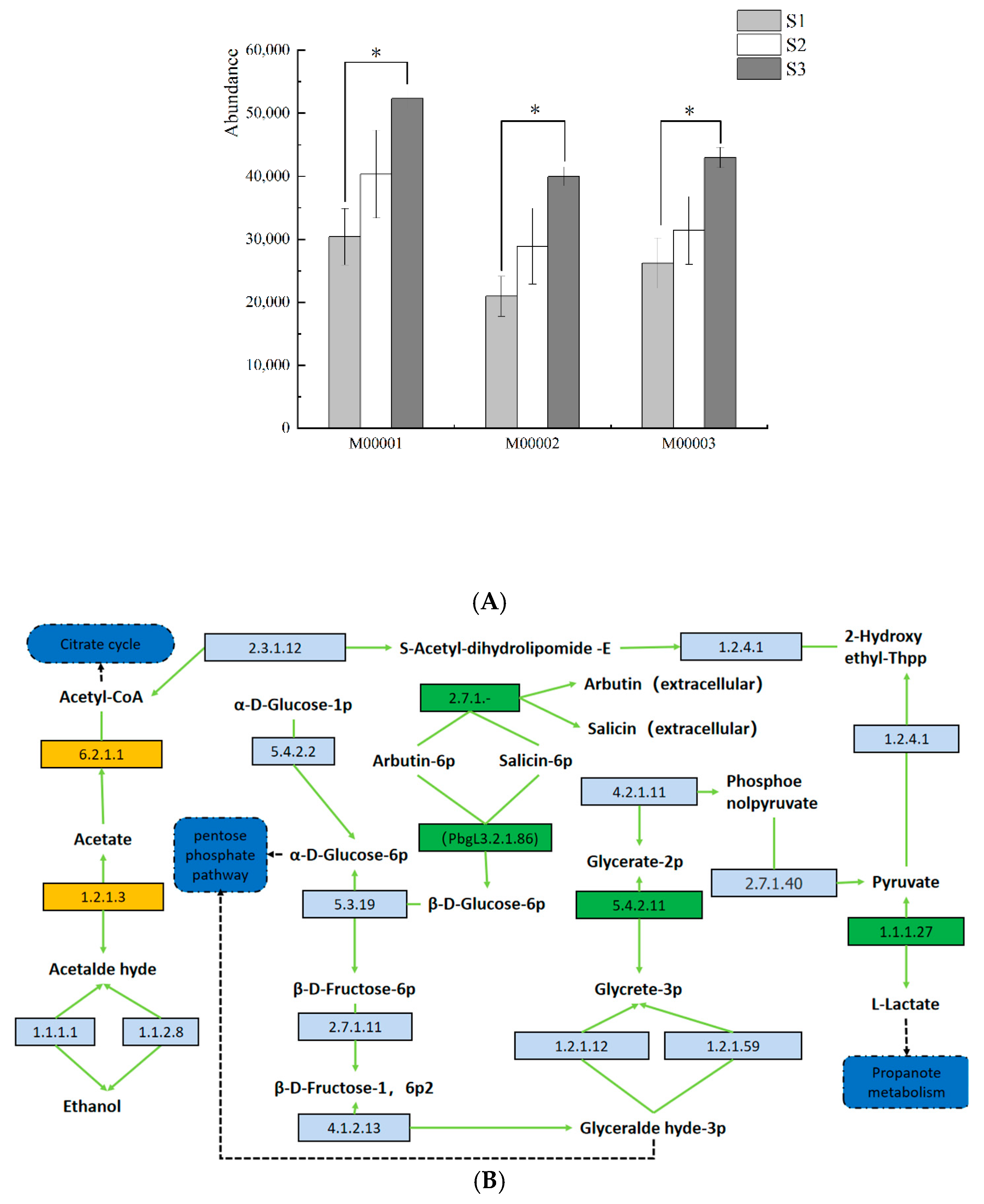
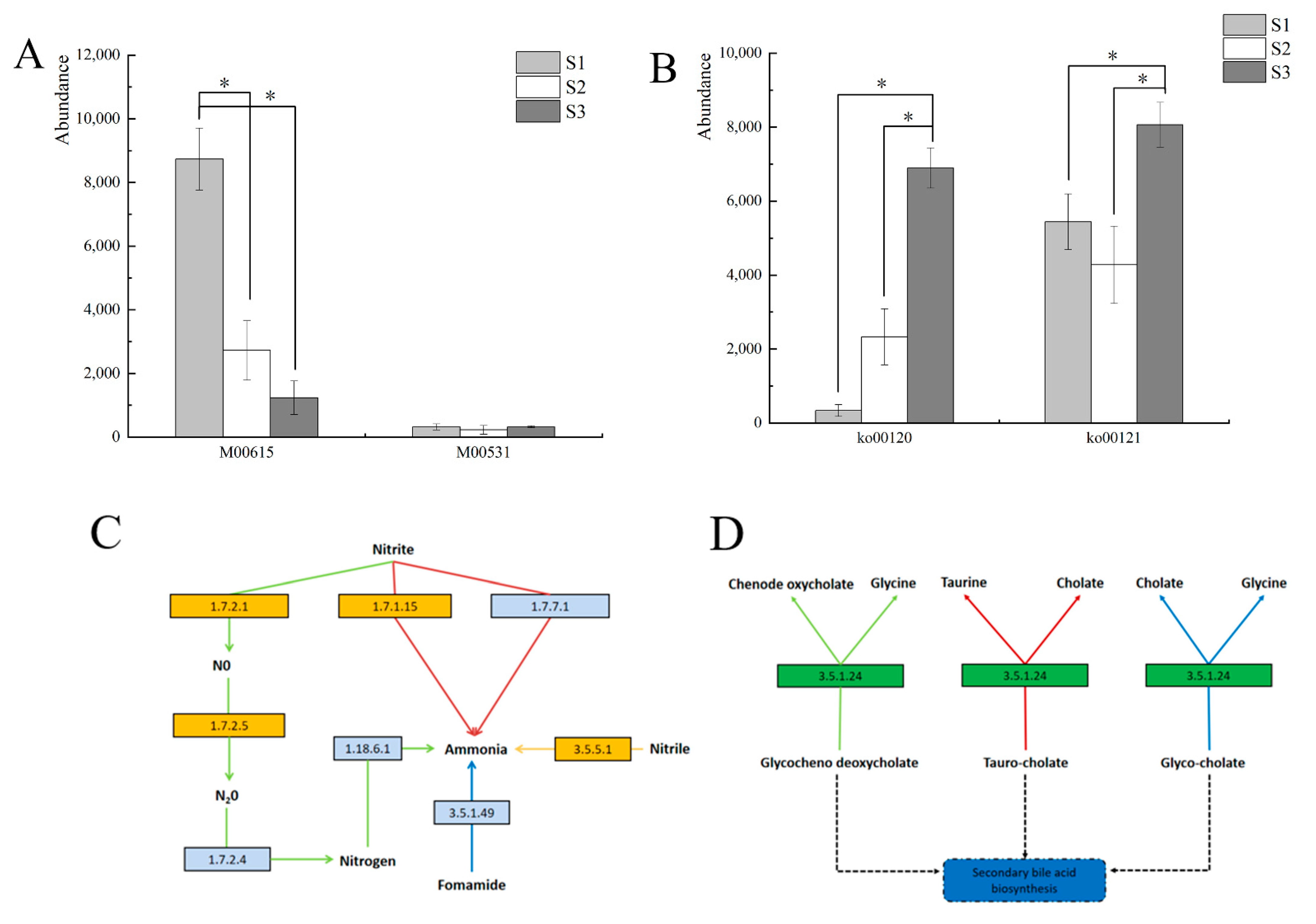
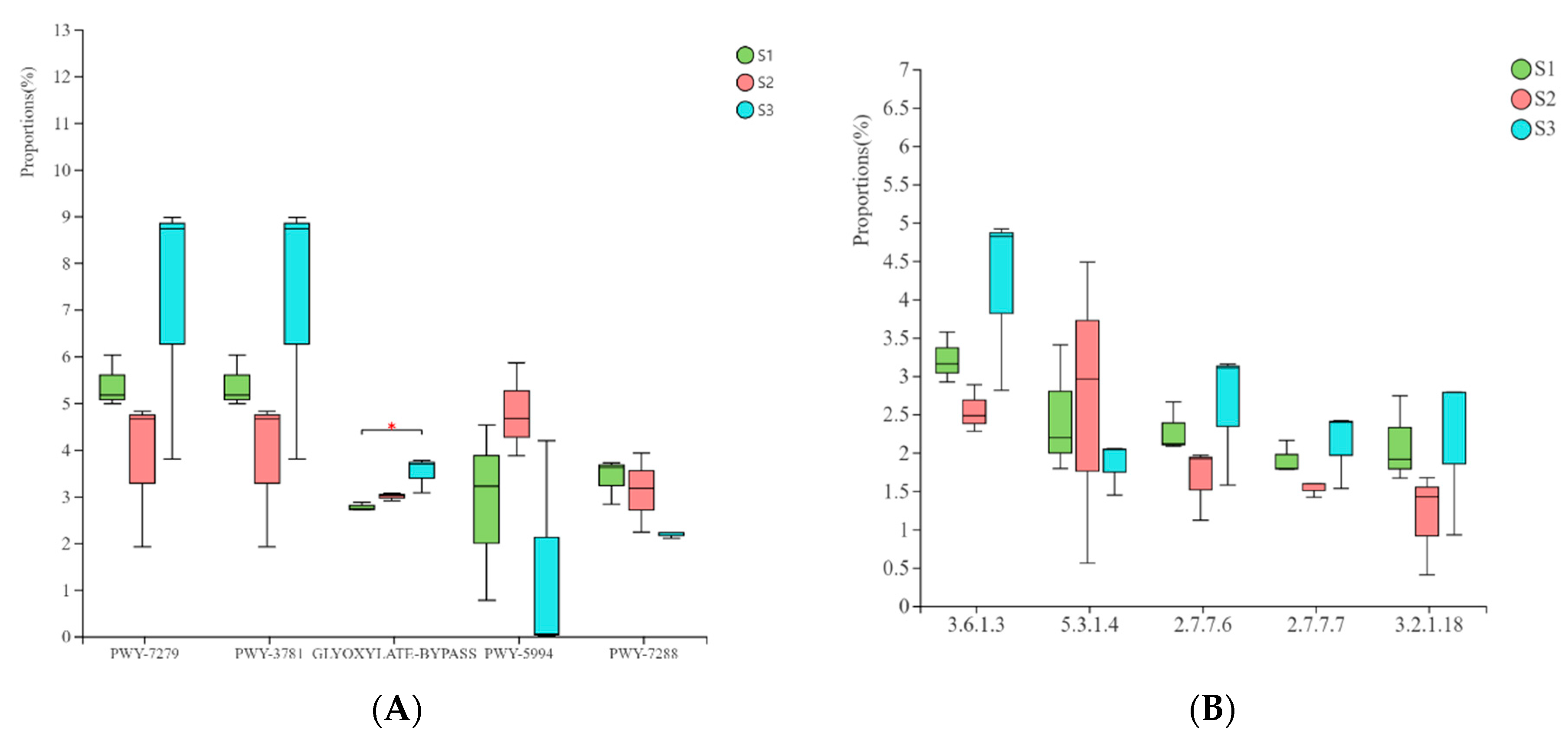
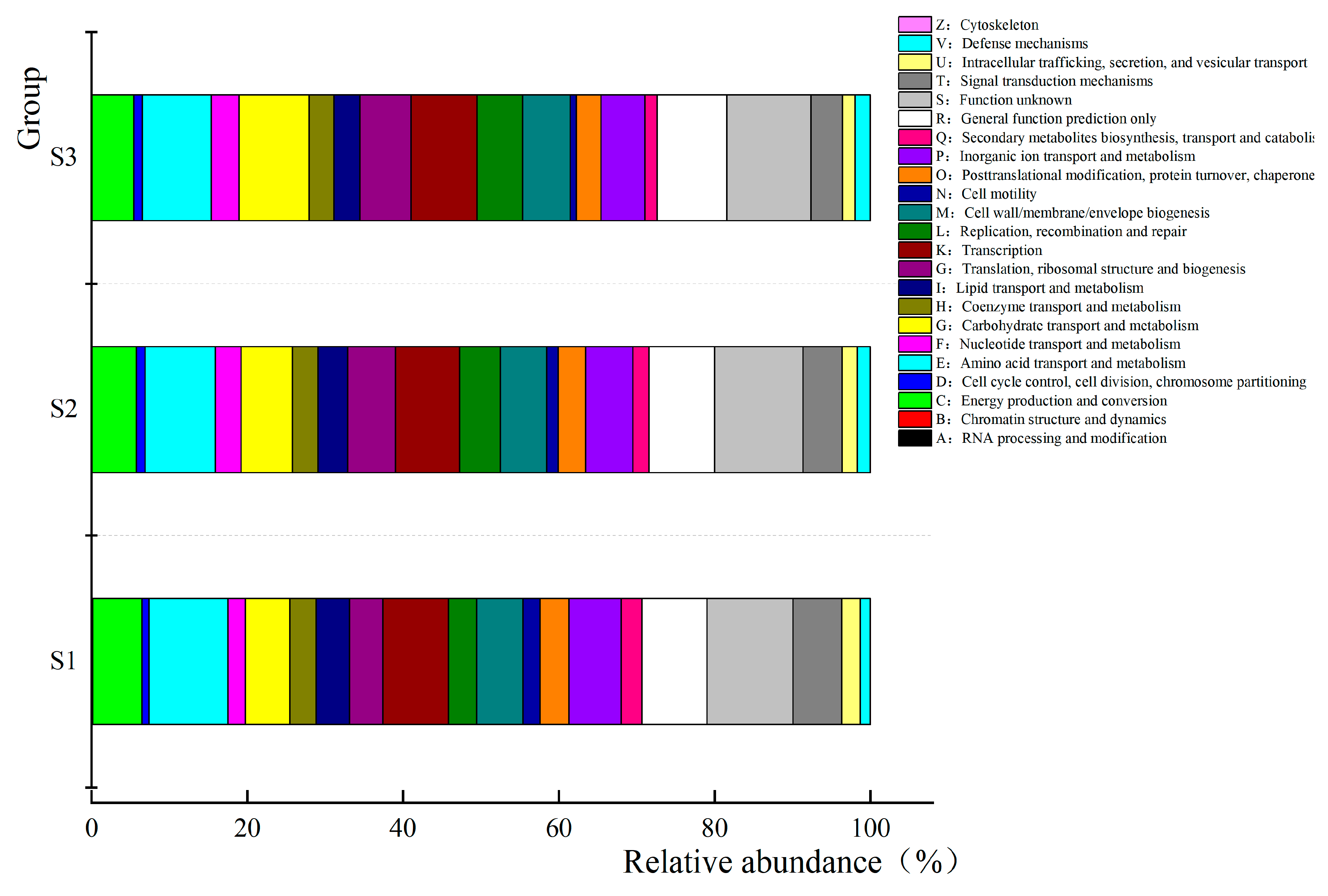
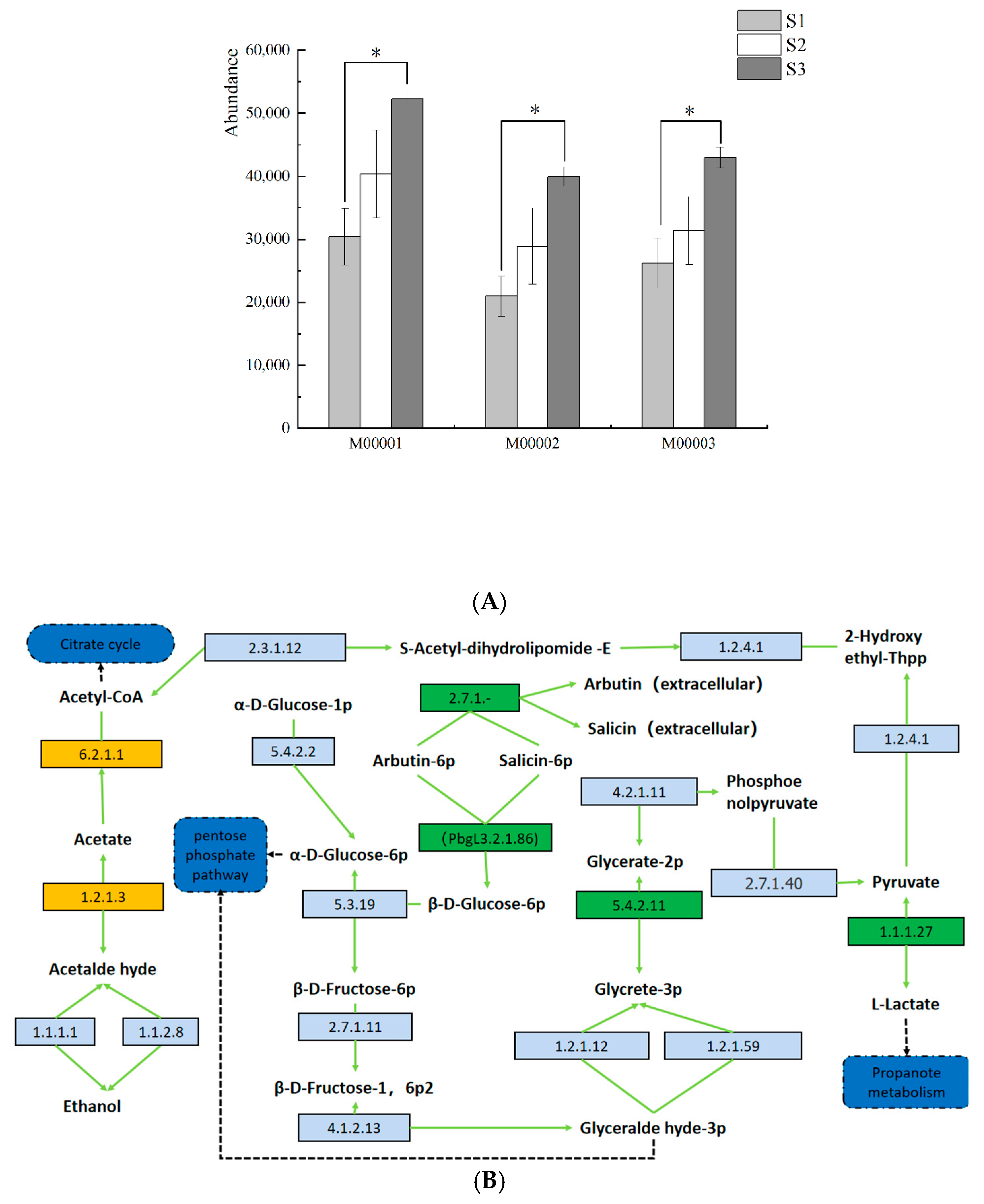
3.6. Prediction and Analysis of Bacterial and Fungal Community Functions in Fermented Paocai
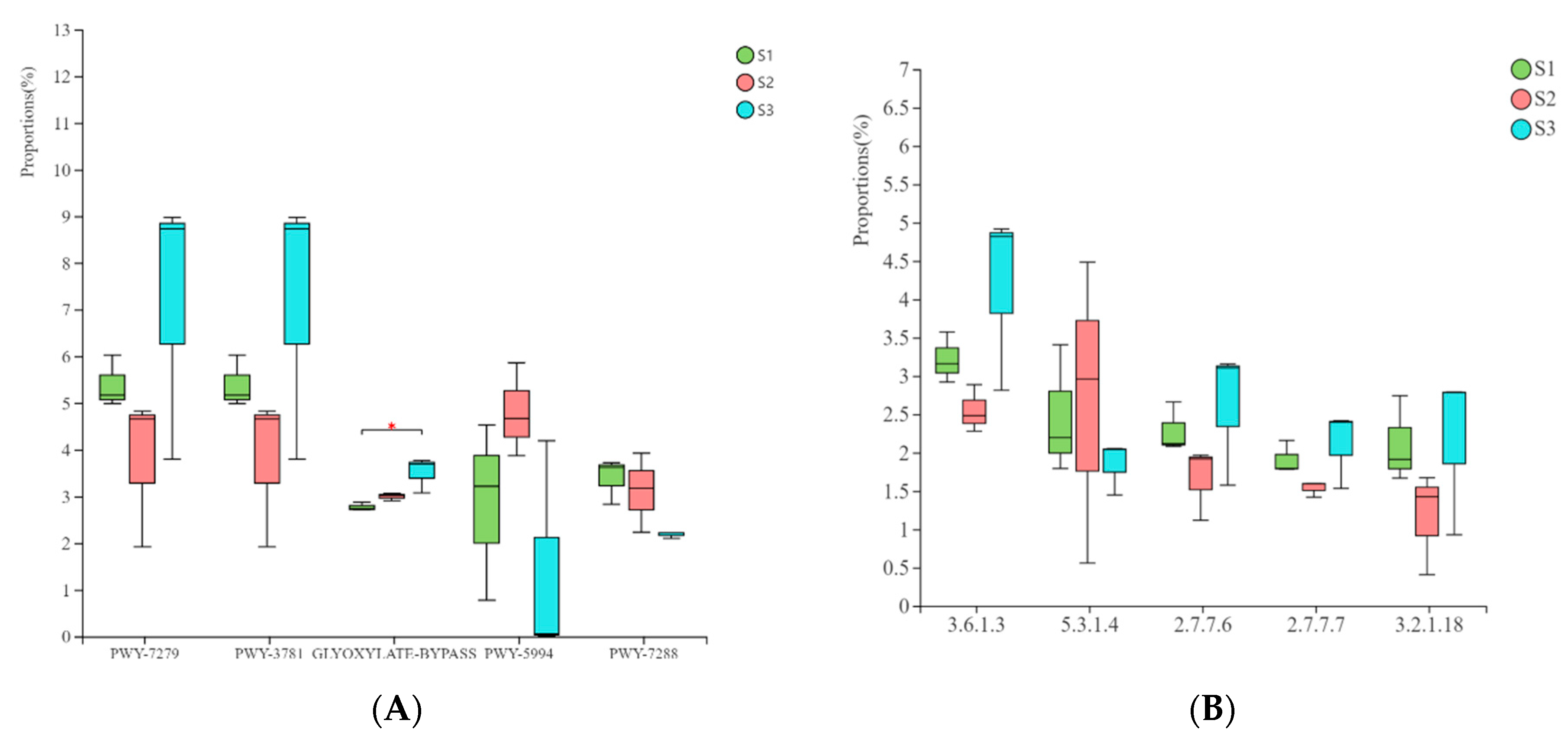
3.7. Ecological Characteristics and Functional Analysis of Fungi in Naturally Fermented Paocai
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ding, W.; Shi, C.; Chen, M. Screening for lactic acid bacteria in traditional fermented Tibetan yak milk and evaluating their probiotic and cholesterol-lowering potentials in rats fed a high-cholesterol diet. J. Funct. Foods 2017, 32, 324–332. [Google Scholar] [CrossRef]
- Liu, Z.; Peng, Z.; Huang, T.; Xiao, Y.; Li, J.; Xie, M.; Xiong, T. Comparison of bacterial diversity in traditionally homemade paocai and Chinese spicy cabbage. Food Microbiol. 2019, 83, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Yang, J.; Hou, Q.; Xu, H.; Zheng, Y.; Zhang, H.; Zhang, L. Assessment of bacterial profiles in aged, home-made Sichuan paocai brine with varying titratable acidity by PacBio SMRT sequencing technology. Food Control. 2017, 78, 14–23. [Google Scholar] [CrossRef]
- Zhao, N.; Zhang, C.; Yang, Q.; Yang, B.; Lu, W.; Li, D.; Liu, X.; Tian, F.; Zhang, H.; Chen, W. Multiple roles of lactic acid bacteria microflora in the formation of marker flavour compounds in traditional chinese paocai. RSC Adv. 2016, 6, 89671–89678. [Google Scholar] [CrossRef]
- De Melo Pereira, G.V.; De Carvalho Neto, D.P.; Maske, B.L.; De Dea Lindner, J.; Vale, A.S.; Favero, G.R.; Viesser, J.; De Carvalho, J.C.; Goes-Neto, A.; Soccol, C.R. An updated review on bacterial community composition of traditional fermented milk products: What next-generation sequencing has revealed so far? Crit. Rev. Food Sci. Nutr. 2020, 62, 1870–1889. [Google Scholar] [CrossRef]
- Kim, M.; Chun, J. Bacterial community structure in kimchi, a Korean fermented vegetable food, as revealed by 16S rRNA gene analysis. Int. J. Food Microbiol. 2005, 103, 91–96. [Google Scholar] [CrossRef]
- GB/T 12456—2008. Determination of Total Acid in Foods. State Administration of Quality Supervision, Inspection and Quarantine of the People’s Republic of China, China National Standardization Administration Committee. China Standards Press: Beijing, China, 2008.
- GB/T 5009.33—2010. Determination of Nitrite and Nitrate in Foods. State Administration of Quality Supervision, Inspection and Quarantine of the People’s Republic of China, China National Standardization Administration Committee. China Standards Press: Beijing, China, 2010.
- GB/T 5009.7—2016. Determination of Reducing Sugar in Foods. State Administration of Quality Supervision, Inspection and Quarantine of the People’s Republic of China, China National Standardization Administration Committee. China Standards Press: Beijing, China, 2016.
- GB/T 5009.5—2016. Determination of Protein in Foods. State Administration of Quality Supervision, Inspection and Quarantine of the People’s Republic of China, China National Standardization Administration Committee. China Standards Press: Beijing, China, 2016.
- GB/T 5009.235—2016. Determination of Amino Acid Nitrogen in Foods. State Administration of Quality Supervision, Inspection and Quarantine of the People’s Republic of China, China National Standardization Administration Committee. China Standards Press: Beijing, China, 2016.
- Azi, F.; Tu, C.; Meng, L.; Zhiyu, L.; Cherinet, M.T.; Ahmadullah, Z.; Dong, M. Metabolite dynamics and phytochemistry of a soy whey-based beverage bio-transformed by water kefir consortium. Food Chem. 2021, 342, 128225. [Google Scholar] [CrossRef]
- Ryu, J.-A.; Kim, E.; Yang, S.-M.; Lee, S.; Yoon, S.-R.; Jang, K.-S.; Kim, H.-Y. High-throughput sequencing of the microbial community associated with the physicochemical properties of meju (dried fermented soybean) and doenjang (traditional Korean fermented soybean paste). LWT 2021, 146, 11473–11482. [Google Scholar] [CrossRef]
- Kirchman, D.L.; Cottrell, M.T.; Lovejoy, C. The structure of bacterial communities in the western Arctic Ocean as revealed by pyrosequencing of 16S rRNA genes. Environ. Microbiol. 2010, 12, 1132–1143. [Google Scholar] [CrossRef]
- Zhang, Q.; Chen, X.; Ding, Y.; Ke, Z.; Zhou, X.; Zhang, J. Diversity and succession of the microbial community and its correlation with lipid oxidation in dry-cured black carp (Mylopharyngodon piceus) during storage. Food Microbiol. 2021, 98, 103686–103730. [Google Scholar] [CrossRef]
- Xiao, H.; Schaefer, D.A.; Yang, X. pH drives ammonia oxidizing bacteria rather than archaea thereby stimulate nitrification under Ageratina adenophora colonization. Soil Biol. Biochem. 2017, 114, 12–19. [Google Scholar] [CrossRef]
- Wang, P.; Chen, Q.; Fang, Z.; Wang, J. Research progress on biological Function and Inhibitors of Intestinal microbial metabolite Biliary saline Hydrolyase. Chin. J. Anim. Nutr. 2022, 34, 1388–1397. [Google Scholar]
- Luo, Q.; Pei, L.; Liang, H.; Yang, J.; Wu, Z.; Zhang, W. Fungal diversity in large-scale fermented kimchi production. China Condiment 2015, 40, 43–46. [Google Scholar]
- Huang, R.; Li, J.; Ming, J.; Ye, M.; Tang, Y.; Wang, D.; Chen, G.; Zhang, Q. Analysis on flavoring substances and microbial community structure of German trial-produced pickles. Food Ferment. Technol. 2021, 57, 51–56. [Google Scholar]
- Sun, J. Morphological and PCR-DGGE Analysis of Bacterial Community Diversity in Natural Fermentation Broth of Sauerkraut; Heilongjiang University: Harbin, China, 2014. [Google Scholar]
- Rao, Y.; Qian, Y.; Tao, Y.; She, X.; Li, Y.; Chen, X.; Guo, S.; Xiang, W.; Liu, L.; Du, H. Characterization of the microbial com-munities and their correlations with chemical profiles in assorted vegetable Sichuan pickles. Food Control. 2020, 113, 107174–107184. [Google Scholar] [CrossRef]
- Jeong, S.H.; Lee, H.J.; Jung, J.Y.; Lee, S.H.; Seo, H.Y.; Park, W.S.; Jeon, C.O. Effects of red pepper powder on microbial communities and metabolites during kimchi fermentation. Int. J. Food Microbiol. 2013, 160, 252–259. [Google Scholar] [CrossRef]
- Stanborough, T.; Fegan, N.; Powell, S.M.; Singh, T.; Tamplin, M.; Chandry, P.S. Genomic and metabolic characterization of spoil-age-associated Pseudomonas species. Int. J. Food Microbiol. 2018, 268, 61–72. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, Y.; Sun, J.; Pan, P.; Liu, Y.; Tian, T. Effects of starter culture inoculation on microbial community diversity and food safety of Chinese Cantonese sausages by high-throughput sequencing. J. Food Sci. Technol. 2021, 58, 931–939. [Google Scholar] [CrossRef]
- Chand, D.; Avinash, V.S.; Yadav, Y.; Pundle, A.V.; Suresh, C.G.; Ramasamy, S. Molecular features of bile salt hydrolases and relevance in human health. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 2981–2991. [Google Scholar] [CrossRef]
- Jia, W.; Xie, G.; Jia, W. Bile acid-microbiota crosstalk in gastrointestinal inflammation and carcinogenesis. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 111–128. [Google Scholar] [CrossRef] [Green Version]
- De Smet, L.V.H.; Vande Woestyne, M.H. Significance of bile salt hydrolytic activities of Lactobacillus. J. Appl. Bacteriol. 1995, 79, 292–301. [Google Scholar] [CrossRef]
- Teneva, D.; Denkova, Z.; Denkova-Kostova, R.; Goranov, B.; Kostov, G.; Slavchev, A.; Hristova-Ivanova, Y.; Uzunova, G.; Degraeve, P. Biological preservation of mayonnaise with Lactobacillus plantarum LBRZ12, dill, and basil essential oils. Food Chem. 2021, 344, 128707. [Google Scholar] [CrossRef] [PubMed]
- Kaur, M.; Singh, H.; Jangra, M.; Kaur, L.; Jaswal, P.; Dureja, C.; Nandanwar, H.; Chaudhuri, S.R.; Raje, M.; Mishra, S.; et al. Lactic acid bacteria isolated from yak milk show probiotic potential. Appl. Microbiol. Biotechnol. 2017, 101, 7635–7652. [Google Scholar] [CrossRef] [PubMed]
- Begley, M.; Hill, C.; Gahan, C.G. Bile salt hydrolase activity in probiotics. Appl. Environ. Microbiol. 2006, 72, 1729–1738. [Google Scholar] [CrossRef] [Green Version]
- Jiachu, L.; Qianwen, C.; Bingkun, L.; Xiao, L. Analysis on diversity of fungal community structure in traditional Laotan naturally fermented pickles. J. Shaanxi Univ. Sci. Technol. 2019, 37, 53–59. [Google Scholar]
- Deasy, T.; Mahony, J.; Neve, H.; Heller, K.J.; Van Sinderen, D. Isolation of a virulent Lactobacillus brevis phage and its application in the control of beer spoilage. J. Food Prot. 2011, 74, 2157–2161. [Google Scholar] [CrossRef]
- Yang, H.; Li, C.; Liu, T.; Zou, H.; Qu, C.; Wu, H. Dynamics and diversity of bacteria during fermentation of traditional Chinese sau-erkraut. Sci. Technol. Food Ind. 2015, 36, 154–165. [Google Scholar]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, P.; Jia, J.; Zhao, H.; Wu, C. Changes and Driving Mechanism of Microbial Community Structure during Paocai Fermentation. Fermentation 2022, 8, 281. https://doi.org/10.3390/fermentation8060281
Yan P, Jia J, Zhao H, Wu C. Changes and Driving Mechanism of Microbial Community Structure during Paocai Fermentation. Fermentation. 2022; 8(6):281. https://doi.org/10.3390/fermentation8060281
Chicago/Turabian StyleYan, Pingmei, Jingjing Jia, Huwei Zhao, and Chendong Wu. 2022. "Changes and Driving Mechanism of Microbial Community Structure during Paocai Fermentation" Fermentation 8, no. 6: 281. https://doi.org/10.3390/fermentation8060281
APA StyleYan, P., Jia, J., Zhao, H., & Wu, C. (2022). Changes and Driving Mechanism of Microbial Community Structure during Paocai Fermentation. Fermentation, 8(6), 281. https://doi.org/10.3390/fermentation8060281