Abstract
Guanidinoacetate (GAA) is a naturally occurring amino acid derivative and the direct precursor of creatine, which is widely used in feed additives and the pharmaceutical industry. The current industrial synthesis of GAA is based on chemical methods, which limits the application of GAA. Here, a biological approach is developed for food safety GAA production via whole-cell biocatalysis by the generally regarded as safe (GRAS) bacterium Bacillus subtilis. First, we introduced a heterologous arginine: glycine amidinotransferase (AgaT) from Amycolatopsis kentuckyensis into B. subtilis and optimized its expression level using strategies including: promoter optimization, ribosome binding site (RBS) and N-terminal coding sequence (NCS) screening. In order to alleviate the waste of arginine and the inhibition of AgaT by ornithine, we optimized the natural ornithine cycle in B. subtilis. At the same time, the first gene in the glycine degradation pathway was knocked out. After optimization using these strategies, the titer of GAA was 4.26 g/L with a productivity of 0.21 g/L/h in 20 h, which provides a new method for the biosynthesis of GAA.
1. Introduction
Guanidinoacetate (GAA) has the physiological functions of promoting insulin secretion, influencing neuromodulation, changing arginine metabolism, etc. It is widely used as an intermediate in pharmaceutical organic synthesis; GAA can promote energy metabolism and improve antioxidant capacity, so it is often used in feed additives and food additives [1,2]. In mammalian organisms, creatine has an important role in the intracellular energy transport process, assisting in providing energy to muscle and nerve cells [3]. Creatine can be synthesized from GAA and S-adenosine methionine by guanidinoacetate N-methyltransferase (GamT, EC: 2.1.1.2). The human body is able to synthesize 60–80% of its own creatine requirements, so additional supplementation from food is required [4]. However, the stability of supplemented creatine is poor, and the utilization rate is low, so nowadays GAA is used to replace creatine for the purpose of increasing tissue load.
Due to the wide application of GAA in food, feed and medicine, a green, environmentally friendly and safe method of GAA production is needed [5]. At present, the synthesis of GAA in industry is based on the chemical synthesis method. However, there are some disadvantages of synthesizing GAA by chemical methods. For example, the end product GAA is easily contaminated, the purification process of GAA is quite tedious, and the production process is not good for the environment.
The whole-cell catalysis method provides the possibility for the efficient and green production of GAA. In prokaryotes, GAA can be synthesized from glycine and arginine by arginine:glycine amidinotransferase (AgaT, EC: 2.1.4.1) [6,7]. AgaT transfers the amidino group of arginine to the amino group of glycine to obtain GAA, while generating the by-product ornithine. Previous studies have shown that the by-product ornithine inhibits AgaT, which is one of the main reasons that limits the synthesis of GAA via whole-cell catalysis [8,9,10]. In order to solve this problem, methods such as optimizing the source of agaT and strengthening the ornithine cycle have been tried in Escherichia coli, which proved the effectiveness of GAA production via whole-cell catalysis [11]. However, the production of food safety compounds by E. coli has many problems, such as the secretion of endotoxins that may cause disease. Here, we aimed to establish a whole-cell catalytic factory of GAA in Bacillus subtilis, which is generally regarded as food safe (GRAS).
As a typical industrial model microorganism of food safety grade, B. subtilis has a clear genetic background and mature gene manipulation technology [12]. Due to its non-pathogenicity and strong protein synthesis ability, it is very suitable for GAA biocatalysis [13]. In this study, we first introduced a heterologous agaT gene from Amycolatopsis kentuckyensis into B. subtilis and constructed a GAA biosynthetic pathway. Next, in order to improve the expression level of AgaT, we adopted strategies such as promoter screening, optimization of ribosome binding site (RBS) and N-terminal coding sequence (NCS). Then, we blocked the conversion of glycine and arginine into other products in B. subtilis, which reduced the waste of arginine and glycine. Since the by-product ornithine can inhibit AgaT, we optimized the natural ornithine cycle in B. subtilis. Finally, a GAA production process using B. subtilis as a whole-cell biocatalyst was demonstrated, which should be more favorable for food-grade GAA production. The strategies used here may be useful for engineering B. subtilis strains for the production of other guanidino compounds or GAA derivatives via whole-cell catalysis.
2. Materials and Methods
2.1. Plasmids and Strains
The plasmids and strains used in this study are listed in Table 1. E. coli JM109 was used as the cloning host, and B. subtilis (B. subtilis 168, B. subtilis WB600) was used as the expression host for this experiment [14]. All strains and plasmids are stored in our laboratory. Gibson assembly was used to construct all plasmids used in this study with pHT01 as backbones [15,16]. To construct the plasmid pHT01-AgaT, first, the agaT gene encoding AgaT from Amycolatopsis kentuckyensis was codon-optimized and synthetized by Suzhou GENEWIZ Biotech Company (Suzhou, China). Then, the codon-optimized agaT gene and different promoters, PlytR, P43, P333, and P566, were used as templates to obtain the gene fragment and promoter fragments, respectively. The different promoters were obtained from our laboratory storage, and the sequences of the promoters are listed in Table 2. Finally, the gene fragment and different promoters’ fragments were cloned into the pHT01 plasmid by Gibson assembly, respectively. Ultimately, we obtained a series of plasmids containing different AgaT expression intensities. They were then extracted from E. coli JM109 that was used for cloning purposes using a plasmid extraction kit purchased from Shanghai Sangon (Shanghai, China) and transported into B. subtilis to construct the strains listed in Table 1. The DNA polymerase and DNA marker were obtained from TAKARA (Dalian, China).

Table 1.
Strains and plasmids used in this study.
Table 2.
Sequences of the promoters used in this study.
For the knockout and overexpression of genes, a homologous recombination box with 1000 bp homology arms was used to perform genome editing in B. subtilis, and the Cre-lox system was used to eliminate resistance selection markers on the genome [17]. For the knockout of the genes on the genome of B. subtilis, we used resistance genes to replace the genes to be knocked out. A PCR product containing lox72- and lox66-specific sites and a cassette encoding zeocin (30 μg/mL) resistance was amplified. Then, 1000 bp homologous arms of the target gene were constructed into a homologous recombination box with the resistance gene cassette. The recombination fragment was obtained by overlap-extension PCR and transformed into B. subtilis strains. For the overexpression of genes, the difference was that we amplified the 1000 bp homologous arms of the argI (accession no. CP053102.1) gene site. The homologous arms, the resistance gene cassette, the promoter Pveg and the gene to be overexpressed were constructed into a homologous recombination box. Other operations were the same as the knockout of the genes, and the sequence of the promoter Pveg is listed in Table 2. In this way, we achieved overexpression of the gene while knocking out the argI gene on the genome of B. subtilis.
2.2. Whole-Cell Catalysis of GAA
The engineered strains were cultured in Luria–Bertani (LB) broth and agar plates. The seed culture was LB broth containing 10 g/L tryptone, 5 g/L yeast extract, and 10 g/L NaCl. The tryptone and yeast extract used in this study were obtained from Thermo Fisher Scientific (Shanghai, China). For the cultivation of B. subtilis cells, the culture of cells was grown in a 50 mL centrifuge tube containing 3 mL LB medium with Chloramphenicol (5 mg/L) at 37 °C for 8 h while shaking at 220 rpm. Then, we inoculated 2.5 mL of the seed culture to the 50 mL fermentation medium (pH 7.0) containing the following: 12 g/L yeast extract, 6 g/L peptone, 6 g/L (NH4)2SO4, 12.5 g/L K2HPO4·3H2O, 2.5 g/L KH2PO4, 3 g/L MgSO4·7H2O and 40 g/L glucose within a 500 mL shake flask. The cells were cultivated at 37 °C for 17 h with Chloramphenicol (5 mg/L) while shaking at 220 rpm. After 17 h of culture, the B. subtilis fermentation broth was centrifuged at 4 °C, 7000× g rpm/min for 10 min to collect the precipitate of the bacterium. The fermentation broth was then washed once with 20 mL of 0.01 M PBS buffer (136.89 mM NaCl, 2.67 mM KCl, 8.1 mM Na2HPO4, 1.76 mM KH2PO4, pH 7.4 adjusted with HCl). Then, all cells were resuspended in OD600 = 60 using PBS buffer containing initial concentrations of 20 g/L arginine and 20 g/L glycine. Five milliliters of the above reaction system was taken and the whole-cell catalysis reaction was performed at 30 °C in a constant temperature shaker at 220 rpm/min.
2.3. Analytical Methods
A liquid chromatograph-mass spectrometer (LC-MS, SHIMADZU) was used to detect the concentrations of GAA on a system equipped with a Waters XBridge NH2 3.5 μm column (4.6 mm × 150 mm) in each sample. The analysis was performed at 30 °C with a mobile phase comprising 75% acetonitrile in water (pH adjusted to 10 with ammonia) at a flow rate of 1 mL/min, and GAA was detected at an OD of 210 nm. This method can also detect Arginine and Glycine simultaneously.
The biomass and fluorescence intensity were measured directly on the 96-well plates, which contained 200 μL of cell culture using a Cytation 3 Multi-Mode Reader (BIOTEK, Cytation 3). The fluorescence intensity was measured at an excitation wavelength of 488 nm and an emission wavelength of 523 nm. OD600 is the absorbance at 600 nm measured on the Cytation 3 Multi-Mode Reader when the amount of liquid in each well of a 96-well plate is 200 μL. The cell concentration was measured following a 50-fold dilution during whole-cell catalysis.
2.4. Design and Flow Screening of the Degenerate Primers
According to the degenerate bases and genetic code tables, we constructed a random mutation library based on pHT01-P566-AgaT-GFP by random A, T, G, and C insertions 13 bp between the −35 and −10 regions of promoter P566 and 35 bp in NCS of agaT via degenerate primer design and PCR. Snapgene 4.3.6 was used for gene sequence analysis and primer design. The PCR reaction condition was 98 °C 3 min, 98 °C 10 s, 55 °C 10 s, 72 °C 3 min, 72 °C 5 min, 12 °C 10 min, and 35 cycles from step 2 to step 4. The random mutation library was transformed into B. subtilis 168.
The organisms were washed using PBS buffer and diluted to an OD600 = 0.1, and single cells with high relative fluorescence intensity were screened by flow cytometry and coated onto plates. Further screening was performed in 96-well plates to detect the biomass and fluorescence intensity of each single colony. Data were analyzed and visualized using FlowJo_V10 (FlowJo, LLC, Ashland, OR, USA) software.
3. Results
3.1. Constructing GAA Biosynthetic Pathway in B. subtilis
To convert glycine and arginine to GAA, agaT was first introduced into B. subtilis (Figure 1). The sequence of agaT was optimized according to the codon preference of B. subtilis. We used plasmid pHT01 as the expression vector to express agaT under the control of a strong promoter PlytR. Then, the plasmid was transformed into wild-type B. subtilis 168, and strain B1-1 was obtained. However, no GAA was detected after whole-cell catalysis. We hypothesized that the low expression level of AgaT was the main limitation [18]. Therefore, we first selected three other strong promoters, P43, P333, and P566, to replace promoter PlytR, resulting in strain B1-2, B1-3, and B1-4, respectively.
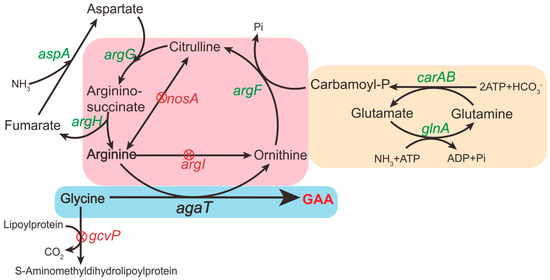
Figure 1.
Schematic presentation of the GAA production in B. subtilis. The green font indicates overexpression of the corresponding genes on the genome of B. subtilis. The red “X” in a red circle indicates deletion of the corresponding genes. The ornithine cycle in B. subtilis is included in the pink box. The heterogeneous GAA synthesis pathway in B. subtilis is included in the blue box. The pathway in the yellow box is to promote the endogenous ornithine cycle in B. subtilis.
To screen for the best promoter that regulates AgaT expression with the highest intensity, we attached a green fluorescent protein (GFP, accession no. AAG29470.1) after AgaT by a short linker (GGAGGAGGATCTGGAGGCGG), and strains B2-1, B2-2, B2-3, and B2-4 were obtained [19] (Figure 2a). We characterized the OD600 and fluorescence intensity in B. subtilis. The expression intensity of AgaT under the control of different promoters was characterized according to the relative fluorescence intensity. We found that the relative fluorescence intensity of AgaT-GFP under the control of the promoter P566 is the highest (Figure 2b). Finally, the agaT under the control of promoter P566 was selected.
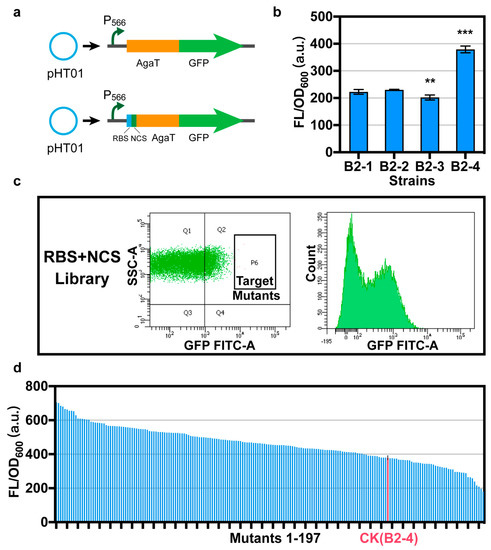
Figure 2.
Optimizing RBS and NCS of agaT. (a) In order to optimize the RBS and NCS of agaT, we attached a gfp after agaT through a linker. Please refer to Table 3 for the starting sequence of RBS and NCS. (b) The relative fluorescence intensity of strains B2-1, B2-2, B2-3, and B2-4 under the control of different promoters. (c) Screening of the random mutant library for efficient mutant using flow cytometry. SSC-A: granularity, FITC-A: GFP fluorescence. (d) Characterization of 197 strains obtained from the library. We detected the relative fluorescence intensity of the 197 strains. Data are shown as mean ± S.D. from three (n = 3) biological replicate experiments, and significance determined from Student’s t-test, where ** p < 0.005, *** p < 0.0005, and N.S. = not significant.
3.2. Optimizing RBS and NCS of agaT
After optimizing the promoter, we still did not detect the production of GAA via whole-cell catalysis. However, the P566 promoter is already the strongest promoter currently in use. Therefore, we assume that the main problem is the low level of translation, which led to the low expression of agaT. RBS and NCS of a gene could significantly impact the expression level in translation by influencing the efficiency of ribosome binding to mRNA and ribosome extension at the initial phase of translation [20]. Optimizing the RBSs and NCSs of genes is an important mechanism for fine-tuning gene expression in bacteria. To promote the expression of agaT in B. subtilis, we optimized the RBS and NCS in the pHT01-P566-AgaT-GFP expression frame. The first step was to construct a random mutation library to screen RBS and NCS for efficiently driving agaT expression. We constructed a random mutation library based on 8 bp random A, T, G, and C substitution in the RBS region and 7 bp synonymous substitution of agaT NCS via degenerate primer design and PCR [21]. After screening a library containing 83,440 mutants using flow cytometry, 197 strains were further characterized using 96-well plates (Figure 2c). We detected the OD600 and fluorescence intensity of all 197 strains (Figure 2d). The positive rate is 77.7%. Eight strains with the highest relative fluorescence intensity detected during rescreening were sequenced. The results showed that these eight single colonies had completely different mutations in both RBS and NCS of agaT (Table 3).
Table 3.
The table shows the sequence of different mutants.
Then, we removed the gfp gene on the eight plasmids and obtained plasmids pHT01-AgaT-1 to pHT01-AgaT-8. We transferred these eight plasmids into B. subtilis 168, and obtained strains B3-1, B3-2, B3-3, B3-4, B3-5, B3-6, B3-7, and B3-8. After whole-cell catalysis, GAA was detected for the first time (Figure 3a). With an initial concentration of 0.871 g/L arginine and 0.504 g/L glycine, the strain with the highest titer was B. subtilis B3-7 containing the plasmid pHT01-AgaT-7 with a titer of 0.062 g/L, followed by B. subtilis B3-1 containing the plasmid pHT01-AgaT-1 with a titer of 0.039 g/L. The above results show that we successfully detected GAA production in B. subtilis by optimizing RBS and NCS.
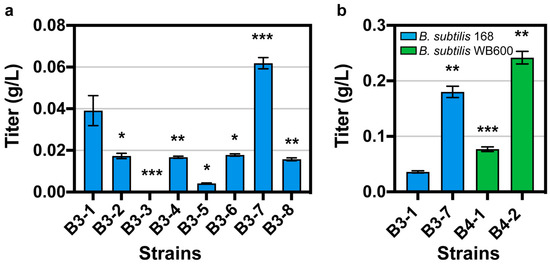
Figure 3.
(a) The whole-cell catalysis results in the eight best mutants. (b) The whole-cell catalysis results in B. subtilis WB600 and B. subtilis 168. Data are shown as mean ± S.D. from three (n = 3) biological replicate experiments, and significance determined from Student’s t-test, where * p < 0.05, ** p < 0.005, *** p < 0.0005, and N.S. = not significant.
Due to the differences in the ability of enzyme expression in different hosts, we compared the production of GAA from arginine and glycine catalyzed by AgaT in different hosts with an initial concentration of 20 g/L arginine and 20 g/L glycine. We transformed the plasmids pHT01-AgaT-1 and pHT01-AgaT-7 into B. subtilis WB600, respectively. We obtained the strains B4-1 and B4-2. It was found that regardless of which plasmid was introduced, GAA production was higher in B. subtilis WB600 than in B. subtilis 168 (Figure 3b). For pHT01-AgaT-7, the production of GAA in WB600 was 1.34-fold that in B. subtilis 168; for pHT01-AgaT-1, the production of GAA in WB600 was 2.13-fold that in B. subtilis 168. After optimizing the host of B. subtilis, the highest titer of GAA was 0.24 g/L.
3.3. Knocking-Out of the Substrate Catabolic Pathway
Blocking the substrate consumption pathway is an effective method to improve the yield of the target product. In the metabolic pathway of B. subtilis, glycine dehydrogenase, encoded by gcvP, is the first gene in the glycine degradation pathway. To make more glycine available for GAA biosynthesis, we knocked out gcvP on the genome of strain B4-2, yielding strain B5-1. Whole-cell catalysis with an initial concentration of 20 g/L arginine and 20 g/L glycine resulted in a GAA production of 0.18 g/L. Knock-out of gcvP on the B. subtilis genome was not as effective as knock-out of argI. We guessed that blocking the cellular utilization of glycine followed by affecting cell growth resulted in affecting GAA synthesis.
In B. subtilis, the ornithine cycle is complete. Arginase, encoded by argI, can catalyze the conversion of arginine to ornithine. Since ornithine has an inhibitory effect on AgaT, it would affect the synthesis of GAA from arginine and glycine catalyzed by AgaT. Therefore, it is essential to knock out argI in the metabolic pathway of B. subtilis. On the one hand, it would alleviate the inhibition of AgaT by ornithine, and on the other hand, it would reduce the consumption of arginine. Knocking out argI on the genome of strain B4-2 increased GAA titer to 0.46 g/L (strain B5-2).
After searching for metabolic pathways of B. subtilis, we found that arginine and citrulline interconvert in the ornithine cycle of B, which is catalyzed by the Nitric-Oxide synthase encoded by nosA. Next, we knocked out the above two or three genes simultaneously. ArgI and gcvP were simultaneously knocked out on the basis of strain B4-2 to obtain strain B6-1. On the basis of strain B4-2, argI and nosA were simultaneously knocked out to obtain strain B6-2. On the basis of strain B4-2, argI, gcvP and nosA were simultaneously knocked out to obtain strain B6-3. The best strain was B6-2, with a titer of 0.91 g/L; the titer of B6-1 was 0.57 g/L, and the titer of B6-3 was 0.84 g/L (Figure 4a). Strain B6-3 had the highest productivity of GAA at 0.56 g/L/h (Figure 4a). The knockout of argI and nosA significantly increased the production of GAA, even better than the simultaneous knockout of argI, nosA and gcvP, suggesting that arginine has a greater effect on GAA biosynthesis than glycine.
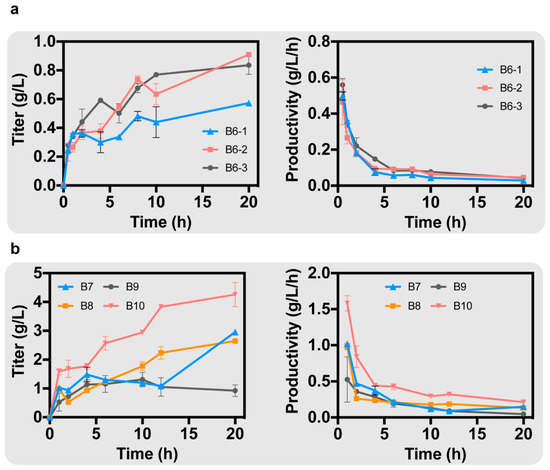
Figure 4.
(a) After knocking out the degradation pathways of glycine and arginine in B. subtilis, the GAA production of different strains was obtained. (b) After enhancing the endogenous ornithine cycle in B. subtilis, the GAA production of different strains was obtained.
3.4. Enhancing Endogenous Ornithine Cycle of B. subtilis
Ornithine can revert to produce arginine in B. subtilis due to the endogenous ornithine cycle. If the metabolic flow of the ornithine cycle is increased to promote the production of arginine from ornithine, it can alleviate the inhibition of AgaT by ornithine on the one hand, and increase the arginine required for GAA biosynthesis on the other hand. Therefore, it was necessary to overexpress genes in the ornithine cycle on the B. subtilis genome. First, the ornithine carbamoyl transferase (EC: 2.1.3.3), encoded by argF, is able to catalyze the conversion of ornithine to citrulline. Overexpressing argF can alleviate the inhibition by ornithine. Meanwhile, carbamoyl-phosphate synthase (EC: 6.3.5.5), encoded by carAB, is able to increase carbamoyl phosphate (CP), which is involved in the synthesis of citrulline [22]. glnA was also overexpressed, which encodes glutamine synthetase (EC: 6.3.1.2) [23]. Glutamine synthetase catalyzes the conversion of glutamate to glutamine and is able to synergize with carbamoyl-phosphate synthase to provide the CP required for citrulline synthesis. On the genome of B. subtilis, argF, carAB, and glnA are on the same gene cluster. Whole-cell catalysis reactions were performed in PBS buffer containing 20 g/L arginine and 20 g/L glycine. Under the regulation of the Promoter Pveg, we simultaneously overexpressed argF, carAB, and glnA in strain B6-2, resulting in strain B7 with a GAA titer of 2.95 g/L (Figure 4b).
In the ornithine cycle, argininosuccinate synthase (EC: 6.3.4.5) and argininosuccinate lyase (EC: 4.3.2.1), encoded by argG and argH, respectively, are on a gene cluster that promotes the production of arginine from citrulline. Overexpression of argG and argH would increase the metabolic flux of the ornithine cycle. Aspartate ammonia-lyase (EC: 4.3.1.1), encoded by aspA, catalyzes the conversion of fumarate to aspartate, providing aspartate required in the ornithine cycle [24]. We overexpressed argG and argH in strain B6-2 under the regulation of the Promoter Pveg, resulting in strain B8. After 20 h of whole-cell catalysis, the production of GAA was 2.45 g/L (Figure 4b). Meanwhile, we overexpressed aspA on strain B7 under the regulation of the Promoter Pveg, resulting in strain B9. The GAA production was only 0.92 g/L, suggesting that overexpression of aspA alone did not promote the synthesis of GAA (Figure 4b).
Finally, we simultaneously overexpressed argF, carAB, glnA, argG, argH and aspA under the regulation of the promoter Pveg in strain B6-2, resulting in strain B10. A total GAA production of 4.26 g/L was achieved in a 20 h whole-cell catalysis with a maximum productivity of 1.58 g/L/h in 1 h (Figure 4b).
4. Conclusions
This study demonstrated the feasibility of synthesizing GAA via whole-cell catalysis in B. subtilis. Due to the advantage of B. subtilis being a food-safe microorganism, we provided a safer and environmentally friendly production method. By optimizing RBS and NCS of agaT and other strategies, as well as optimizing the metabolic pathway in B. subtilis WB600, we obtained a recombinant B. subtilis with a highest GAA production of 4.26 g/L after a 20 h whole-cell catalysis. Compared to the currently reported GAA production of 8.61 g/L with a maximum productivity of 0.35 g/L/h after a 22 h whole-cell catalysis in E. coli, we obtained a higher maximum production intensity of 1.58 g/L/h [11]. This method has the advantages of easy operation and stable production, which provides a new strategy to realize the industrial production of GAA.
Author Contributions
Conceptualization, Y.L., L.L. and X.L.; methodology, K.Y., R.T. and L.Z.; validation, Y.L., R.T. and K.Y.; formal analysis, R.T. and K.Y.; investigation, Y.L.; resources, Y.L., K.Y. and R.T.; data curation, K.Y. and R.T.; writing—original draft preparation, K.Y. and R.T.; writing—review and editing, Y.L.; visualization, K.Y. and R.T.; supervision, Y.L. and L.L.; project administration, Y.L. and L.L.; funding acquisition, Y.L., K.Y. and R.T. contributed equally to this work. All authors have read and agreed to the published version of the manuscript.
Funding
This study was financially supported by the National Key Research and Development Program of China (2020YFA0908300), National Natural Science Foundation of China (32172349), Foundation for Innovative Research Groups of the National Natural Science Foundation of China (32021005), Natural Science Foundation of Jiangsu Province (BK20200085), and Key Research and Development Program of Jiangsu Province (BE2019628).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All data information is available in this paper.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Larsen, S.D.; Connell, M.A.; Cudahy, M.M.; Evans, B.R.; May, P.D.; Meglasson, M.D.; O’Sullivan, T.J.; Schostarez, H.J.; Sih, J.C.; Stevens, F.C.; et al. Synthesis and biological activity of analogues of the antidiabetic/antiobesity agent 3-guanidinopropionic acid: Discovery of a novel aminoguanidinoacetic acid antidiabetic agent. J. Med. Chem. 2001, 44, 1217–1230. [Google Scholar] [CrossRef] [PubMed]
- Michiels, J.; Maertens, L.; Buyse, J.; Lemme, A.; Rademacher, M.; Dierick, N.A.; De Smet, S. Supplementation of guanidinoacetic acid to broiler diets: Effects on performance, carcass characteristics, meat quality, and energy metabolism. Poult. Sci. 2012, 91, 402–412. [Google Scholar] [CrossRef] [PubMed]
- da Silva, R.P.; Clow, K.; Brosnan, J.T.; Brosnan, M.E. Synthesis of guanidinoacetate and creatine from amino acids by rat pancreas. Br. J. Nutr. 2014, 111, 571–577. [Google Scholar] [CrossRef] [PubMed]
- Appleyard, G.; Woods, D.D. The pathway of creatine catabolism by Pseudomonas ovalis. J. Gen. Microbiol. 1956, 14, 351–365. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yan, Z.; Yan, Z.; Liu, S.; Yin, Y.; Yang, T.; Chen, Q. Regulative Mechanism of Guanidinoacetic Acid on Skeletal Muscle Development and Its Application Prospects in Animal Husbandry: A Review. Front. Nutr. 2021, 8, 714567. [Google Scholar] [CrossRef] [PubMed]
- Barón-Sola, Á.; Sanz-Alférez, S.; Del Campo, F.F. First evidence of accumulation in cyanobacteria of guanidinoacetate, a precursor of the toxin cylindrospermopsin. Chemosphere 2015, 119, 1099–1104. [Google Scholar] [CrossRef] [PubMed]
- da Silva, R.P.; Nissim, I.; Brosnan, M.E.; Brosnan, J.T. Creatine synthesis: Hepatic metabolism of guanidinoacetate and creatine in the rat in vitro and in vivo. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E256–E261. [Google Scholar] [CrossRef]
- Ostojic, S.M. Nutraceuticals for L-Arginine:glycine Amidinotransferase Modulation: An Overview. Curr. Top. Nutraceutical Res. 2021, 19, 235–239. [Google Scholar] [CrossRef]
- Muenchhoff, J.; Siddiqui, K.S.; Poljak, A.; Raftery, M.J.; Barrow, K.D.; Neilan, B.A. A novel prokaryotic L-arginine:glycine amidinotransferase is involved in cylindrospermopsin biosynthesis. FEBS J. 2010, 277, 3844–3860. [Google Scholar] [CrossRef]
- Humm, A.; Fritsche, E.; Steinbacher, S.; Huber, R. Crystal structure and mechanism of human L-arginine:glycine amidinotransferase: A mitochondrial enzyme involved in creatine biosynthesis. EMBO J. 1997, 16, 3373–3385. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhou, H.; Tao, Y.; Lin, B. Reconstitution of the Ornithine Cycle with Arginine:Glycine Amidinotransferase to Engineer Escherichia coli into an Efficient Whole-Cell Catalyst of Guanidinoacetate. ACS Synth. Biol. 2020, 9, 2066–2075. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Tian, R.; Shen, Q.; Liu, Y.; Liu, L.; Li, J.; Du, G. Pathway Engineering of Bacillus subtilis for Enhanced N-Acetylneuraminic Acid Production via Whole-Cell Biocatalysis. Biotechnol. J. 2019, 14, e1800682. [Google Scholar] [CrossRef] [PubMed]
- van Dijl, J.M.; Hecker, M. Bacillus subtilis: From soil bacterium to super-secreting cell factory. Microb. Cell Fact. 2013, 12, 3. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Liu, Y.; Lv, X.; Li, J.; Du, G.; Liu, L. CAMERS-B: CRISPR/Cpf1 assisted multiple-genes editing and regulation system for Bacillus subtilis. Biotechnol. Bioeng. 2020, 117, 1817–1825. [Google Scholar] [CrossRef] [PubMed]
- Gibson, D.G. Enzymatic assembly of overlapping DNA fragments. In Methods in Enzymology; Academic Press: London, UK, 2011; Volume 498, pp. 349–361. [Google Scholar]
- Nguyen, H.D.; Phan, T.T.; Schumann, W. Expression vectors for the rapid purification of recombinant proteins in Bacillus subtilis. Curr. Microbiol. 2007, 55, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Yu, H.J.; Hong, Q.; Li, S.P. Cre/lox system and PCR-based genome engineering in Bacillus subtilis. Appl. Environ. Microbiol. 2008, 74, 5556–5562. [Google Scholar] [CrossRef]
- Alper, H.; Fischer, C.; Nevoigt, E.; Stephanopoulos, G. Tuning genetic control through promoter engineering. Proc. Natl. Acad. Sci. USA 2005, 102, 12678–12683. [Google Scholar] [CrossRef]
- Zhang, X.; Cao, Y.; Liu, Y.; Liu, L.; Li, J.; Du, G.; Chen, J. Development and optimization of N-acetylneuraminic acid biosensors in Bacillus subtilis. Biotechnol. Appl. Biochem. 2020, 67, 693–705. [Google Scholar] [CrossRef]
- Tian, R.; Liu, Y.; Chen, J.; Li, J.; Liu, L.; Du, G.; Chen, J. Synthetic N-terminal coding sequences for fine-tuning gene expression and metabolic engineering in Bacillus subtilis. Metab. Eng. 2019, 55, 131–141. [Google Scholar] [CrossRef]
- Tian, R.; Liu, Y.; Cao, Y.; Zhang, Z.; Li, J.; Liu, L.; Du, G.; Chen, J. Titrating bacterial growth and chemical biosynthesis for efficient N-acetylglucosamine and N-acetylneuraminic acid bioproduction. Nat. Commun. 2020, 11, 5078. [Google Scholar] [CrossRef]
- Johnson, J.L.; West, J.K.; Nelson, A.D.; Reinhart, G.D. Resolving the fluorescence response of Escherichia coli carbamoyl phosphate synthetase: Mapping intra-and intersubunit conformational changes. Biochemistry 2007, 46, 387–397. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Du, J.; Wu, J.; Dai, G.; Wang, C.; Zhou, X.; Song, M.; Li, J.; Li, J. Metabolism of Escherichia coli is interfered by Bacillus subtilis glnA gene. Chin. J. Biotechnol. 2009, 25, 626–631. [Google Scholar]
- Papierz, M.; Gadomska, G.; Sobierajski, B.; Chmiel, A. Selection and activation of Escherichia coli strains for L-aspartic acid biosynthesis. Pol. J. Microbiol. 2007, 56, 71–76. [Google Scholar] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).