Hydrogen-Cycling during Solventogenesis in Clostridium acetobutylicum American Type Culture Collection (ATCC) 824 Requires the [NiFe]-Hydrogenase for Energy Conservation

 ,
,  ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Bacterial Strain Propagation
2.2. Construction of HypF/HypD Mutant
2.3. Construction of Complement Strains
2.4. Sample Collection for RNA Seq Data
2.5. RNA Extraction, Purification, and rRNA Depletion
2.6. Sequencing Library Preparation
2.7. RNA-Seq Data Analysis
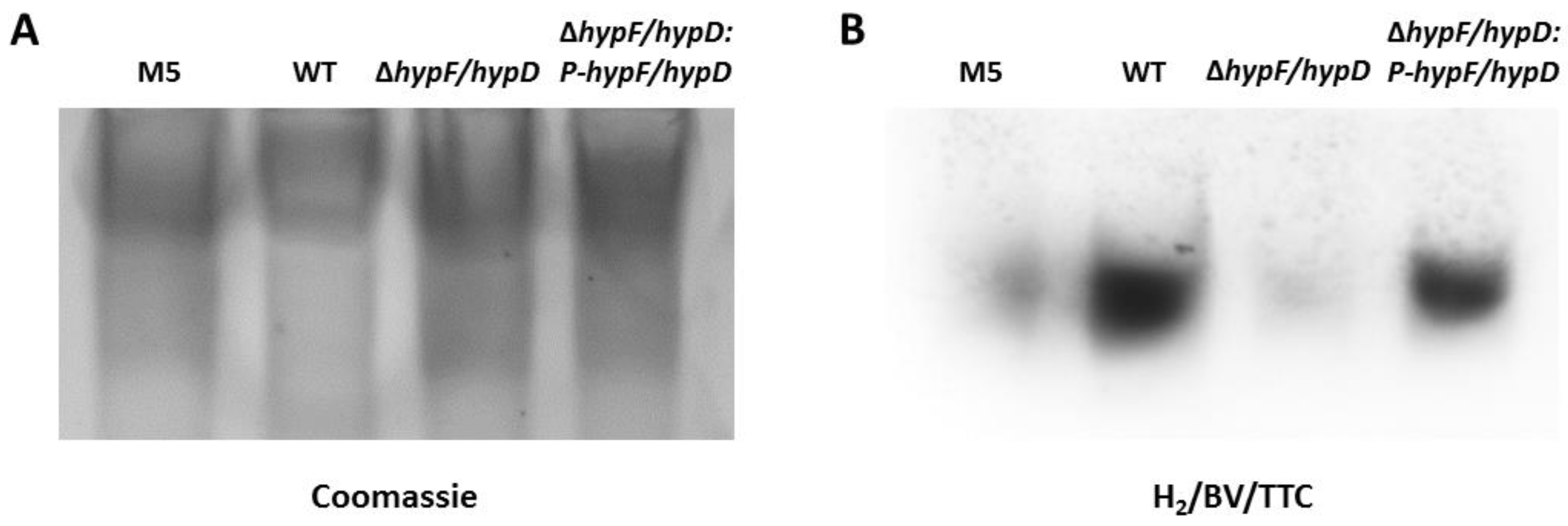
2.8. Hydrogenase Zymogram
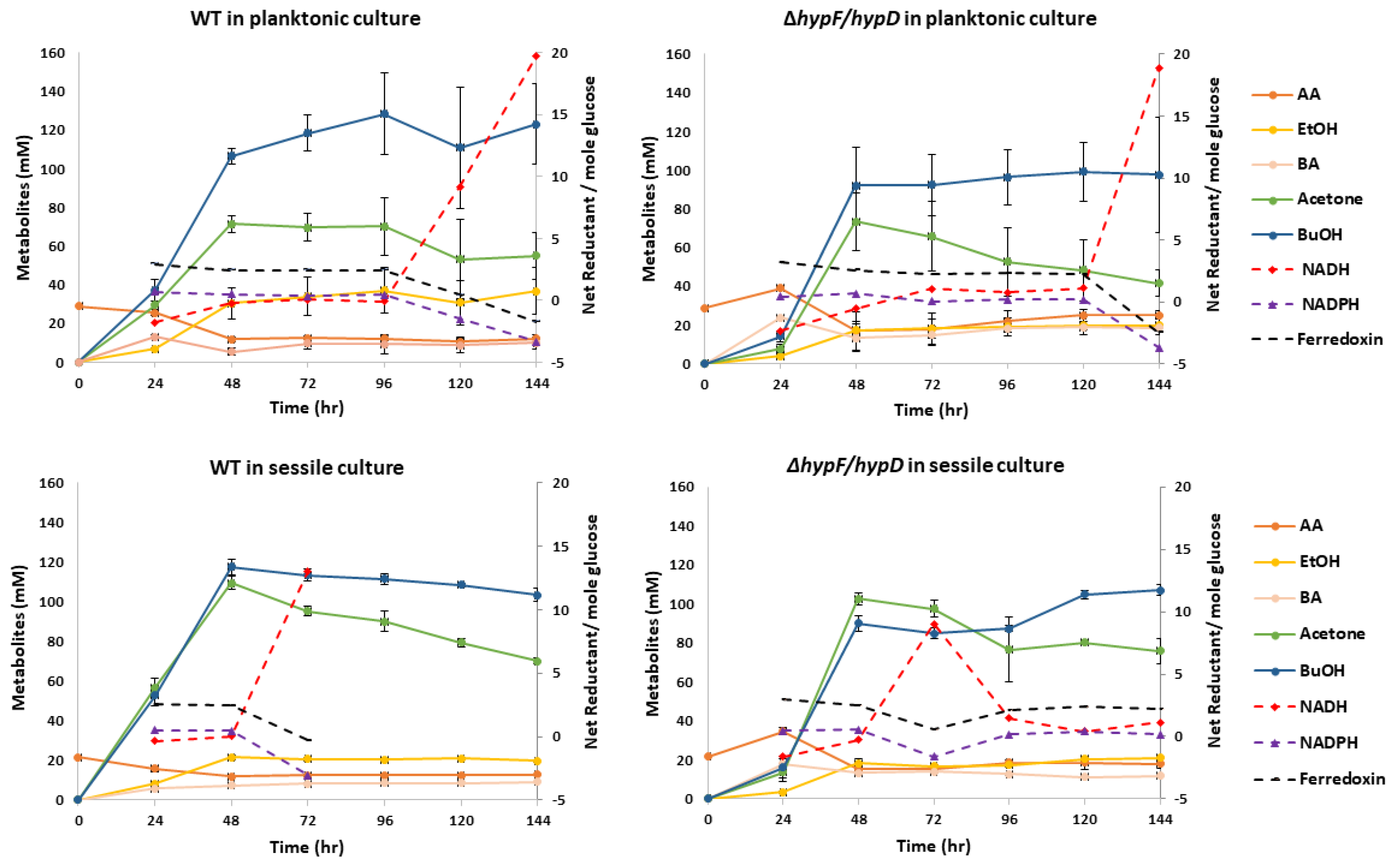
2.9. Growth and Metabolite Studies
2.10. Reduction of Palladium
2.11. Sample Preparation for TEM
2.12. TEM Imaging of Samples
2.13. Energy Filtered TEM
2.14. Reduction Potential Calculations
3. Results
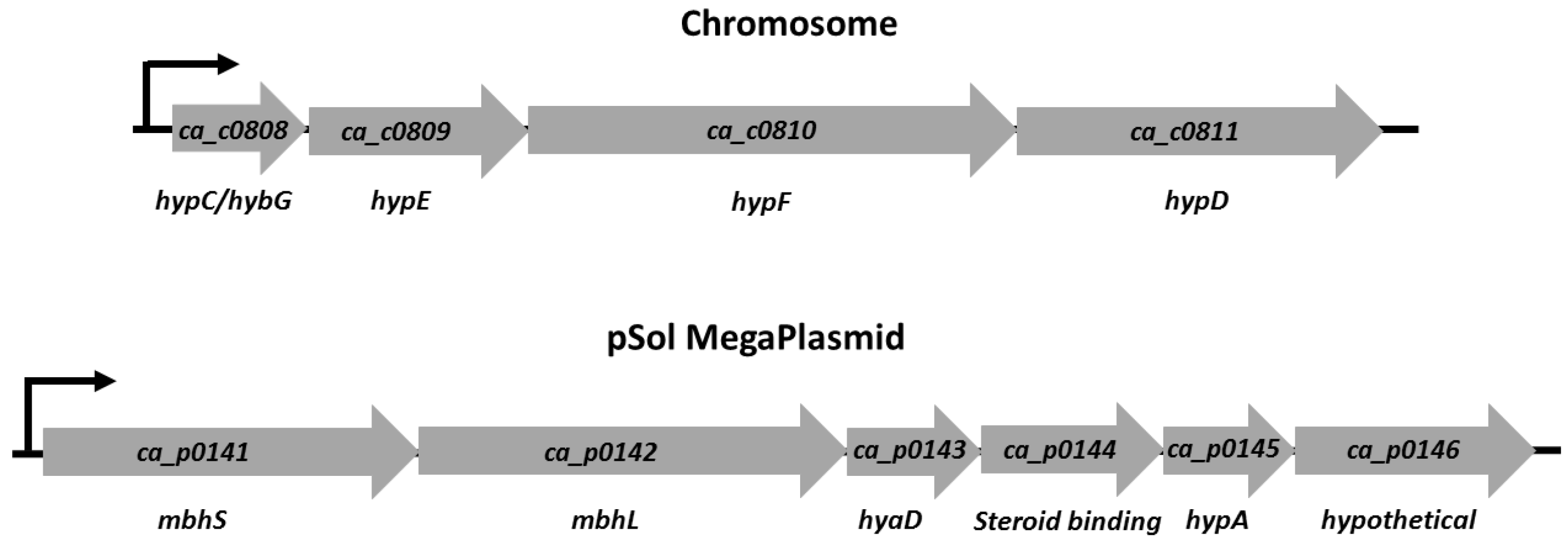
3.1. [NiFe]-Hydrogenase Operon Structure
3.2. Disruption of [NiFe]-Hydrogenase Maturation
3.3. Contribution of [NiFe]-Hydrogenase to Metabolic Output
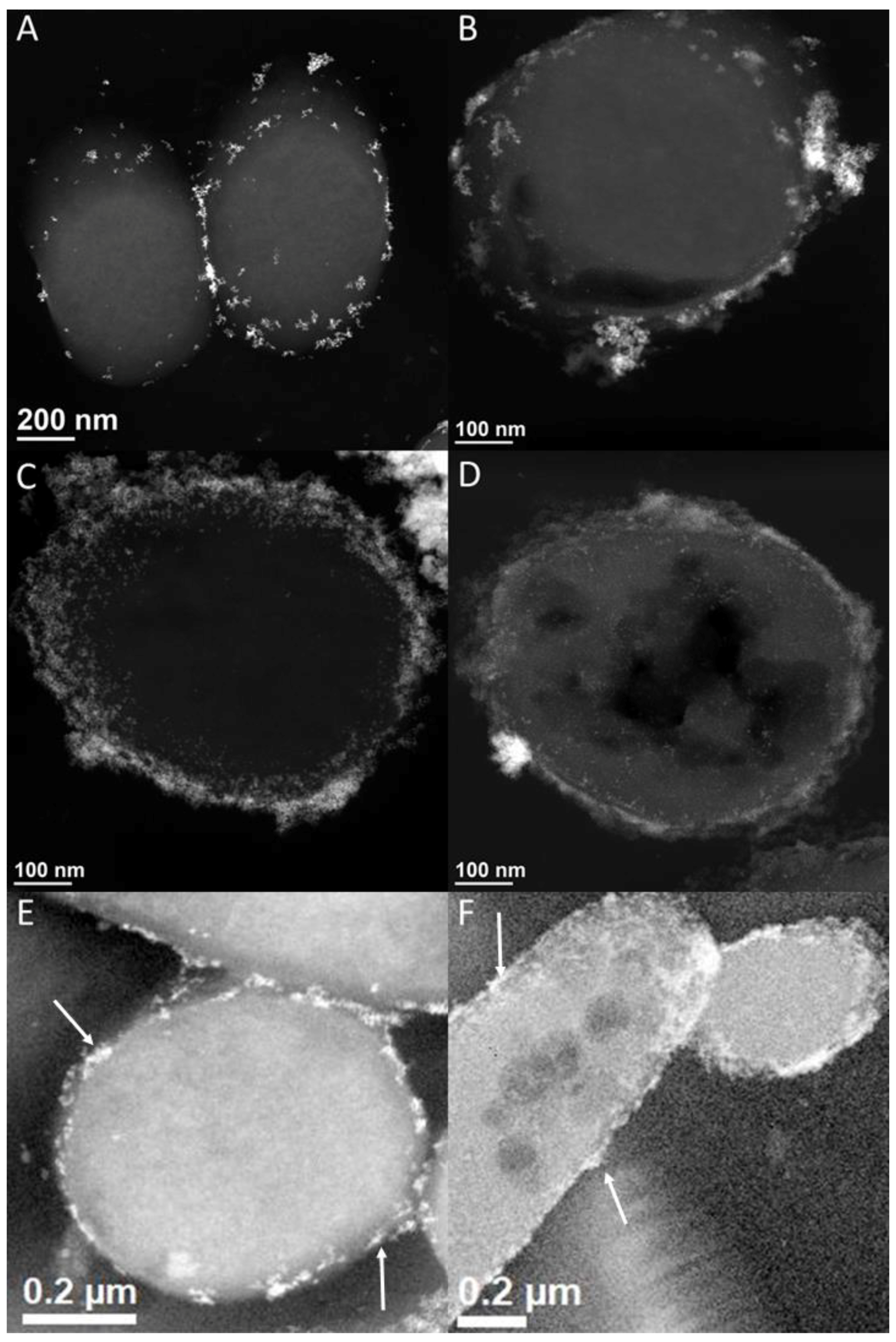
3.4. [NiFe]-Hydrogenase Location
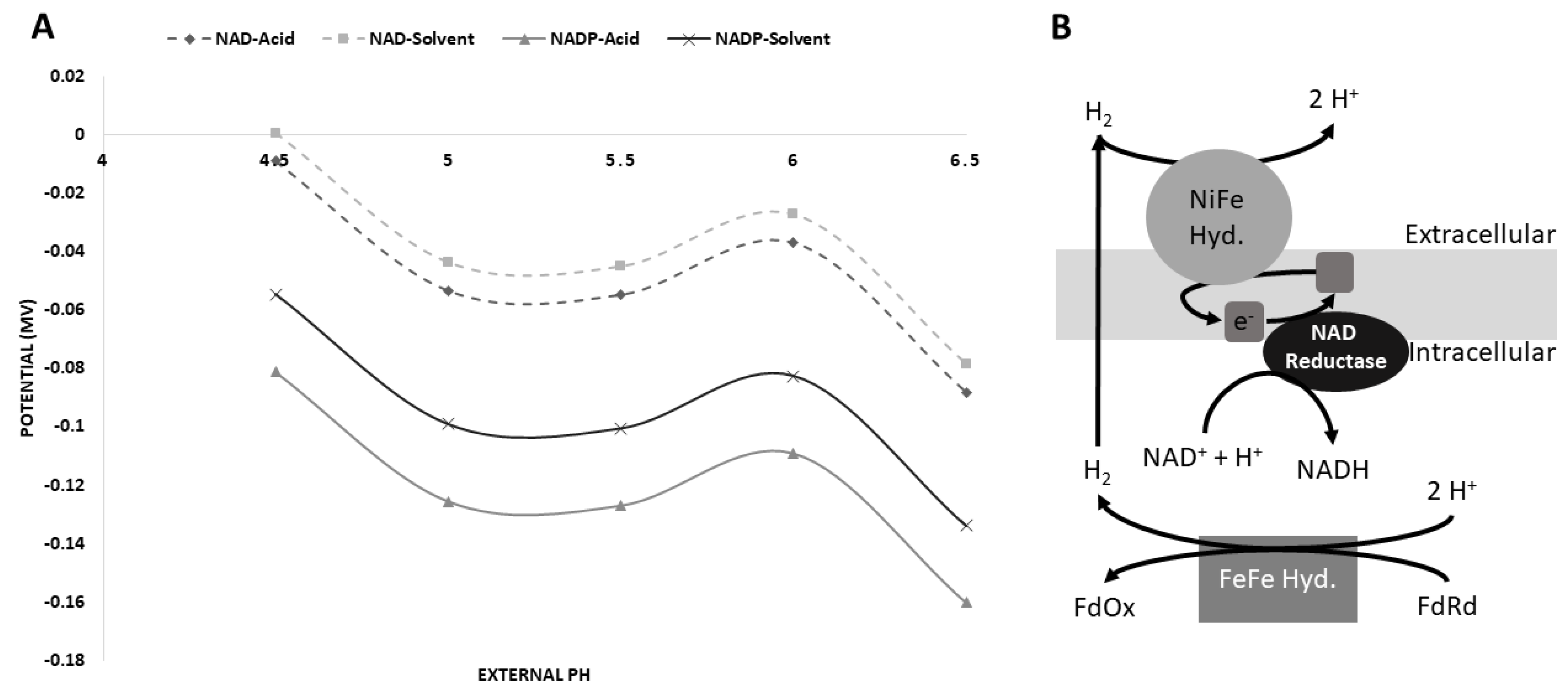
3.5. Possible [NiFe]-Hydrogenase Function
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Jones, D.T.; Woods, D.R. Acetone-butanol fermentation revisited. Microbiol. Rev. 1986, 50, 484–524. [Google Scholar] [PubMed]
- Gapes, J.R. The economics of acetone-butanol fermentation: Theoretical and market considerations. J. Mol. Microbiol. Biotechnol. 2000, 2, 27–32. [Google Scholar] [PubMed]
- Keis, S.; Shaheen, R.; Jones, D.T. Emended descriptions of Clostridium acetobutylicum and Clostridium beijerinckii, and descriptions of Clostridium saccharoperbutylacetonicum sp. nov. and Clostridium saccharobutylicum sp. nov. Int. J. Syst. Evol. Microbiol. 2001, 51, 2095–2103. [Google Scholar] [CrossRef] [PubMed]
- Girbal, L.; Croux, C.; Vasconcelos, I.; Soucaille, P. Regulation of metabolic shifts in Clostridium-acetobutylicum ATCC-824. FEMS Microbiol. Rev. 1995, 17, 287–297. [Google Scholar] [CrossRef]
- Cai, X.; Servinsky, M.; Kiel, J.; Sund, C.; Bennett, G.N. Analysis of redox responses during TNT transformation by Clostridium acetobutylicum ATCC 824 and mutants exhibiting altered metabolism. Appl. Microbiol. Biotechnol. 2013, 97, 4651–4663. [Google Scholar] [CrossRef] [PubMed]
- Servinsky, M.D.; Liu, S.; Gerlach, E.S.; Germane, K.L.; Sund, C.J. Fermentation of oxidized hexose derivatives by Clostridium acetobutylicum. Microb. Cell Fact. 2014, 13, 139. [Google Scholar] [CrossRef] [PubMed]
- Lutke-Eversloh, T.; Bahl, H. Metabolic engineering of Clostridium acetobutylicum: Recent advances to improve butanol production. Curr. Opin. Biotechnol. 2011, 22, 634–647. [Google Scholar] [CrossRef] [PubMed]
- Girbal, L.; Vasconcelos, I.; Soucaille, P. Transmembrane pH of Clostridium acetobutylicum is inverted (more acidic inside) when the in vivo activity of hydrogenase is decreased. J. Bacteriol. 1994, 176, 6146–6147. [Google Scholar] [CrossRef] [PubMed]
- Calusinska, M.; Happe, T.; Joris, B.; Wilmotte, A. The surprising diversity of clostridial hydrogenases: A comparative genomic perspective. Microbiology 2010, 156, 1575–1588. [Google Scholar] [CrossRef] [PubMed]
- Demuez, M.; Cournac, L.; Guerrini, O.; Soucaille, P.; Girbal, L. Complete activity profile of Clostridium acetobutylicum [FeFe]-hydrogenase and kinetic parameters for endogenous redox partners. FEMS Microbiol. Lett. 2007, 275, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Yoo, M.; Bestel-Corre, G.; Croux, C.; Riviere, A.; Meynial-Salles, I.; Soucaille, P. A quantitative system-scale characterization of the metabolism of Clostridium acetobutylicum. mBio 2015, 6, e01808–e01815. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Xu, J.; Wang, Y.; Chen, Y.; Shen, X.; Niu, H.; Guo, T.; Ying, H. Comparative transcriptomic analysis of Clostridium acetobutylicum biofilm and planktonic cells. J. Biotechnol. 2016, 218, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, S.; Kosaka, T.; Hirakawa, H.; Matsuura, K.; Yoshino, S.; Furukawa, K. Metabolic engineering for solvent productivity by downregulation of the hydrogenase gene cluster hupCBA in Clostridium saccharoperbutylacetonicum strain N1-4. Appl. Microbiol. Biotechnol. 2008, 78, 483–493. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.W.; Bennett, G.N.; Rudolph, F.B. Isolation and characterization of mutants of Clostridium acetobutylicum ATCC 824 deficient in acetoacetyl-coenzyme a: Acetate/butyrate: Coenzyme a-transferase (EC 2.8. 3.9) and in other solvent pathway enzymes. Appl. Environ. Microbiol. 1989, 55, 970–976. [Google Scholar] [PubMed]
- Al-Shorgani, N.K.N.; Kalil, M.S.; Ali, E.; Yusoff, W.M.W.; Hamid, A.A. Enhancement of biobutanol production by butyric acid addition using Clostridium saccharoperbutylacetonicum N1-4 (ATCC 13564). Biotechnology 2012, 11, 326–332. [Google Scholar]
- Servinsky, M.D.; Kiel, J.T.; Dupuy, N.F.; Sund, C.J. Transcriptional analysis of differential carbohydrate utilization by Clostridium acetobutylicum. Microbiology 2010, 156, 3478–3491. [Google Scholar] [CrossRef] [PubMed]
- Heap, J.T.; Pennington, O.J.; Cartman, S.T.; Carter, G.P.; Minton, N.P. The clostron: A universal gene knock-out system for the genus clostridium. J. Microbiol. Methods 2007, 70, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Perutka, J.; Wang, W.; Goerlitz, D.; Lambowitz, A.M. Use of computer-designed group II introns to disrupt Escherichia coli DExH/D-box protein and DNA helicase genes. J. Mol. Biol. 2004, 336, 421–439. [Google Scholar] [CrossRef] [PubMed]
- Heap, J.T.; Kuehne, S.A.; Ehsaan, M.; Cartman, S.T.; Cooksley, C.M.; Scott, J.C.; Minton, N.P. The clostron: Mutagenesis in clostridium refined and streamlined. J. Microbiol. Methods 2010, 80, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Servinsky, M.D.; Julin, D.A. Effect of a recd mutation on DNA damage resistance and transformation in Deinococcus radiodurans. J. Bacteriol. 2007, 189, 5101–5107. [Google Scholar] [CrossRef] [PubMed]
- Tyurin, M.; Padda, R.; Huang, K.X.; Wardwell, S.; Caprette, D.; Bennett, G.N. Electrotransformation of Clostridium acetobutylicum ATCC 824 using high-voltage radio frequency modulated square pulses. J. Appl. Microbiol. 2000, 88, 220–227. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- McClure, R.; Balasubramanian, D.; Sun, Y.; Bobrovskyy, M.; Sumby, P.; Genco, C.A.; Vanderpool, C.K.; Tjaden, B. Computational analysis of bacterial RNA-Seq data. Nucleic Acids Res. 2013, 41, e140. [Google Scholar] [CrossRef] [PubMed]
- Pinske, C.; Jaroschinsky, M.; Sargent, F.; Sawers, G. Zymographic differentiation of [NiFe]-hydrogenases 1, 2 and 3 of Escherichia coli K-12. BMC Microbiol. 2012, 12, 1. [Google Scholar] [CrossRef] [PubMed]
- Finch, A.S.; Mackie, T.D.; Sund, C.J.; Sumner, J.J. Metabolite analysis of Clostridium acetobutylicum: Fermentation in a microbial fuel cell. Bioresour. Technol. 2011, 102, 312–315. [Google Scholar] [CrossRef] [PubMed]
- Yong, P.; Rowson, N.A.; Farr, J.P.G.; Harris, I.R.; Macaskie, L.E. Bioreduction and biocrystallization of palladium by Desulfovibrio desulfuricans NCIMB 8307. Biotechnol. Bioeng. 2002, 80, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Gibbins, L.N.; Forsberg, C.W. Transmembrane pH gradient and membrane potential in Clostridium acetobutylicum during growth under acetogenic and solventogenic conditions. Appl. Environ. Microbiol. 1985, 50, 1043–1047. [Google Scholar] [PubMed]
- Amador-Noguez, D.; Feng, X.J.; Fan, J.; Roquet, N.; Rabitz, H.; Rabinowitz, J.D. Systems-level metabolic flux profiling elucidates a complete, bifurcated tricarboxylic acid cycle in Clostridium acetobutylicum. J. Bacteriol. 2010, 192, 4452–4461. [Google Scholar] [CrossRef] [PubMed]
- Mao, F.L.; Dam, P.; Chou, J.; Olman, V.; Xu, Y. Door: A database for prokaryotic operons. Nucleic Acids Res. 2009, 37, D459–D463. [Google Scholar] [CrossRef] [PubMed]
- Dam, P.; Olman, V.; Harris, K.; Su, Z.C.; Xu, Y. Operon prediction using both genome-specific and general genomic information. Nucleic Acids Res. 2007, 35, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Tjaden, B. De novo assembly of bacterial transcriptomes from RNA-Seq data. Genome Biol. 2015, 16, 1. [Google Scholar] [CrossRef] [PubMed]
- Caspi, R.; Billington, R.; Ferrer, L.; Foerster, H.; Fulcher, C.A.; Keseler, I.M.; Kothari, A.; Krummenacker, M.; Latendresse, M.; Mueller, L.A.; et al. The metacyc database of metabolic pathways and enzymes and the biocyc collection of pathway/genome databases. Nucleic Acids Res. 2016, 44, D471–D480. [Google Scholar] [CrossRef] [PubMed]
- Boratyn, G.M.; Schaffer, A.A.; Agarwala, R.; Altschul, S.F.; Lipman, D.J.; Madden, T.L. Domain enhanced lookup time accelerated BLAST. Biol. Direct 2012, 7, 12. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.; Zaretskaya, I.; Raytselis, Y.; Merezhuk, Y.; McGinnis, S.; Madden, T.L. NCBI BLAST: A better web interface. Nucleic Acids Res. 2008, 36, W5–W9. [Google Scholar] [CrossRef] [PubMed]
- Muller, A.; MacCallum, R.M.; Sternberg, M.J.E. Benchmarking PSI-BLAST in genome annotation. J. Mol. Biol. 1999, 293, 1257–1271. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Paschos, A.; Bauer, A.; Zimmermann, A.; Zehelein, E.; Bock, A. Hypf, a carbamoyl phosphate-converting enzyme involved in [NiFe]-hydrogenase maturation. J. Biol. Chem. 2002, 277, 49945–49951. [Google Scholar] [CrossRef] [PubMed]
- Odom, J.M.; Peck, H.D., Jr. Hydrogenase, electron-transfer proteins, and energy coupling in the sulfate-reducing bacteria Desulfovibrio. Annu. Rev. Microbiol. 1984, 38, 551–592. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; San, K.-Y.; Bennett, G.N. Cofactor engineering for advancing chemical biotechnology. Curr. Opin. Biotechnol. 2013, 24, 994–999. [Google Scholar] [CrossRef] [PubMed]
- Vasconcelos, I.; Girbal, L.; Soucaille, P. Regulation of carbon and electron flow in Clostridium-acetobutylicum grown in chemostat culture at neutral ph on mixtures of glucose and glycerol. J. Bacteriol. 1994, 176, 1443–1450. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Chen, Y.; Li, A.; Ding, F.Y.; Zhou, T.; He, Y.; Li, B.B.; Niu, H.Q.; Lin, X.Q.; Xie, J.J.; et al. Enhanced butanol production by modulation of electron flow in Clostridium acetobutylicum B3 immobilized by surface adsorption. Bioresour. Technol. 2013, 129, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Yang, Z.J.; Wang, P.; Niu, H.Q.; Zhuang, W.; Chen, Y.; Wu, J.L.; Zhu, C.J.; Ying, H.J.; Ouyang, P.K. Towards acetone-uncoupled biofuels production in solventogenic clostridium through reducing power conservation. Metab. Eng. 2018, 47, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Mikheenko, I.P.; Rousset, M.; Dementin, S.; Macaskie, L.E. Bioaccumulation of palladium by Desulfovibrio fructosivorans wild-type and hydrogenase-deficient strains. Appl. Environ. Microbiol. 2008, 74, 6144–6146. [Google Scholar] [CrossRef] [PubMed]
- Frielingsdorf, S.; Schubert, T.; Pohlmann, A.; Lenz, O.; Friedrich, B. A trimeric supercomplex of the oxygen-tolerant membrane-bound [NIFE]-hydrogenase from ralstonia eutropha H16. Biochemistry 2011, 50, 10836–10843. [Google Scholar] [CrossRef] [PubMed]
- Pauss, A.; Andre, G.; Perrier, M.; Guiot, S.R. Liquid-to-gas mass transfer in anaerobic processes: Inevitable transfer limitations of methane and hydrogen in the biomethanation process. Appl. Environ. Microbiol. 1990, 56, 1636–1644. [Google Scholar] [PubMed]
- Terracciano, J.S.; Kashket, E.R. Intracellular conditions required for initiation of solvent production by Clostridium acetobutylicum. Appl. Environ. Microbiol. 1986, 52, 86–91. [Google Scholar] [PubMed]
- Magnan, D.T.C.; San, K.Y.; Bennett, G.N. Modification of butyrate and butanol production in Clostridium acetobutylicum by introduction of a ferredoxin oxidoreductase. In Proceedings of the Rice University: SIMB Annual Meeting, New Orleans, LA, USA, 24–28 July 2016. [Google Scholar]
- Iddar, A.; Valverde, F.; Serrano, A.; Soukri, A. Expression, purification, and characterization of recombinant nonphosphorylating NADP-dependent glyceraldehyde-3-phosphate dehydrogenase from Clostridium acetobutylicum. Protein Expr. Purif. 2002, 25, 519–526. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Glucose Consumed | Acetate | Acetone | Acetoin | Ethanol | Butyrate | Butanol | |
|---|---|---|---|---|---|---|---|
| WT agit. | 76.9 | 28.8 | 3.4 | 1.3 | 1.9 | 43.7 | 5.7 |
| ΔhypF/hypD agit. | 63.4 | 31.5 | 0.0 | 0.9 | 1.9 | 41.9 | 1.6 |
| WT stat. | 106 | 24.9 | 17.9 | 2.9 | 5.4 | 27.2 | 36.4 |
| ΔhypF/hypD stat. | 63.7 | 27.6 | 0.0 | 1.7 | 2.8 | 41.8 | 5.33 |
| N = 2 | Concentrations in mM Time = 24 h | ||||||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Germane, K.L.; Liu, S.; Gerlach, E.S.; Savage, A.M.; Renberg, R.L.; Zu, T.N.K.; Dong, H.; Walck, S.D.; Servinsky, M.D.; Sund, C.J. Hydrogen-Cycling during Solventogenesis in Clostridium acetobutylicum American Type Culture Collection (ATCC) 824 Requires the [NiFe]-Hydrogenase for Energy Conservation. Fermentation 2018, 4, 55. https://doi.org/10.3390/fermentation4030055
Germane KL, Liu S, Gerlach ES, Savage AM, Renberg RL, Zu TNK, Dong H, Walck SD, Servinsky MD, Sund CJ. Hydrogen-Cycling during Solventogenesis in Clostridium acetobutylicum American Type Culture Collection (ATCC) 824 Requires the [NiFe]-Hydrogenase for Energy Conservation. Fermentation. 2018; 4(3):55. https://doi.org/10.3390/fermentation4030055
Chicago/Turabian StyleGermane, Katherine L., Sanchao Liu, Elliot S. Gerlach, Alice M. Savage, Rebecca L. Renberg, Theresah N. K. Zu, Hong Dong, Scott D. Walck, Matthew D. Servinsky, and Christian J. Sund. 2018. "Hydrogen-Cycling during Solventogenesis in Clostridium acetobutylicum American Type Culture Collection (ATCC) 824 Requires the [NiFe]-Hydrogenase for Energy Conservation" Fermentation 4, no. 3: 55. https://doi.org/10.3390/fermentation4030055
APA StyleGermane, K. L., Liu, S., Gerlach, E. S., Savage, A. M., Renberg, R. L., Zu, T. N. K., Dong, H., Walck, S. D., Servinsky, M. D., & Sund, C. J. (2018). Hydrogen-Cycling during Solventogenesis in Clostridium acetobutylicum American Type Culture Collection (ATCC) 824 Requires the [NiFe]-Hydrogenase for Energy Conservation. Fermentation, 4(3), 55. https://doi.org/10.3390/fermentation4030055