
Composition Divergence and Synergistic Mechanisms in Microbial Communities During Multi-Varietal Wine Co-Fermentation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Determination of Volatile Organic Compounds
2.3. Extraction of DNA
2.4. Bioinformatics Analysis
3. Results and Discussion
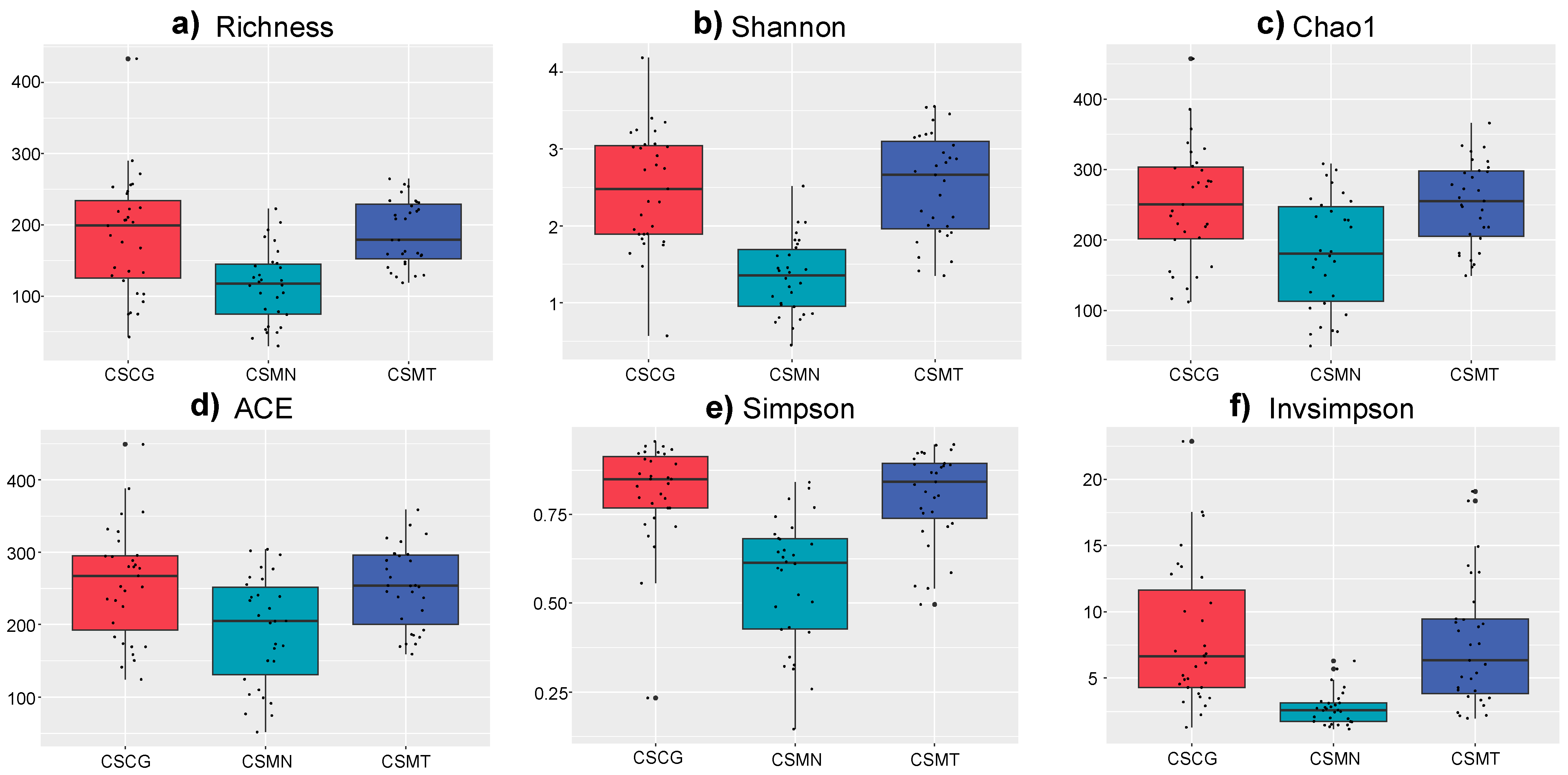
3.1. Microbial Community Structure in Single Fermentation and Different Co-Fermentation Processes
3.2. Relationship Between Volatile Organic Compounds and Microbial Communities in Different Fermentation Processes
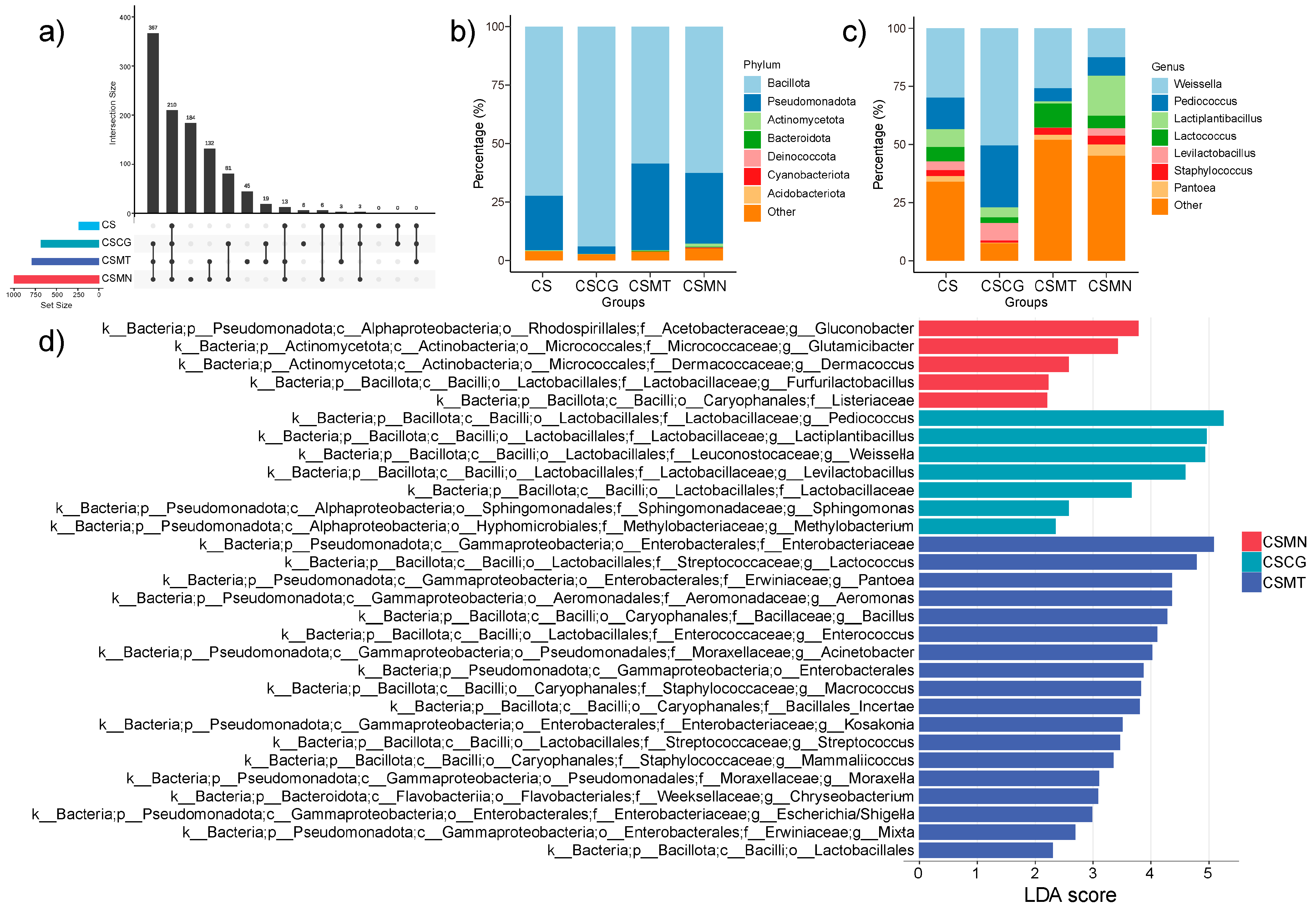
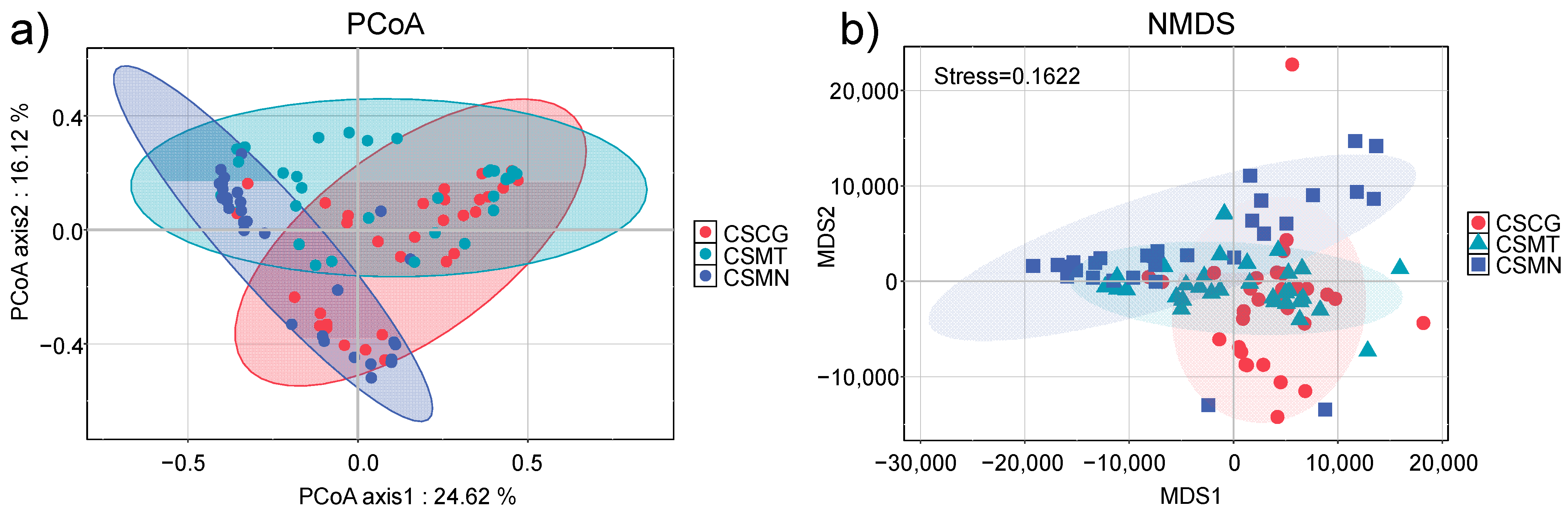
3.3. Differences in and Distribution of Microbial Communities Across Different Co-Fermentation Processes
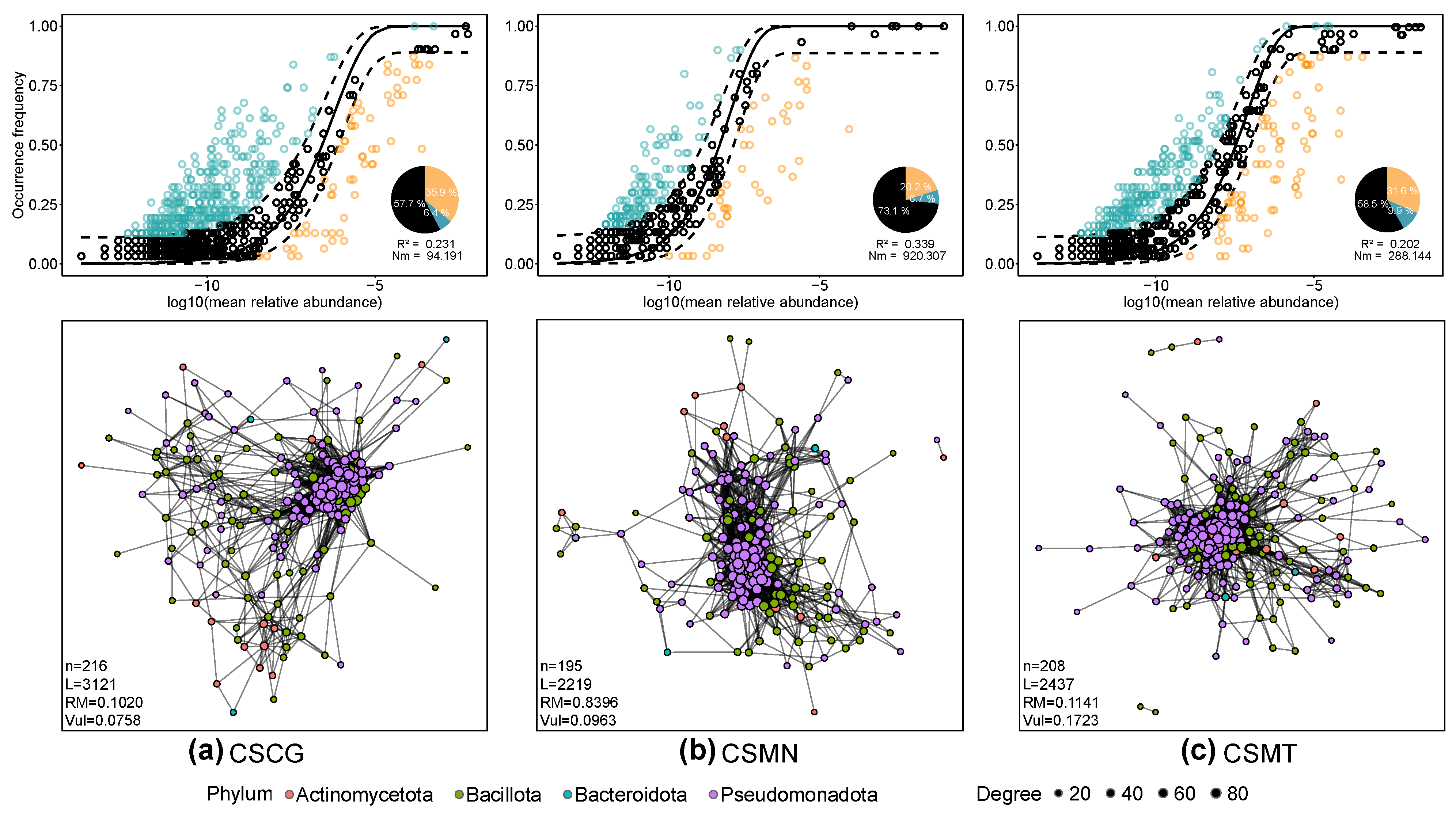
3.4. Ecological Characteristics of Core Microbial Communities in Three Co-Fermentation Processes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Abrahamse, C.E.; Bartowsky, E.J. Timing of Malolactic Fermentation Inoculation in Shiraz Grape Must and Wine: Influence on Chemical Composition. World J. Microbiol. Biotechnol. 2012, 28, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Burns, K.N.; Kluepfel, D.A.; Strauss, S.L.; Bokulich, N.A.; Cantu, D.; Steenwerth, K.L. Vineyard Soil Bacterial Diversity and Composition Revealed by 16S rRNA Genes: Differentiation by Geographic Features. Soil Biol. Biochem. 2015, 91, 232–247. [Google Scholar] [CrossRef]
- Chalvantzi, I.; Banilas, G.; Tassou, C.; Nisiotou, A. Biogeographical Regionalization of Wine Yeast Communities in Greece and Environmental Drivers of Species Distribution at a Local Scale. Front. Microbiol. 2021, 12, 705001. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Lei, X.; Jiang, J.; Qin, Y.; Jiang, L.; Liu, Y.-L. Microbial Diversity on Grape Epidermis and Wine Volatile Aroma in Spontaneous Fermentation Comprehensively Driven by Geography, Subregion, and Variety. Int. J. Food Microbiol. 2023, 404, 110315. [Google Scholar] [CrossRef]
- Gao, F.; Chen, J.; Xiao, J.; Cheng, W.; Zheng, X.; Wang, B.; Shi, X. Microbial Community Composition on Grape Surface Controlled by Geographical Factors of Different Wine Regions in Xinjiang, China. Food Res. Int. 2019, 122, 348–360. [Google Scholar] [CrossRef]
- Gao, J.; Geng, H.; Chai, R.; Wu, T.; Huang, W.; You, Y.; Zhan, J. Fungal Community Composition and Its Relationship with Volatile Compounds during Spontaneous Fermentation of Cabernet Sauvignon from Two Chinese Wine-Growing Regions. Foods 2024, 13, 106. [Google Scholar] [CrossRef]
- Guo, J.; Zhao, X.; Shi, J. Correlation of Microbial Community Structure and Volatile Flavor Compounds during Corn Yellow Wine Fermentation. Biotechnol. Prog. 2024, 40, e3408. [Google Scholar] [CrossRef]
- Kioroglou, D.; Mas, A.; Portillo, M.C. High-Throughput Sequencing Approach to Analyze the Effect of Aging Time and Barrel Usage on the Microbial Community Composition of Red Wines. Front. Microbiol. 2020, 11, 562560. [Google Scholar] [CrossRef]
- Knight, S.J.; Karon, O.; Goddard, M.R. Small Scale Fungal Community Differentiation in a Vineyard System. Food Microbiol. 2020, 87, 103358. [Google Scholar] [CrossRef]
- Liang, L.; Ma, Y.; Jiang, Z.; Sam, F.E.; Peng, S.; Li, M.; Wang, J. Dynamic Analysis of Microbial Communities and Flavor Properties in Merlot Wines Produced from Inoculation and Spontaneous Fermentation. Food Res. Int. 2023, 164, 112379. [Google Scholar] [CrossRef]
- Liu, D.; Chen, Q.; Zhang, P.; Chen, D.; Howell, K.S. The Fungal Microbiome Is an Important Component of Vineyard Ecosystems and Correlates with Regional Distinctiveness of Wine. mSphere 2020, 5, e00534-20. [Google Scholar] [CrossRef]
- Liu, D.; Legras, J.-L.; Zhang, P.; Chen, D.; Howell, K. Diversity and Dynamics of Fungi during Spontaneous Fermentations and Association with Unique Aroma Profiles in Wine. Int. J. Food Microbiol. 2021, 338, 108983. [Google Scholar] [CrossRef]
- Liu, D.; Zhang, P.; Chen, D.; Howell, K. From the Vineyard to the Winery: How Microbial Ecology Drives Regional Distinctiveness of Wine. Front. Microbiol. 2019, 10, 2679. [Google Scholar] [CrossRef]
- Liu, Q.; Hao, N.; Mi, L.; Peng, S.; Marie-Colette, A.K.; Zhao, X.; Wang, J. From Microbial Communities to Aroma Profiles: A Comparative Study of Spontaneous Fermentation in Merlot and Cabernet Sauvignon Wines. Food Chem. X 2025, 26, 102317. [Google Scholar] [CrossRef] [PubMed]
- Martiniuk, J.T.; Hamilton, J.; Dodsworth, T.; Measday, V. Grape-Associated Fungal Community Patterns Persist from Berry to Wine on a Fine Geographical Scale. FEMS Yeast Res. 2023, 23, foac067. [Google Scholar] [CrossRef] [PubMed]
- Nelson, J.; Coleman, R.; Gravesen, P.; Silacci, M.; Velasquez, A.; Marinelli, K.; Boulton, R. Analysis of a Commercial Red Wine Fermentation Dataset with a Wine Kinetic Model. Fermentation 2025, 11, 4. [Google Scholar] [CrossRef]
- Edgar, R.C. UPARSE: Highly Accurate OTU Sequences from Microbial Amplicon Reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef]
- Dixon, P. VEGAN, a Package of R Functions for Community Ecology. J. Veg. Sci. 2003, 14, 927–930. [Google Scholar] [CrossRef]
- Liu, C.; Cui, Y.; Li, X.; Yao, M. Microeco: An R Package for Data Mining in Microbial Community Ecology. FEMS Microbiol. Ecol. 2021, 97, fiaa255. [Google Scholar] [CrossRef]
- Wen, T.; Xie, P.; Yang, S.; Niu, G.; Liu, X.; Ding, Z.; Xue, C.; Liu, Y.-X.; Shen, Q.; Yuan, J. ggClusterNet: An R Package for Microbiome Network Analysis and Modularity-Based Multiple Network Layouts. iMeta 2022, 1, e32. [Google Scholar] [CrossRef]
- Piao, H.; Hawley, E.; Kopf, S.; DeScenzo, R.; Sealock, S.; Henick-Kling, T.; Hess, M. Insights into the Bacterial Community and Its Temporal Succession during the Fermentation of Wine Grapes. Front. Microbiol. 2015, 6, 809. [Google Scholar] [CrossRef] [PubMed]
- Sumerta, I.N.; Ruan, X.; Howell, K. The Forgotten Wine: Understanding Palm Wine Fermentation and Composition. Int. J. Food Microbiol. 2025, 429, 111022. [Google Scholar] [CrossRef]
- Ohwofasa, A.; Tian, B.; Torrico, D.; Dhami, M.; Winefield, C.; On, S.L.W. Differences in Organic Pinot Noir Wine Production Systems Correlated with Microbiome Analysis, Sensory Characteristics and Volatile Composition. Food Biosci. 2024, 61, 104562. [Google Scholar] [CrossRef]
- Peng, Q.; Zheng, H.; Quan, L.; Li, S.; Huang, J.; Li, J.; Xie, G. Development of a Flavor-Oriented Synthetic Microbial Community for Pour-over Rice Wine: A Comprehensive Microbial Community Analysis. Food Microbiol. 2025, 126, 104677. [Google Scholar] [CrossRef]
- Varo, M.A.; Martín-Gómez, J.; Merida, J.; Serratosa, M.P. Exploring the Impact of Temperature and Fermentation Time on the Evolution of Bioactive Compounds, Antioxidant Activity, and Color Evolution in Blueberry Wines. ACS Food Sci. Technol. 2024, 4, 1301–1309. [Google Scholar] [CrossRef]
- Tian, P.; Wan, J.; Yin, T.; Liu, L.; Ren, H.; Cai, H.; Liu, X.; Zhang, H. Acidity, Sugar, and Alcohol Contents during the Fermentation of Osmanthus-Flavored Sweet Rice Wine and Microbial Community Dynamics. PeerJ 2025, 13, e18826. [Google Scholar] [CrossRef]
- Walker, R.S.K.; Pretorius, I.S. Synthetic Biology for the Engineering of Complex Wine Yeast Communities. Nat. Food 2022, 3, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Li, S.; Zhao, H.; Gu, P.; Chen, Y.; Zhang, B.; Zhu, B. Acetaldehyde Released by Lactobacillus plantarum Enhances Accumulation of Pyranoanthocyanins in Wine during Malolactic Fermentation. Food Res. Int. 2018, 108, 254–263. [Google Scholar] [CrossRef]
- Xie, G.; Wang, L.; Gao, Q.; Yu, W.; Hong, X.; Zhao, L.; Zou, H. Microbial Community Structure in Fermentation Process of Shaoxing Rice Wine by Illumina-Based Metagenomic Sequencing. J. Sci. Food Agric. 2013, 93, 3121–3125. [Google Scholar] [CrossRef]
- Xu, J.-Z.; Zhang, Y.-Y.; Zhang, W.-G. Correlation between Changes in Flavor Compounds and Microbial Community Ecological Succession in the Liquid Fermentation of Rice Wine. World J. Microbiol. Biotechnol. 2023, 40, 17. [Google Scholar] [CrossRef]
- Yuan, Y.; Yang, Z.; Liang, Z.; Wu, Q.; Yan, Y.; Chen, S.; Li, X.; Ai, L.; Ni, L.; Lv, X. The Regulatory Effects of Microbial Community on the Formation of Higher Alcohols and Volatile Flavor Components in Hongqu Rice Wine Brewing. Food Biosci. 2023, 56, 103142. [Google Scholar] [CrossRef]
- Zhang, T.; Liao, Z.; Li, Z.; Liu, Y.; Liu, Y.; Song, Y.; Qin, Y. Dynamic Changes in Dissolved Oxygen Concentration, Microbial Communities, and Volatile Compounds during Industrial Oak-Barrel Fermentation of Sauvignon Blanc Wine. Food Res. Int. 2024, 197, 115250. [Google Scholar] [CrossRef]
- Zhao, B.; Chen, L.; Zhang, M.; Nie, C.; Yang, Q.; Yu, K.; Xia, Y. Electric-Inducive Microbial Interactions in a Thermophilic Anaerobic Digester Revealed by High-Throughput Sequencing of Micron-Scale Single Flocs. Environ. Sci. Technol. 2023, 57, 4367–4378. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Yang, J.; Yan, Y. Composition Divergence and Synergistic Mechanisms in Microbial Communities During Multi-Varietal Wine Co-Fermentation. Fermentation 2025, 11, 349. https://doi.org/10.3390/fermentation11060349
Zhang Y, Yang J, Yan Y. Composition Divergence and Synergistic Mechanisms in Microbial Communities During Multi-Varietal Wine Co-Fermentation. Fermentation. 2025; 11(6):349. https://doi.org/10.3390/fermentation11060349
Chicago/Turabian StyleZhang, Yuhan, Jiao Yang, and Yuxi Yan. 2025. "Composition Divergence and Synergistic Mechanisms in Microbial Communities During Multi-Varietal Wine Co-Fermentation" Fermentation 11, no. 6: 349. https://doi.org/10.3390/fermentation11060349
APA StyleZhang, Y., Yang, J., & Yan, Y. (2025). Composition Divergence and Synergistic Mechanisms in Microbial Communities During Multi-Varietal Wine Co-Fermentation. Fermentation, 11(6), 349. https://doi.org/10.3390/fermentation11060349