
Density Functional Theory Analysis of the Impact of Boron Concentration and Surface Oxidation in Boron-Doped Graphene for Sodium and Aluminum Storage


Abstract
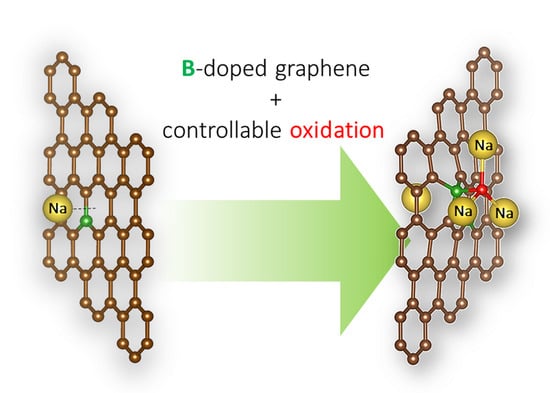
:
1. Introduction
2. Methods
3. Results
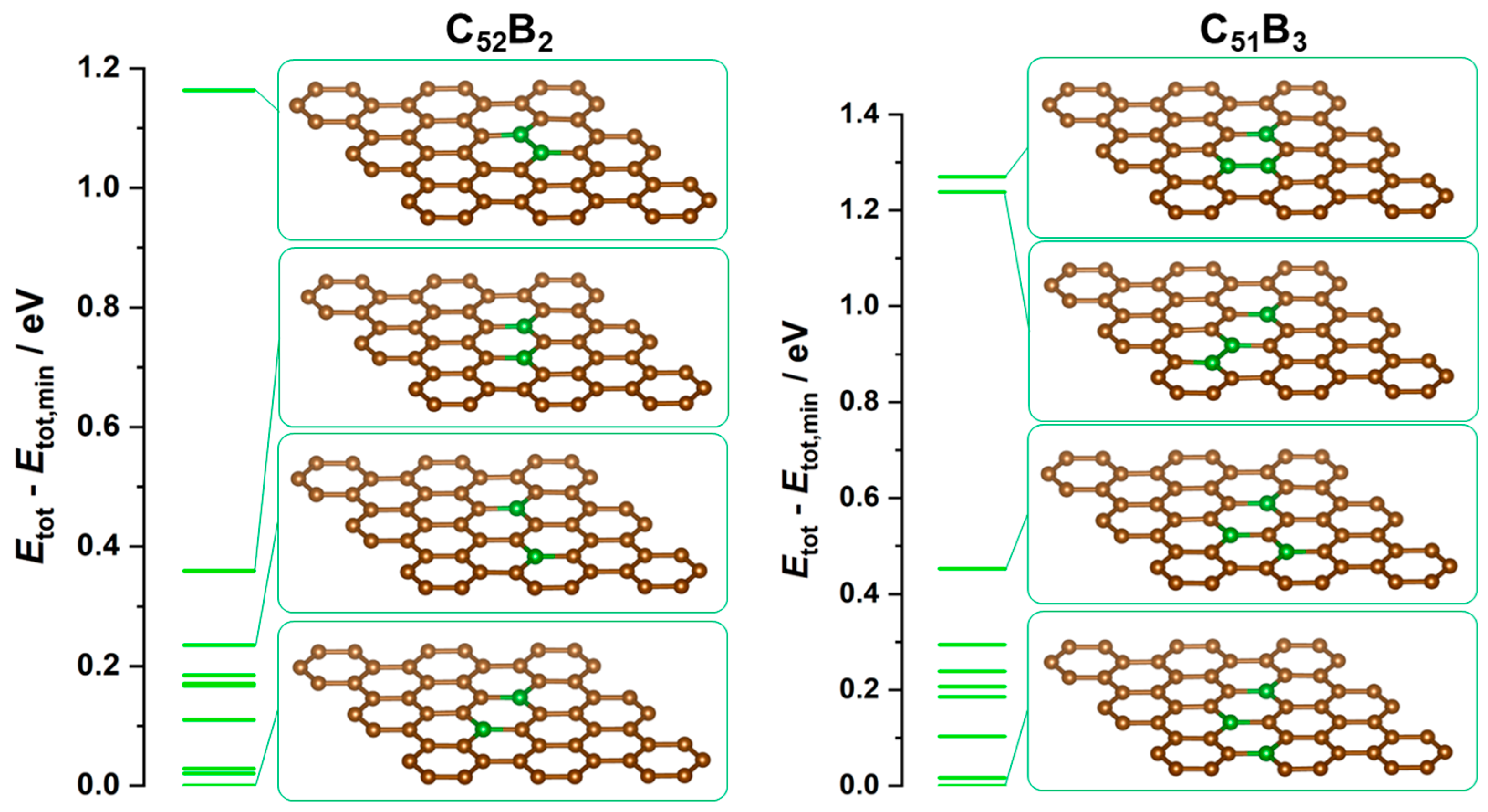
3.1. Graphene Doping by Boron
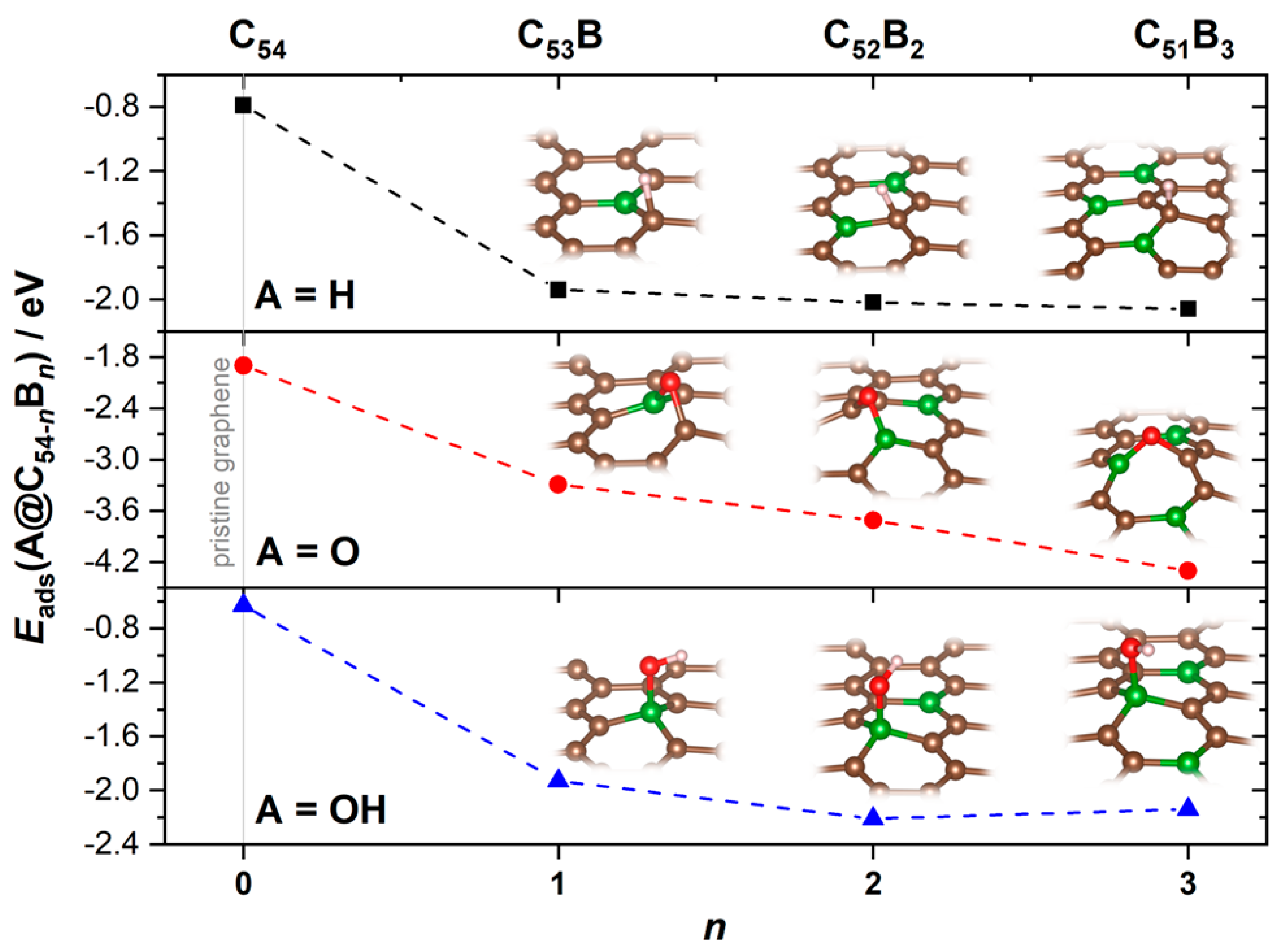
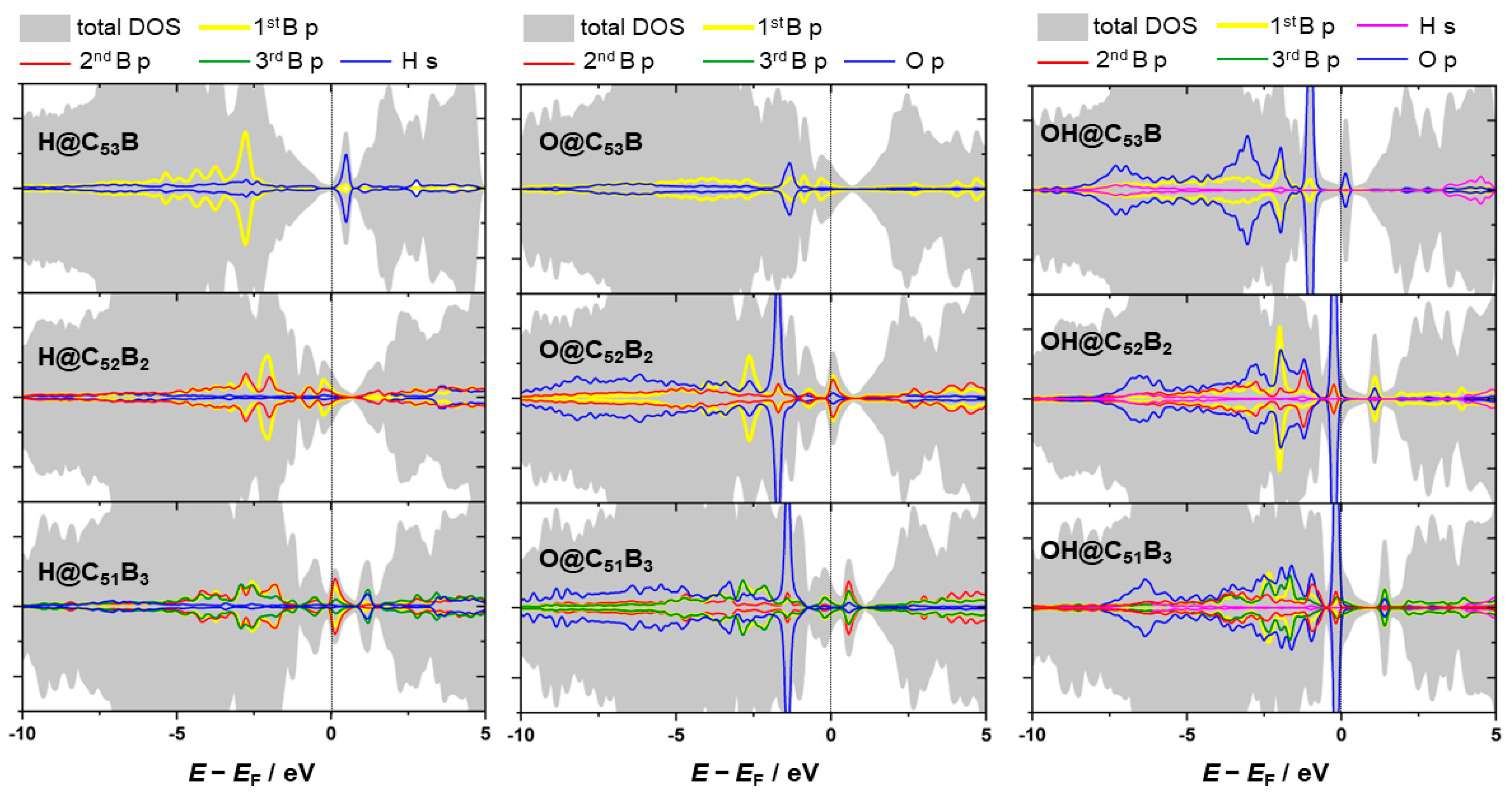
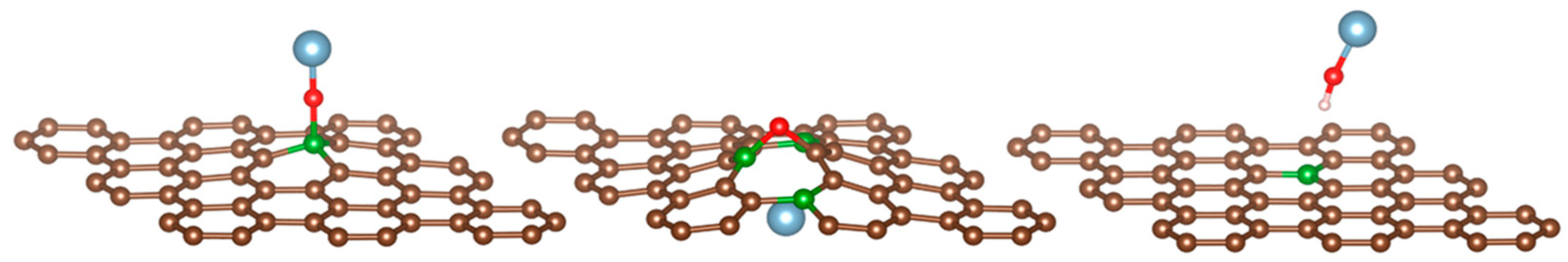
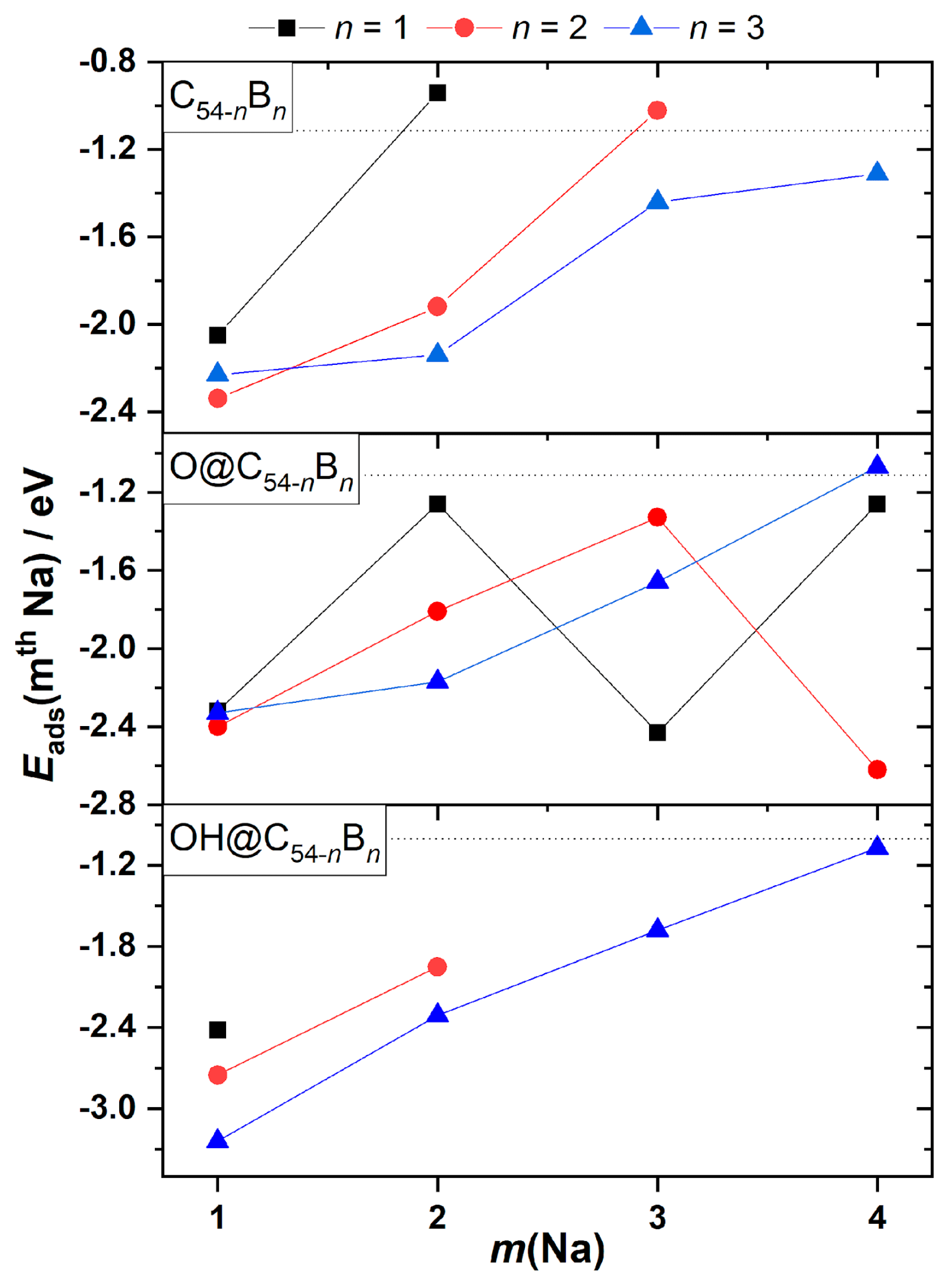
3.2. Reactivity and Oxidation
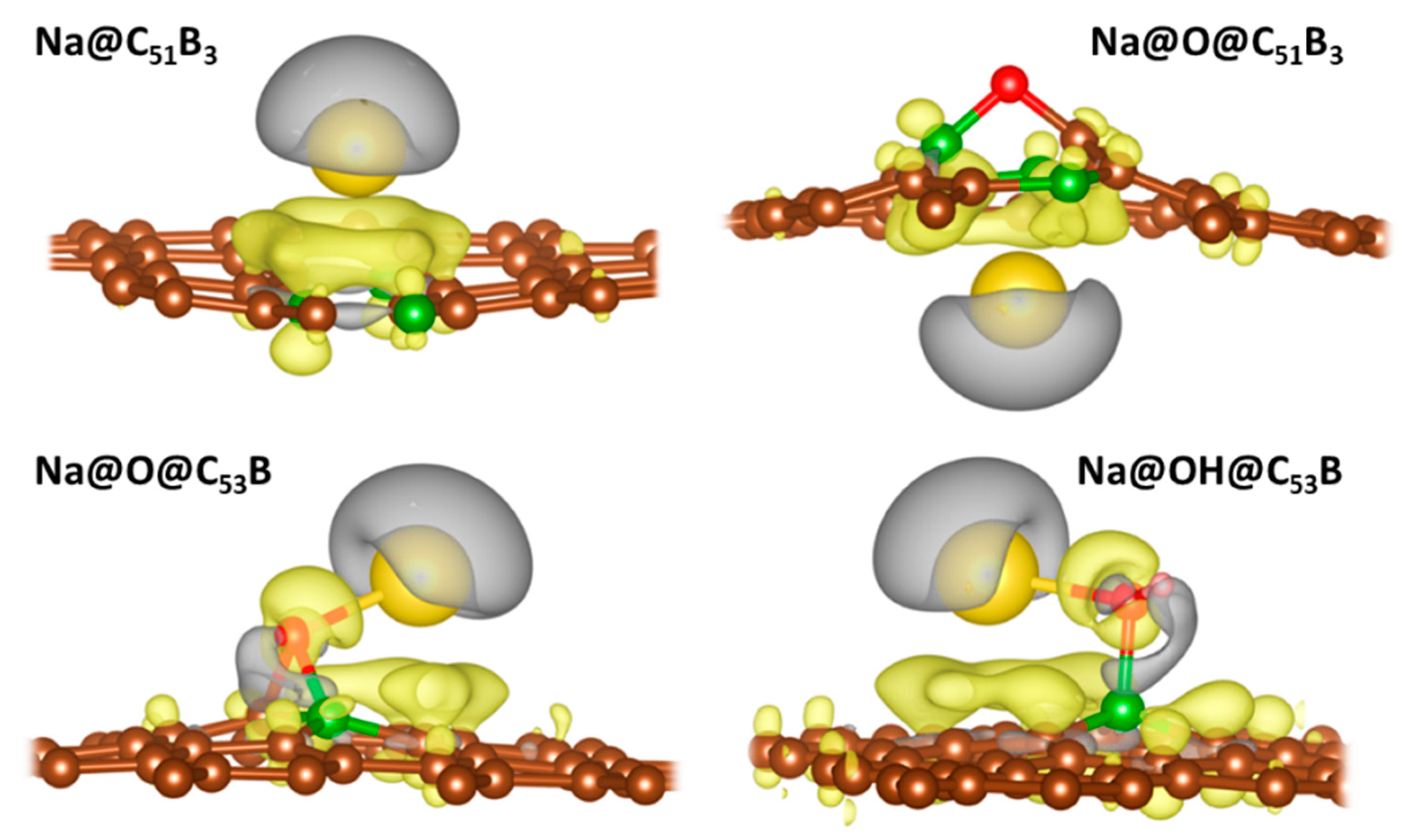
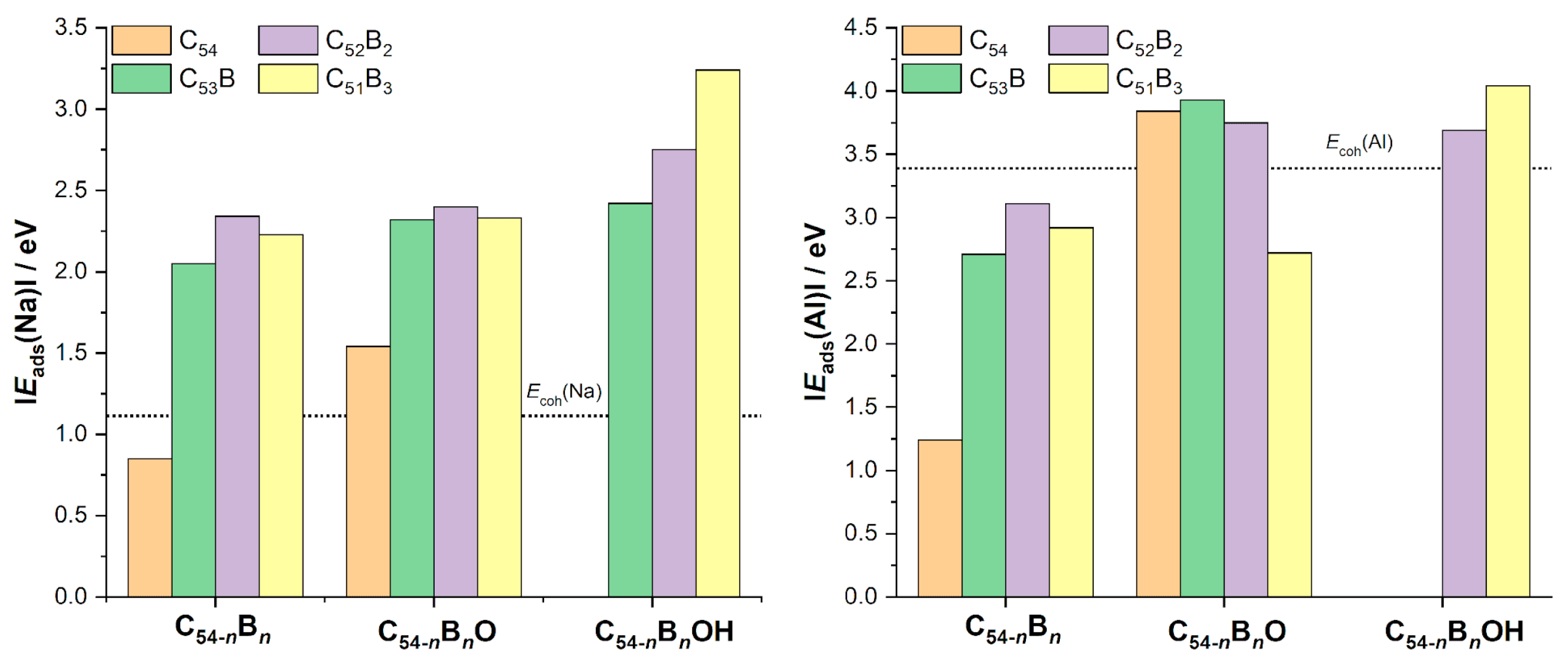
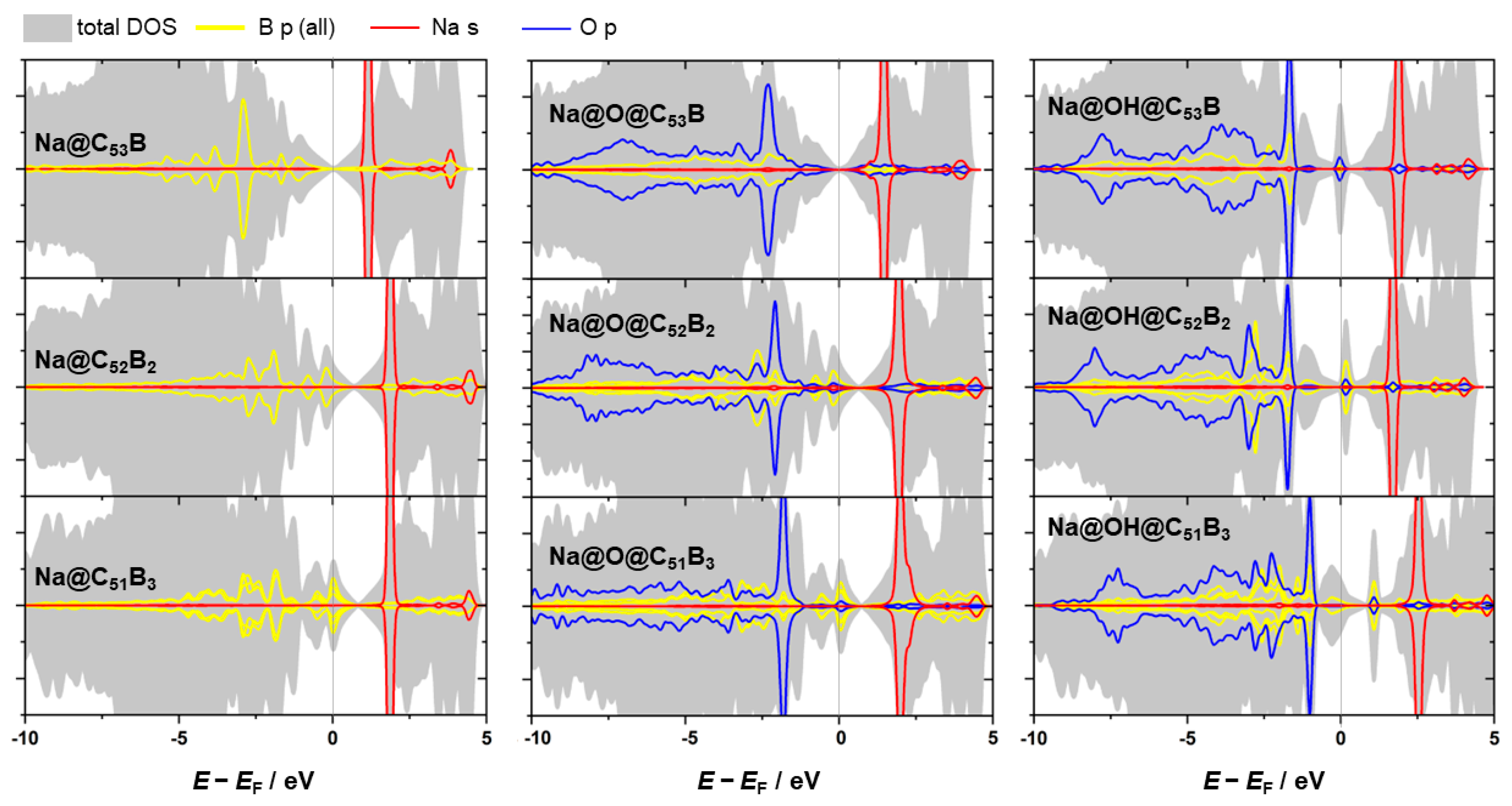
3.3. Metal Adsorption
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.; Katsnelson, M.I.; Grigorieva, I.V.; Dubonos, S.V.; Firsov, A.A. Two-Dimensional Gas of Massless Dirac Fermions in Graphene. Nature 2005, 438, 197–200. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liang, T.; Tu, R.; Lai, W.; Liu, Y. Redistribution of π and σ Electrons in Boron-Doped Graphene from DFT Investigation. Appl. Surf. Sci. 2019, 481, 344–352. [Google Scholar] [CrossRef]
- Brownson, D.A.C.; Kampouris, D.K.; Banks, C.E. An Overview of Graphene in Energy Production and Storage Applications. J. Power Sources 2011, 196, 4873–4885. [Google Scholar] [CrossRef]
- Shanmugam, S.; Nachimuthu, S.; Subramaniam, V. DFT Study of Adsorption of Ions on Doped and Defective Graphene. Mater. Today Commun. 2020, 22, 100714. [Google Scholar] [CrossRef]
- Ali, S.; Lone, B. Adsorption of Cytosine on Si and Ge Doped Graphene: A DFT Study. Mater. Today Proc. 2023, 80, 774–781. [Google Scholar] [CrossRef]
- Tiwari, S.K.; Sahoo, S.; Wang, N.; Huczko, A. Graphene Research and Their Outputs: Status and Prospect. J. Sci. Adv. Mater. Devices 2020, 5, 10–29. [Google Scholar] [CrossRef]
- Soldano, C.; Mahmood, A.; Dujardin, E. Production, Properties and Potential of Graphene. Carbon 2010, 48, 2127–2150. [Google Scholar] [CrossRef]
- Agnoli, S.; Favaro, M. Doping Graphene with Boron: A Review of Synthesis Methods, Physicochemical Characterization, and Emerging Applications. J. Mater. Chem. A 2016, 4, 5002–5025. [Google Scholar] [CrossRef]
- Liu, L.; Qing, M.; Wang, Y.; Chen, S. Defects in Graphene: Generation, Healing, and Their Effects on the Properties of Graphene: A Review. J. Mater. Sci. Technol. 2015, 31, 599–606. [Google Scholar] [CrossRef]
- Yang, G.; Li, L.; Lee, W.B.; Ng, M.C. Structure of Graphene and Its Disorders: A Review. Sci. Technol. Adv. Mater. 2018, 19, 613–648. [Google Scholar] [CrossRef]
- Riyaz, M.; Garg, S.; Kaur, N.; Goel, N. Boron Doped Graphene as Anode Material for Mg Ion Battery: A DFT Study. Comput. Theor. Chem. 2022, 1214, 113757. [Google Scholar] [CrossRef]
- Dobrota, A.S.; Pašti, I.A.; Mentus, S.V.; Skorodumova, N.V. A DFT Study of the Interplay between Dopants and Oxygen Functional Groups over the Graphene Basal Plane-Implications in Energy-Related Applications. Phys. Chem. Chem. Phys. 2017, 19, 8530–8540. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhou, Y.; Yu, Y.; Chen, B.-Y.; Hong, J. Exploring Catalytic Performance of Boron-Doped Graphene Electrode for Electrochemical Degradation of Acetaminophen. Appl. Surf. Sci. 2020, 508, 145111. [Google Scholar] [CrossRef]
- Etacheri, V.; Marom, R.; Elazari, R.; Salitra, G.; Aurbach, D. Challenges in the Development of Advanced Li-Ion Batteries: A Review. Energy Environ. Sci. 2011, 4, 3243–3262. [Google Scholar] [CrossRef]
- Liang, Y.; Dong, H.; Aurbach, D.; Yao, Y. Current Status and Future Directions of Multivalent Metal-Ion Batteries. Nat. Energy 2020, 5, 646–656. [Google Scholar] [CrossRef]
- Pašti, I.A.; Jovanović, A.; Dobrota, A.S.; Mentus, S.V.; Johansson, B.; Skorodumova, N.V. Atomic Adsorption on Pristine Graphene along the Periodic Table of Elements—From PBE to Non-Local Functionals. Appl. Surf. Sci. 2018, 436, 433–440. [Google Scholar] [CrossRef]
- Lin, M.C.; Gong, M.; Lu, B.; Wu, Y.; Wang, D.Y.; Guan, M.; Angell, M.; Chen, C.; Yang, J.; Hwang, B.J.; et al. An Ultrafast Rechargeable Aluminium-Ion Battery. Nature 2015, 520, 324–328. [Google Scholar] [CrossRef]
- Zhou, W.; Jia, J.; Lu, J.; Yang, L.; Hou, D.; Li, G.; Chen, S. Recent Developments of Carbon-Based Electrocatalysts for Hydrogen Evolution Reaction. Nano Energy 2016, 28, 29–43. [Google Scholar] [CrossRef]
- Zhang, T.; Li, C.; Wang, F.; Noori, A.; Mousavi, M.F.; Xia, X.; Zhang, Y. Recent Advances in Carbon Anodes for Sodium-Ion Batteries. Chem. Rec. 2022, 22, e202200083. [Google Scholar] [CrossRef]
- Benti, N.E.; Tiruye, G.A.; Mekonnen, Y.S. Boron and Pyridinic Nitrogen-Doped Graphene as Potential Catalysts for Rechargeable Non-Aqueous Sodium–Air Batteries. RSC Adv. 2020, 10, 21387–21398. [Google Scholar] [CrossRef]
- Panchakarla, L.S.; Subrahmanyam, K.S.; Saha, S.K.; Govindaraj, A.; Krishnamurthy, H.R.; Waghmare, U.V.; Rao, C.N.R. Synthesis, Structure, and Properties of Boron- and Nitrogen-Doped Graphene. Adv. Mater. 2009, 21, 4726–4730. [Google Scholar] [CrossRef]
- Yu, X.; Han, P.; Wei, Z.; Huang, L.; Gu, Z.; Peng, S.; Ma, J.; Zheng, G. Boron-Doped Graphene for Electrocatalytic N2 Reduction. Joule 2018, 2, 1610–1622. [Google Scholar] [CrossRef]
- Rao, C.N.R.; Gopalakrishnan, K.; Govindaraj, A. Synthesis, Properties and Applications of Graphene Doped with Boron, Nitrogen and Other Elements. Nano Today 2014, 9, 324–343. [Google Scholar] [CrossRef]
- Liu, Y.; Artyukhov, V.I.; Liu, M.; Harutyunyan, A.R.; Yakobson, B.I. Feasibility of Lithium Storage on Graphene and Its Derivatives. J. Phys. Chem. Lett. 2013, 4, 1737–1742. [Google Scholar] [CrossRef]
- Hao, Q.; Xia, X.; Lei, W.; Wang, W.; Qiu, J. Facile Synthesis of Sandwich-like Polyaniline/Boron-Doped Graphene Nano Hybrid for Supercapacitors. Carbon 2015, 81, 552–563. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, Z.Y.; Liang, R.P.; Li, Y.H.; Qiu, J.D. Boron-Doped Graphene Quantum Dots for Selective Glucose Sensing Based on the “Abnormal” Aggregation-Induced Photoluminescence Enhancement. Anal. Chem. 2014, 86, 4423–4430. [Google Scholar] [CrossRef]
- Choudhuri, I.; Patra, N.; Mahata, A.; Ahuja, R.; Pathak, B. B-N@Graphene: Highly Sensitive and Selective Gas Sensor. J. Phys. Chem. C 2015, 119, 24827–24836. [Google Scholar] [CrossRef]
- Fan, Z.; Li, Y.; Li, X.; Fan, L.; Zhou, S.; Fang, D.; Yang, S. Surrounding Media Sensitive Photoluminescence of Boron-Doped Graphene Quantum Dots for Highly Fluorescent Dyed Crystals, Chemical Sensing and Bioimaging. Carbon 2014, 70, 149–156. [Google Scholar] [CrossRef]
- Ling, C.; Mizuno, F. Boron-Doped Graphene as a Promising Anode for Na-Ion Batteries. Phys. Chem. Chem. Phys. 2014, 16, 10419–10424. [Google Scholar] [CrossRef]
- Zhang, Y.; Qin, J.; Batmunkh, M.; Zhong, Y.L. Facile Synthesis of Boron-Doped Reduced Electrochemical Graphene Oxide for Sodium Ion Battery Anode. JOM 2021, 73, 2531–2539. [Google Scholar] [CrossRef]
- Du, Y.; Zhang, B.; Kang, R.; Zhou, W.; Zhang, W.; Jin, H.; Wan, J.; Zhang, J.; Chen, G. Boron-Doping-Induced Defect Engineering Enables High Performance of a Graphene Cathode for Aluminum Batteries. Inorg. Chem. Front. 2022, 9, 925–934. [Google Scholar] [CrossRef]
- Diklić, N.P.; Dobrota, A.S.; Pašti, I.A.; Mentus, S.V.; Johansson, B.; Skorodumova, N.V. Sodium Storage via Single Epoxy Group on Graphene—The Role of Surface Doping. Electrochim. Acta 2019, 297, 523–528. [Google Scholar] [CrossRef]
- Dobrota, A.S.; Pašti, I.A.; Mentus, S.V.; Johansson, B.; Skorodumova, N.V. Functionalized Graphene for Sodium Battery Applications: The DFT Insights. Electrochim. Acta 2017, 250, 185–195. [Google Scholar] [CrossRef]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. QUANTUM ESPRESSO: A Modular and Open-Source Software Project for Quantum Simulations of Materials. J. Phys. Condens. Matter 2009, 21, 395502. [Google Scholar] [CrossRef] [PubMed]
- Giannozzi, P.; Andreussi, O.; Brumme, T.; Bunau, O.; Buongiorno Nardelli, M.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Cococcioni, M.; et al. Advanced Capabilities for Materials Modelling with Quantum ESPRESSO. J. Phys. Condens. Matter 2017, 29, 465901. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S. Semiempirical GGA-Type Density Functional Constructed with a Long-Range Dispersion Correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Monkhorst, H.J.; Pack, J.D. Special Points for Brillouin-Zone Integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Momma, K.; Izumi, F. VESTA 3 for Three-Dimensional Visualization of Crystal, Volumetric and Morphology Data. J. Appl. Crystallogr. 2011, 44, 1272–1276. [Google Scholar] [CrossRef]
- Dobrota, A.S.; Pašti, I.A.; Skorodumova, N.V. Oxidized Graphene as an Electrode Material for Rechargeable Metal-Ion Batteries—A DFT Point of View. Electrochim. Acta 2015, 176, 1092–1099. [Google Scholar] [CrossRef]
- Dobrota, A.S.; Gutić, S.; Kalijadis, A.; Baljozović, M.; Mentus, S.V.; Skorodumova, N.V.; Pašti, I.A. Stabilization of Alkali Metal Ions Interaction with OH-Functionalized Graphene: Via Clustering of OH Groups-Implications in Charge Storage Applications. RSC Adv. 2016, 6, 57910–57919. [Google Scholar] [CrossRef]
- Cuxart, M.G.; Perilli, D.; Tömekce, S.; Deyerling, J.; Haag, F.; Muntwiler, M.; Allegretti, F.; Di Valentin, C.; Auwärter, W. Spatial Segregation of Substitutional B Atoms in Graphene Patterned by the Moiré Superlattice on Ir(111). Carbon 2023, 201, 881–890. [Google Scholar] [CrossRef]
- Pašti, I.A.; Jovanović, A.; Dobrota, A.S.; Mentus, S.V.; Johansson, B.; Skorodumova, N.V. Atomic Adsorption on Graphene with a Single Vacancy: Systematic DFT Study through the Periodic Table of Elements. Phys. Chem. Chem. Phys. 2018, 20, 858–865. [Google Scholar] [CrossRef] [PubMed]
- Miwa, R.H.; Martins, T.B.; Fazzio, A. Hydrogen Adsorption on Boron Doped Graphene: An Ab Initio Study. Nanotechnology 2008, 19, 155708. [Google Scholar] [CrossRef] [PubMed]
- Sathe, B.R.; Zou, X.; Asefa, T. Metal-Free B-Doped Graphene with Efficient Electrocatalytic Activity for Hydrogen Evolution Reaction. Catal. Sci. Technol. 2014, 4, 2023–2030. [Google Scholar] [CrossRef]
- Dobrota, A.S.; Pašti, I.A.; Mentus, S.V.; Johansson, B.; Skorodumova, N.V. Altering the Reactivity of Pristine, N- and P-Doped Graphene by Strain Engineering: A DFT View on Energy Related Aspects. Appl. Surf. Sci. 2020, 514, 145937. [Google Scholar] [CrossRef]
- Oh, J.S.; Kim, K.N.; Yeom, G.Y. Graphene Doping Methods and Device Applications. J. Nanosci. Nanotechnol. 2014, 14, 1120–1133. [Google Scholar] [CrossRef]
- Kittel, C. Introduction to Solid State Physics, 8th ed.; Wiley & Sons: Hoboken, NJ, USA, 2004. [Google Scholar]
- Kaxiras, E. Atomic and Electronic Structure of Solids; Cambridge University Press: Cambridge, UK, 2010. [Google Scholar]
- Yao, L.H.; Cao, M.S.; Yang, H.J.; Liu, X.J.; Fang, X.Y.; Yuan, J. Adsorption of Na on Intrinsic, B-Doped, N-Doped and Vacancy Graphenes: A First-Principles Study. Comput. Mater. Sci. 2014, 85, 179–185. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | Eb(B)/eV | d(C-B)/Å | ∆q(B)/e |
|---|---|---|---|
| C53B | –12.90 | 1.49 | −0.1 |
| C52B2 | –12.46 | 1.51 | −0.2 |
| C51B3 | –12.15 | 1.51 | −0.2 |
| m(Al) | Eads(mth Al@subs)/eV | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| C53B | C52B2 | C51B3 | O@ C53B | O@ C52B2 | O@ C51B3 | OH@ C53B | OH@ C52B2 | OH@ C51B3 | |
| 1 | −2.71 | −3.11 | −2.92 | −3.93 | −3.75 | −2.72 | −3.80 | −3.69 | −4.05 |
| 2 | - | - | - | - | −3.70 | - | - | - | −2.81 |
| Subs | 2Na | 3Na | 4Na | |||
|---|---|---|---|---|---|---|
| Eads,int /eV | E0(Na+/Na@subs) /V | Eads,int /eV | E0(Na+/Na@subs) /V | Eads,int /eV | E0(Na+/Na@subs) /V | |
| C53B | –1.50 | –2.34 | * | * | * | * |
| C52B2 | –2.13 | –1.71 | * | * | * | * |
| C51B3 | –2.19 | –1.65 | –1.94 | –1.90 | –1.78 | –2.06 |
| O@C53B | –1.79 | –2.05 | –2.00 | –1.84 | –1.82 | –2.02 |
| O@C52B2 | –2.10 | –1.74 | –1.84 | –2.00 | –2.04 | –1.80 |
| O@C51B3 | –2.25 | –1.59 | –2.05 | –1.79 | * | * |
| OH@C52B2 | –2.35 | –1.49 | * | * | * | * |
| OH@C51B3 | –2.77 | –1.07 | –2.41 | –1.43 | * | * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ritopečki, M.S.; Skorodumova, N.V.; Dobrota, A.S.; Pašti, I.A. Density Functional Theory Analysis of the Impact of Boron Concentration and Surface Oxidation in Boron-Doped Graphene for Sodium and Aluminum Storage. C 2023, 9, 92. https://doi.org/10.3390/c9040092
Ritopečki MS, Skorodumova NV, Dobrota AS, Pašti IA. Density Functional Theory Analysis of the Impact of Boron Concentration and Surface Oxidation in Boron-Doped Graphene for Sodium and Aluminum Storage. C. 2023; 9(4):92. https://doi.org/10.3390/c9040092
Chicago/Turabian StyleRitopečki, Milica S., Natalia V. Skorodumova, Ana S. Dobrota, and Igor A. Pašti. 2023. "Density Functional Theory Analysis of the Impact of Boron Concentration and Surface Oxidation in Boron-Doped Graphene for Sodium and Aluminum Storage" C 9, no. 4: 92. https://doi.org/10.3390/c9040092
APA StyleRitopečki, M. S., Skorodumova, N. V., Dobrota, A. S., & Pašti, I. A. (2023). Density Functional Theory Analysis of the Impact of Boron Concentration and Surface Oxidation in Boron-Doped Graphene for Sodium and Aluminum Storage. C, 9(4), 92. https://doi.org/10.3390/c9040092