

Electrocatalytic Enhancement of CO Methanation at the Metal–Electrolyte Interface Studied Using In Situ X-ray Photoelectron Spectroscopy
 , , , , , , and
, , , , , , and 
Abstract
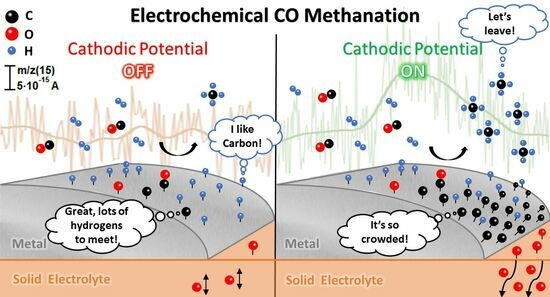
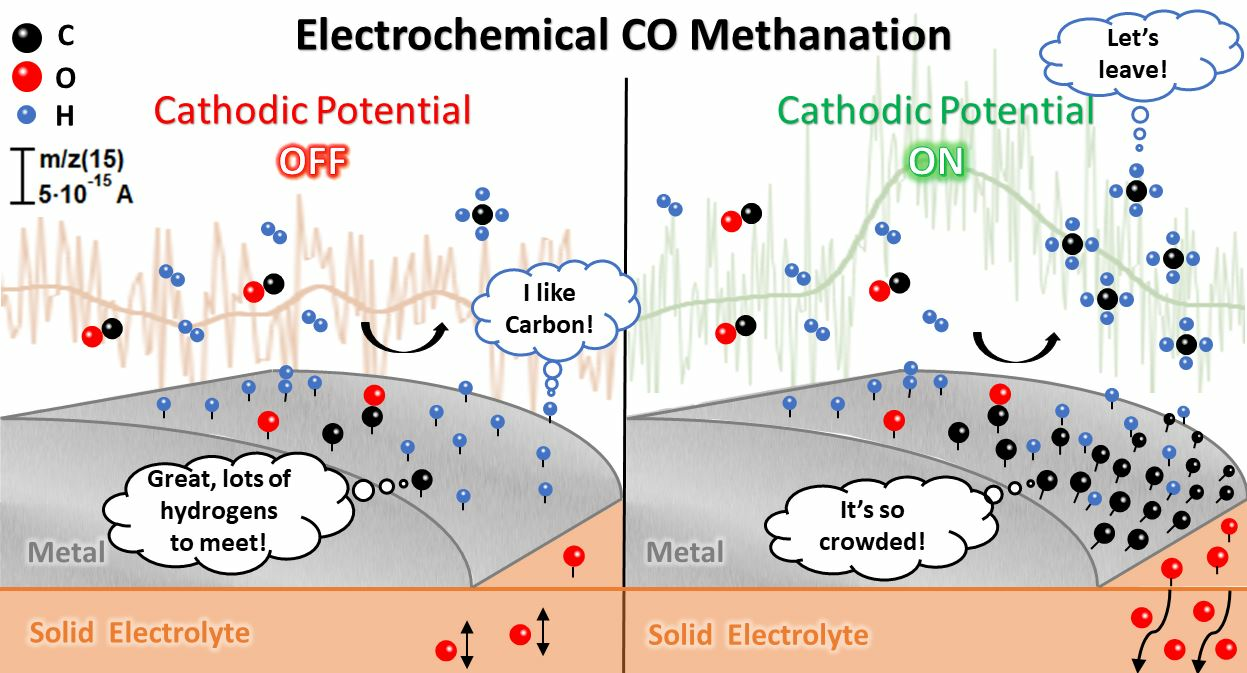
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Experimental Methods
3. Results and Discussion
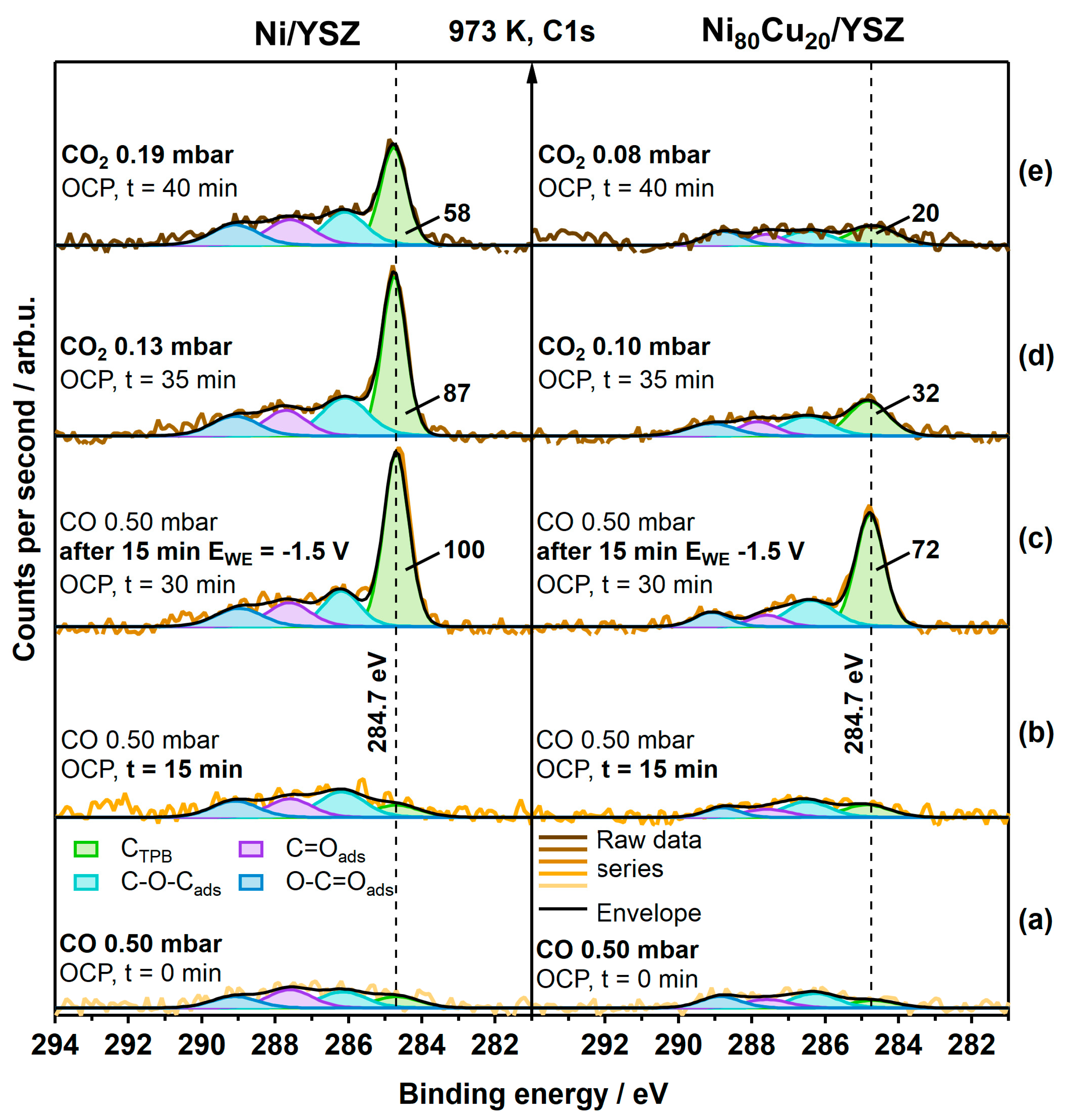
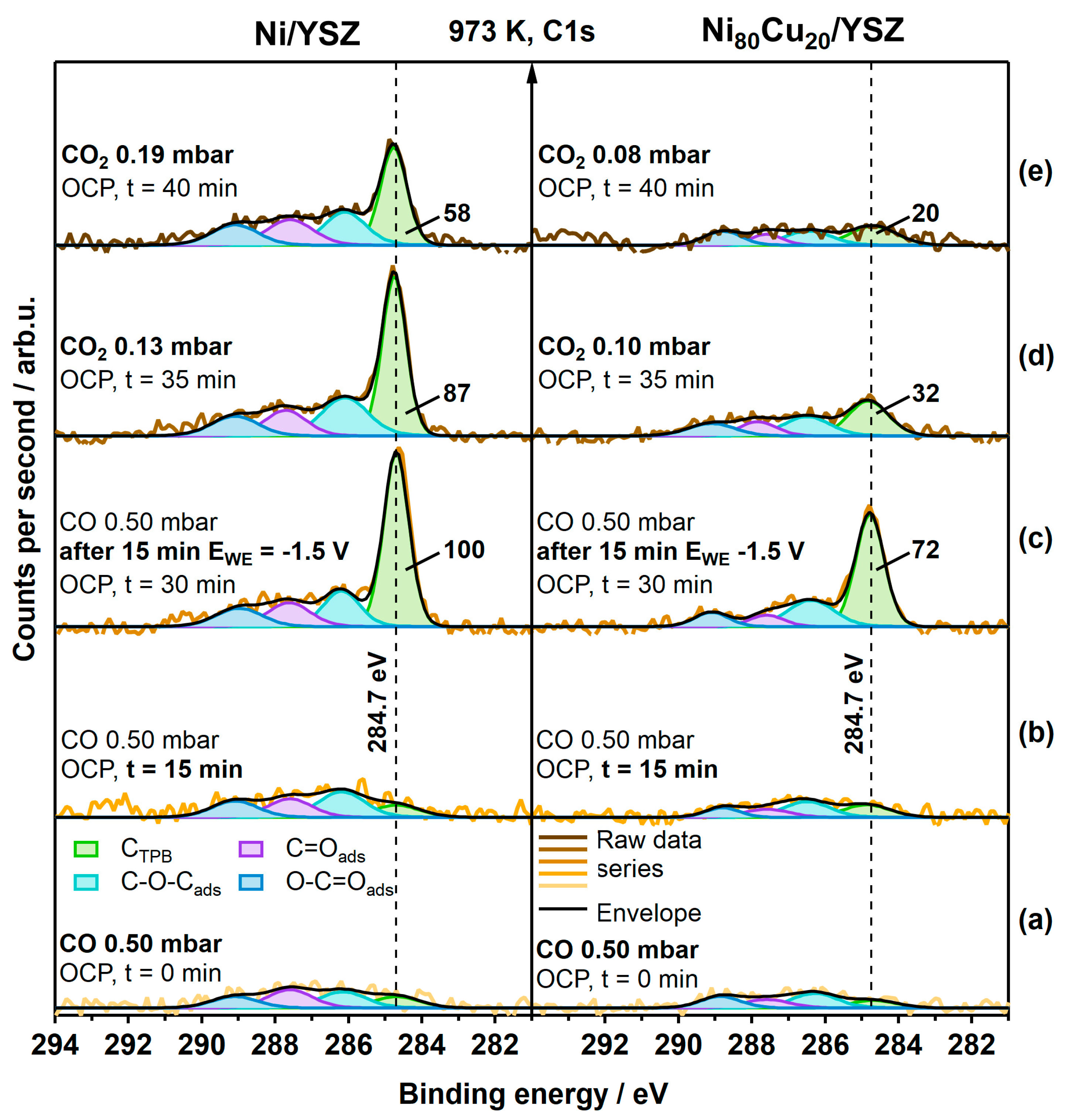
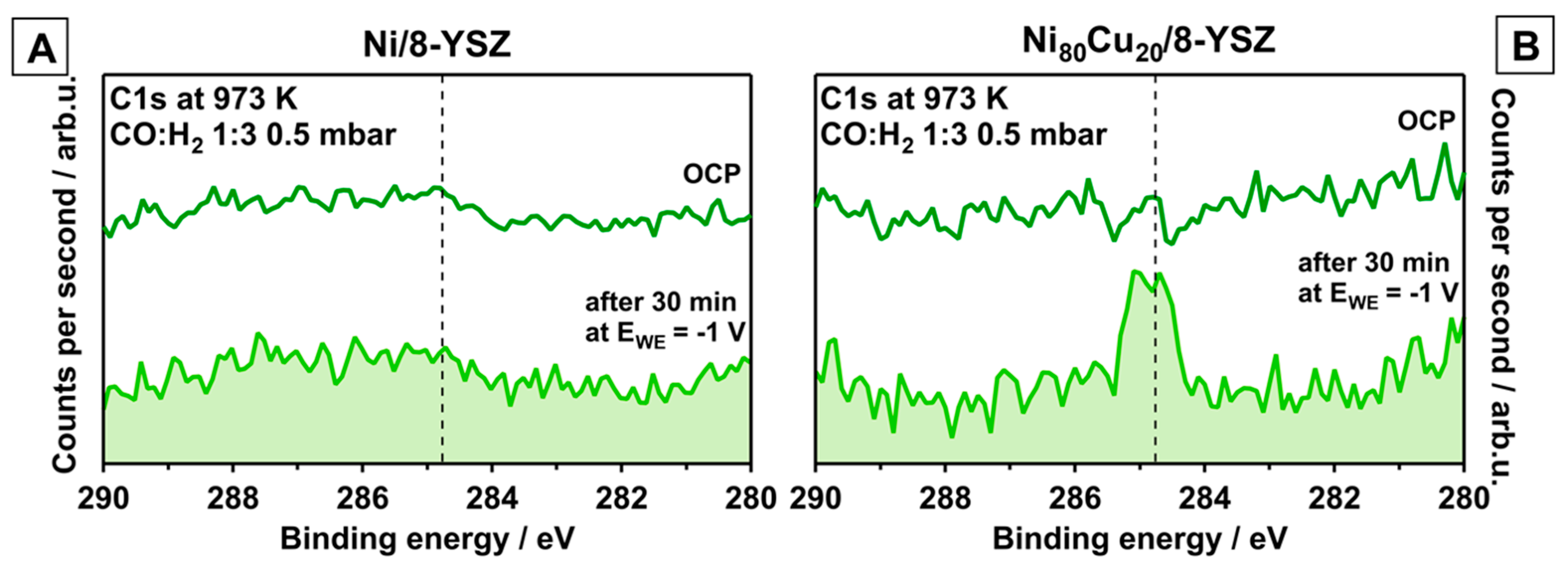
3.1. Carbon-Active Electrodes
- (a)
- Clean-off reaction with Hads toward CH4, which is a function of the available active hydrogen species;
- (b)
- Dissolution of C atoms in the (bi)metal bulk;
- (c)
- Accumulation of graphitic-like carbon.
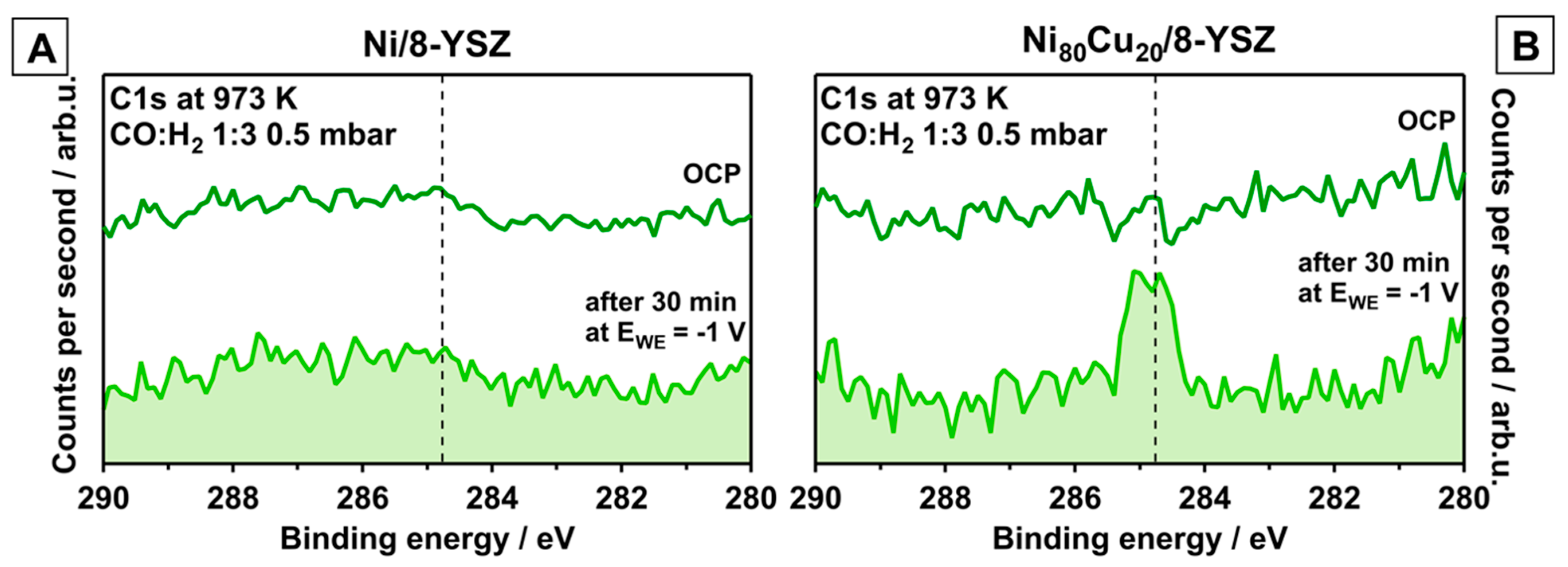
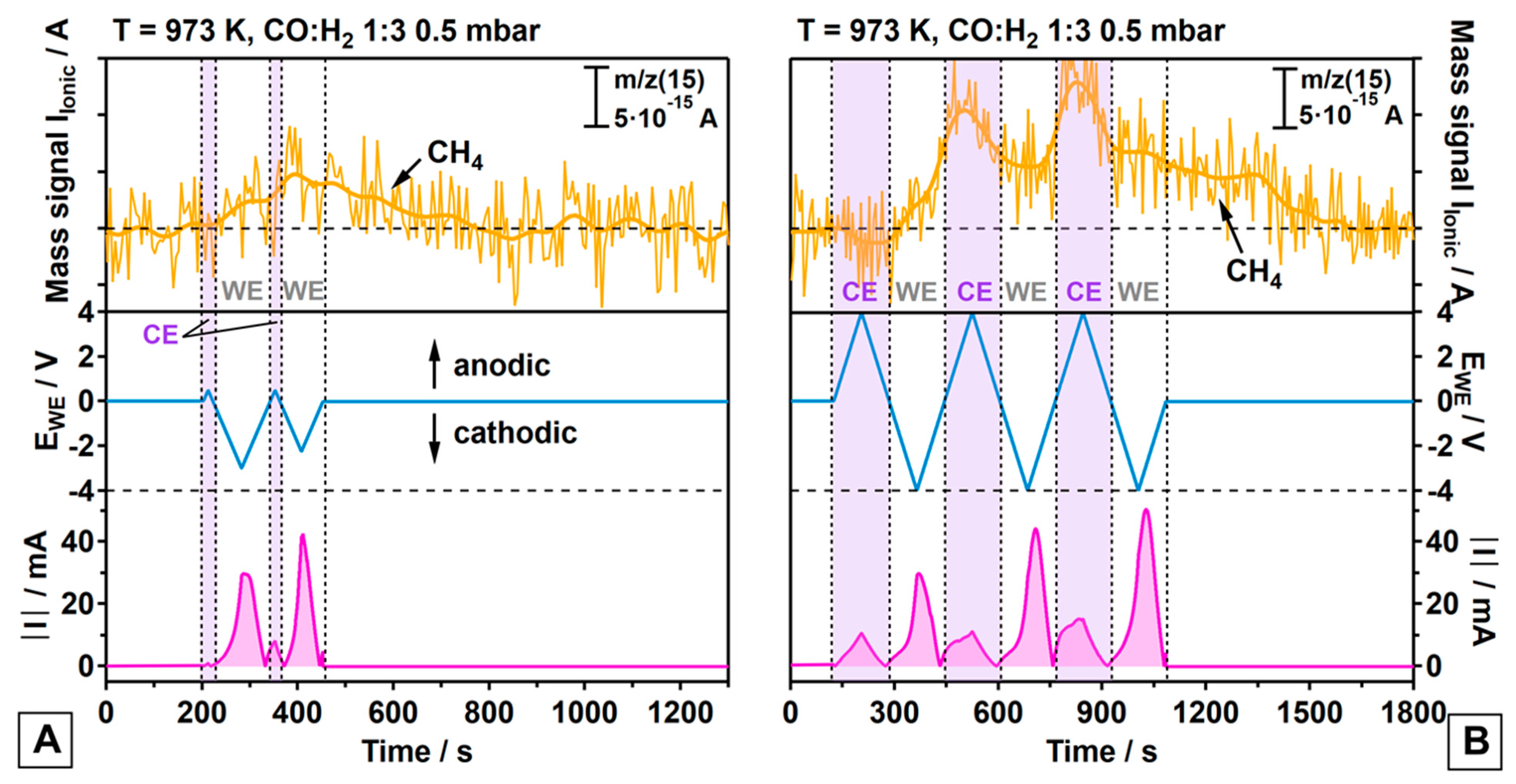
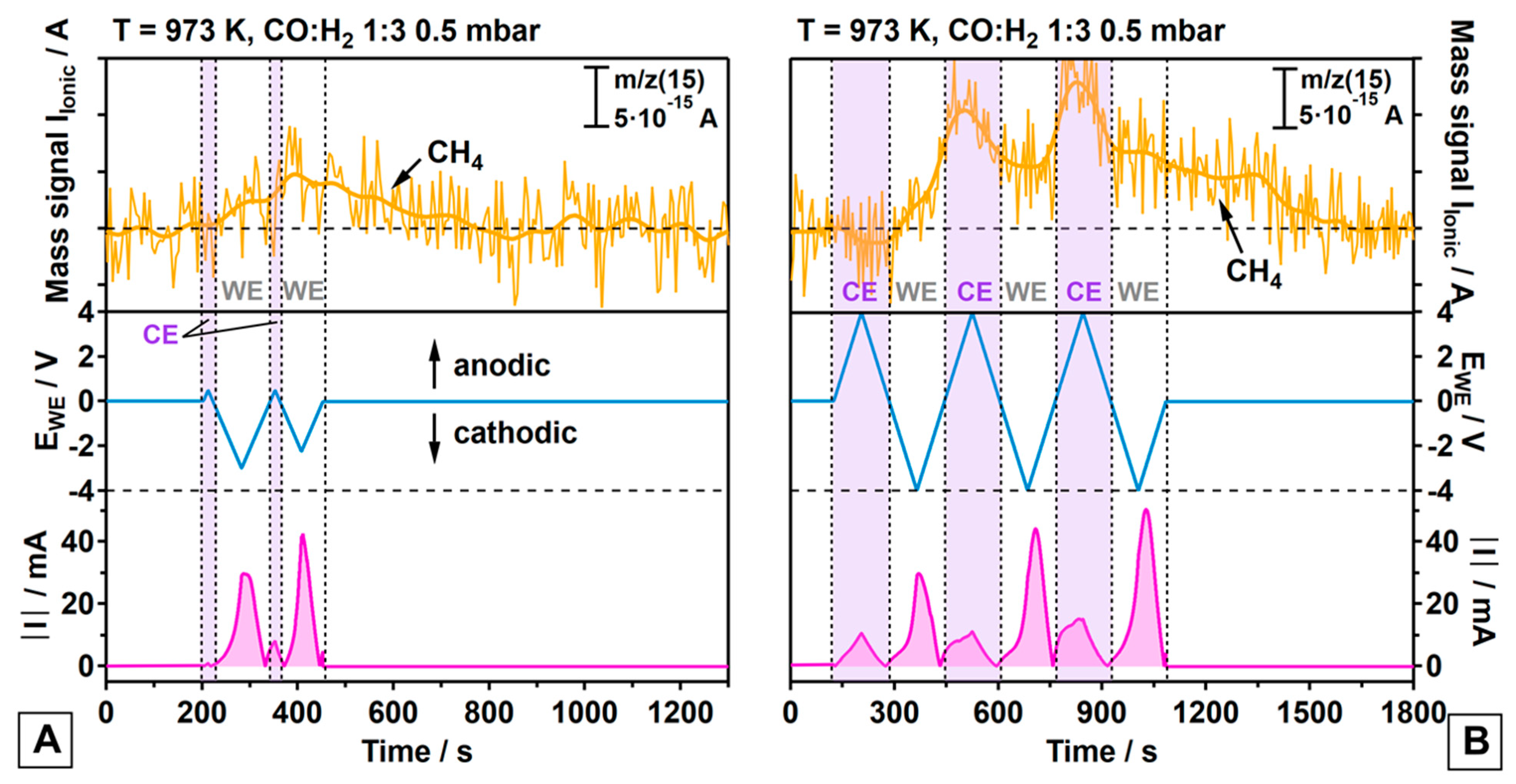
3.2. Electrochemically Promoted Methane Formation
- (a)
- On both the WE and the CE, the methanation reaction is promoted via cathodic polarization triggering reaction (3);
- (b)
- Ongoing reaction (6) suggests the contribution of a faradaic enhancement in activity on both electrodes via an increase in the surface carbon activity (a(C), compare equation (7));
- (c)
- Methane formation is much more promoted on the Pt/GDC-10 CE than on the WE, irrespective of Ni or Ni80Cu20 being used as the metallic WE phase;
- (d)
- Methane promotion at the CE is associated with a much smaller charge transfer for the CO electrolysis reaction (3) as compared to the WE, suggesting a relatively higher efficiency in the hydrogenation of surface C;
- (e)
- The first cathodic polarization cycle of the CE yields no measurable CH4 formation but is expected to activate the CE for CH4 formation for the second cycle. It appears likely that the oxygen-ion buffering GDC requires sufficient oxygen-ion withdrawal in a first cycle to activate the TPB in terms of generating a proper oxygen vacancy concentration;
- (f)
- Once the CE is activated, the time-response of formation of additional CH4 from CTPB is much faster on the CE, as deduced from the synchronicity of cathodic CE polarization and the accelerated CH4 increase;
- (g)
- CH4 formation on the WE starts immediately at the beginning of the first cathodic polarization cycle, but then increases rather slowly and is associated with a smaller amount of CH4 yielded from CTPB, especially in view of the much larger integral charge transfer. The response of CH4 formation to the alternating potential is very sluggish on the WE as compared to the CE.
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zheng, Y.; Wang, J.; Yu, B.; Zhang, W.; Chen, J.; Qiao, J.; Zhang, J. A review of high temperature co-electrolysis of H2O and CO2 to produce sustainable fuels using solid oxide electrolysis cells (SOECs): Advanced materials and technology. Chem. Soc. Rev. 2017, 46, 1427–1463. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Wang, H.; Shi, Y.; Cai, N. Performance and methane production characteristics of H2O–CO2 co-electrolysis in solid oxide electrolysis cells. Int. J. Hydrogen Energy 2013, 38, 11104–11109. [Google Scholar] [CrossRef]
- Gao, J.; Wang, Y.; Ping, Y.; Hu, D.; Xu, G.; Gu, F.; Su, F. A thermodynamic analysis of methanation reactions of carbon oxides for the production of synthetic natural gas. RSC Adv. 2012, 2, 2358–2368. [Google Scholar] [CrossRef]
- Vayenas, C.G.; Bebelis, S.; Neophytides, S. Non-Faradaic electrochemical modification of catalytic activity. J. Phys. Chem. 1988, 92, 5083–5085. [Google Scholar] [CrossRef]
- Katsaounis, A. Recent developments and trends in the electrochemical promotion of catalysis (EPOC). J. Appl. Electrochem. 2010, 40, 885–902. [Google Scholar] [CrossRef]
- Imbihl, R. Electrochemical promotion of catalytic reactions. Prog. Surf. Sci. 2010, 85, 241–278. [Google Scholar] [CrossRef]
- Bebelis, S.; Karasali, H.; Vayenas, C.G. Electrochemical promotion of CO2 hydrogenation on Rh/YSZ electrodes. J. Appl. Electrochem. 2008, 38, 1127–1133. [Google Scholar] [CrossRef]
- Giehr, A.; Maier, L.; Schunk, S.A.; Deutschmann, O. Thermodynamic Considerations on the Oxidation State of Co/γ-Al2O3 and Ni/γ-Al2O3 Catalysts under Dry and Steam Reforming Conditions. ChemCatChem 2018, 10, 751–757. [Google Scholar] [CrossRef]
- Pakhare, D.; Spivey, J. A review of dry (CO2) reforming of methane over noble metal catalysts. Chem. Soc. Rev. 2014, 43, 7813–7837. [Google Scholar] [CrossRef]
- Degerman, D.; Lömker, P.; Goodwin, C.M.; Shipilin, M.; García-Martínez, F.; Schlueter, C.; Nilsson, A.; Amann, P. State of the Surface During CO Hydrogenation over Ni(111) and Ni(211) Probed by Operando X-ray Photoelectron Spectroscopy. J. Phys. Chem. C 2023, 127, 4021–4032. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, Y.; Lin, Y.; Yang, Z.; Su, D.; Han, M.; Chen, F. Direct-methane solid oxide fuel cells with hierarchically porous Ni-based anode deposited with nanocatalyst layer. Nano Energy 2014, 10, 1–9. [Google Scholar] [CrossRef]
- Qu, J.; Wang, W.; Chen, Y.; Deng, X.; Shao, Z. Stable direct-methane solid oxide fuel cells with calcium-oxide-modified nickel-based anodes operating at reduced temperatures. Appl. Energy 2016, 164, 563–571. [Google Scholar] [CrossRef]
- An, W.; Zeng, X.C.; Turner, C.H. First-principles study of methane dehydrogenation on a bimetallic Cu/Ni(111) surface. J. Chem. Phys. 2009, 131, 174702. [Google Scholar] [CrossRef] [PubMed]
- Song, K.; Lu, M.; Xu, S.; Chen, C.; Zhan, Y.; Li, D.; Au, C.; Jiang, L.; Tomishige, K. Effect of alloy composition on catalytic performance and coke-resistance property of Ni-Cu/Mg(Al)O catalysts for dry reforming of methane. Appl. Catal. B 2018, 239, 324–333. [Google Scholar] [CrossRef]
- An, W.; Gatewood, D.; Dunlap, B.; Turner, C.H. Catalytic activity of bimetallic nickel alloys for solid-oxide fuel cell anode reactions from density-functional theory. J. Power Sources 2011, 196, 4724–4728. [Google Scholar] [CrossRef]
- Zambaldi, P.; Haug, L.; Penner, S.; Klötzer, B. Dry Reforming of Methane on NiCu and NiPd Model Systems: Optimization of Carbon Chemistry. Catalysts 2022, 12, 311. [Google Scholar] [CrossRef]
- Lee, G.-J.; Lee, J.-H.; Lee, D.; Park, K.-I.; Jeong, C.K.; Park, J.-J.; Lee, M.-K. Synthesis and characterization of carbon-coated Cu-Ni alloy nanoparticles and their application in conductive films. Appl. Surf. Sci. 2021, 566, 150672. [Google Scholar] [CrossRef]
- Luyten, L.J.M.; Von Eck, M.; Von Grondelle, J.; Von Hooff, J.H.C. Hydrogenation of carbon monoxide over silica supported nickel-copper and ruthenium-copper catalysts. J. Phys. Chem. 1978, 82, 2000–2002. [Google Scholar] [CrossRef]
- Hatta, A.H.; Jalil, A.A.; Hassan, N.S.; Hamid, M.Y.S.; Rahman, A.F.A.; Teh, L.P.; Prasetyoko, D. A review on recent bimetallic catalyst development for synthetic natural gas production via CO methanation. Int. J. Hydrogen Energy 2022, 47, 30981–31002. [Google Scholar] [CrossRef]
- Tsiotsias, A.I.; Charisiou, N.D.; Yentekakis, I.V.; Goula, M.A. Bimetallic Ni-Based Catalysts for CO2 Methanation: A Review. Nanomaterials 2021, 11, 28. [Google Scholar] [CrossRef]
- Schmider, D.; Maier, L.; Deutschmann, O. Reaction Kinetics of CO and CO2 Methanation over Nickel. Ind. Eng. Chem. Res. 2021, 60, 5792–5805. [Google Scholar] [CrossRef]
- Wensheng, X.; Haiyou, W.; Huilin, W.; Qianer, Z. An energetics study on syngas(CO+H2) methanation reaction on Ni, Cu and Ni-Cu alloy surfaces by bond-order conservation model. Acta Chim. Sinica 1998, 56, 773–779. [Google Scholar]
- Luo, Y.; Li, W.; Shi, Y.; Ye, X.; Wang, S.; Cai, N. Methane Synthesis Characteristics of H2O/CO2 Co-Electrolysis in Tubular Solid Oxide Electrolysis Cells. ECS Trans. 2015, 68, 3465. [Google Scholar] [CrossRef]
- Fleig, J.; Baumann, F.S.; Brichzin, V.; Kim, H.R.; Jamnik, J.; Cristiani, G.; Habermeier, H.U.; Maier, J. Thin Film Microelectrodes in SOFC Electrode Research. Fuel Cells 2006, 6, 284–292. [Google Scholar] [CrossRef]
- Vannice, M.A. The catalytic synthesis of hydrocarbons from H2CO mixtures over the group VIII metals: II. The kinetics of the methanation reaction over supported metals. J. Catal. 1975, 37, 462–473. [Google Scholar] [CrossRef]
- Barbieri, P.F.; de Siervo, A.; Carazzolle, M.F.; Landers, R.; Kleiman, G.G. XPS and XAES study of Ag–Pd and Cu–Ni alloys: Spectra, shifts and electronic structure information. J. Electron. Spectrosc. Relat. Phenom. 2004, 135, 113–118. [Google Scholar] [CrossRef]
- Biesinger, M.C. Advanced analysis of copper X-ray photoelectron spectra. Surf. Interface Anal. 2017, 49, 1325–1334. [Google Scholar] [CrossRef]
- Biesinger, M.C. Accessing the robustness of adventitious carbon for charge referencing (correction) purposes in XPS analysis: Insights from a multi-user facility data review. Appl. Surf. Sci. 2022, 597, 153681. [Google Scholar] [CrossRef]
- Zegkinoglou, I.; Pielsticker, L.; Han, Z.-K.; Divins, N.J.; Kordus, D.; Chen, Y.-T.; Escudero, C.; Pérez-Dieste, V.; Zhu, B.; Gao, Y.; et al. Surface Segregation in CuNi Nanoparticle Catalysts During CO2 Hydrogenation: The Role of CO in the Reactant Mixture. J. Phys. Chem. C 2019, 123, 8421–8428. [Google Scholar] [CrossRef]
- Linstrom, P. NIST Chemistry WebBook, NIST Standard Reference Database 69; National Institute of Standards and Testing (NIST): Gaithersburg, MD, USA, 1997. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thurner, C.W.; Haug, L.; Winkler, D.; Griesser, C.; Leitner, M.; Moser, T.; Werner, D.; Thaler, M.; Scheibel, L.A.; Götsch, T.; et al. Electrocatalytic Enhancement of CO Methanation at the Metal–Electrolyte Interface Studied Using In Situ X-ray Photoelectron Spectroscopy. C 2023, 9, 106. https://doi.org/10.3390/c9040106
Thurner CW, Haug L, Winkler D, Griesser C, Leitner M, Moser T, Werner D, Thaler M, Scheibel LA, Götsch T, et al. Electrocatalytic Enhancement of CO Methanation at the Metal–Electrolyte Interface Studied Using In Situ X-ray Photoelectron Spectroscopy. C. 2023; 9(4):106. https://doi.org/10.3390/c9040106
Chicago/Turabian StyleThurner, Christoph W., Leander Haug, Daniel Winkler, Christoph Griesser, Matthias Leitner, Toni Moser, Daniel Werner, Marco Thaler, Lucas A. Scheibel, Thomas Götsch, and et al. 2023. "Electrocatalytic Enhancement of CO Methanation at the Metal–Electrolyte Interface Studied Using In Situ X-ray Photoelectron Spectroscopy" C 9, no. 4: 106. https://doi.org/10.3390/c9040106
APA StyleThurner, C. W., Haug, L., Winkler, D., Griesser, C., Leitner, M., Moser, T., Werner, D., Thaler, M., Scheibel, L. A., Götsch, T., Carbonio, E., Kunze-Liebhäuser, J., Portenkirchner, E., Penner, S., & Klötzer, B. (2023). Electrocatalytic Enhancement of CO Methanation at the Metal–Electrolyte Interface Studied Using In Situ X-ray Photoelectron Spectroscopy. C, 9(4), 106. https://doi.org/10.3390/c9040106