Modifying Electronic and Elastic Properties of 2-Dimensional [110] Diamond by Nitrogen Substitution

 , ,
, ,  and
and 
Abstract
1. Introduction
2. Computational Method
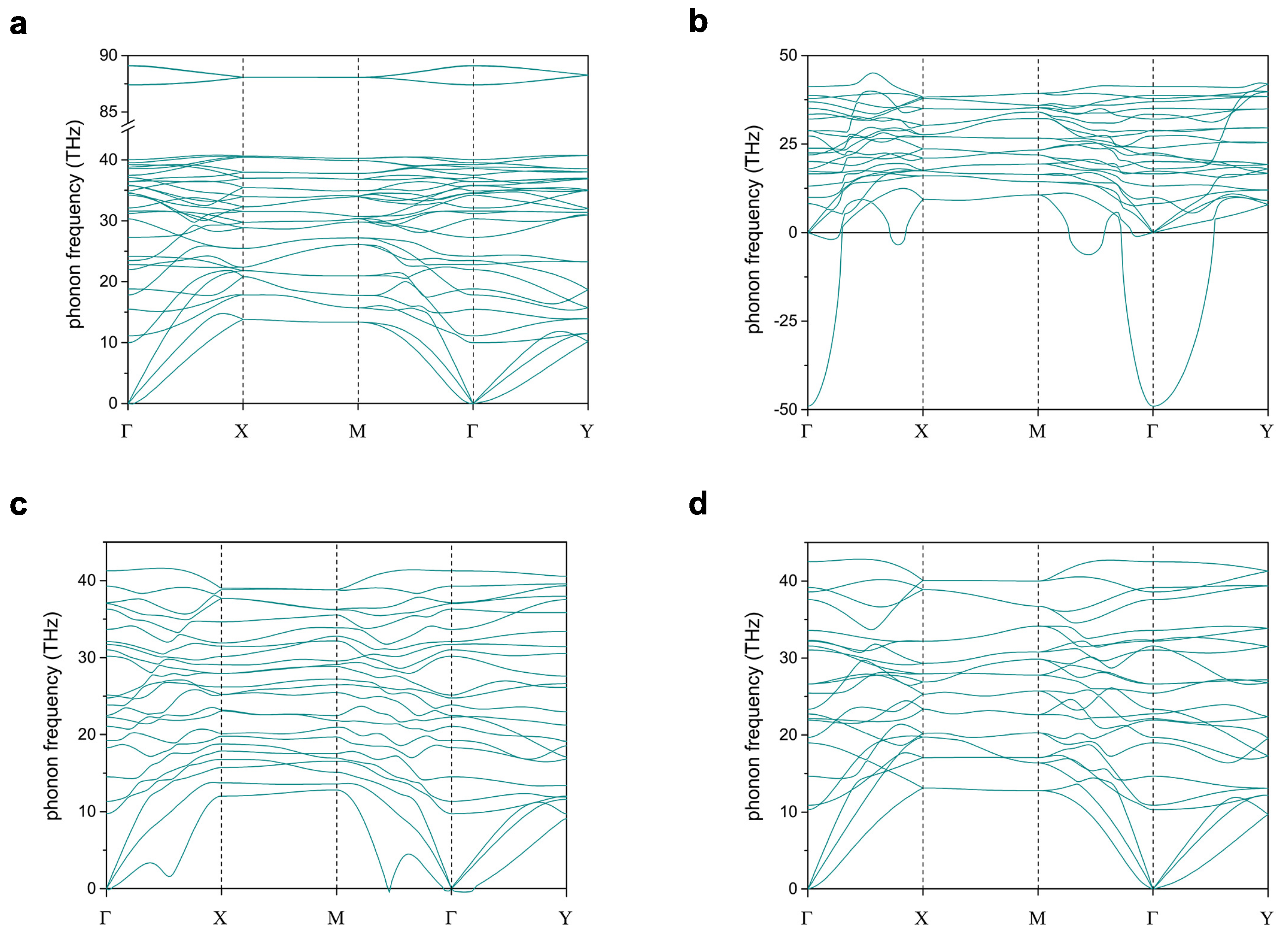
3. Results
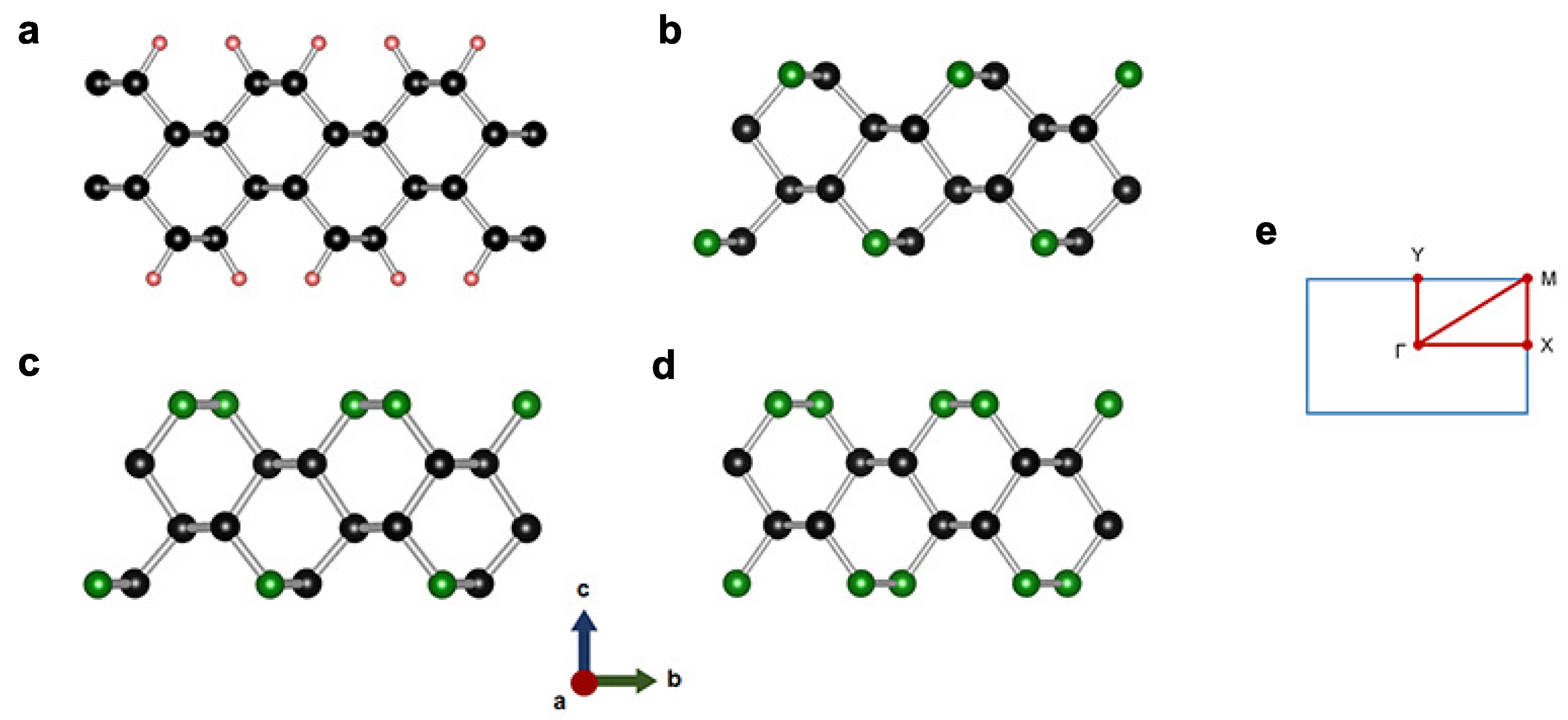
3.1. 2-Dimensional Structures
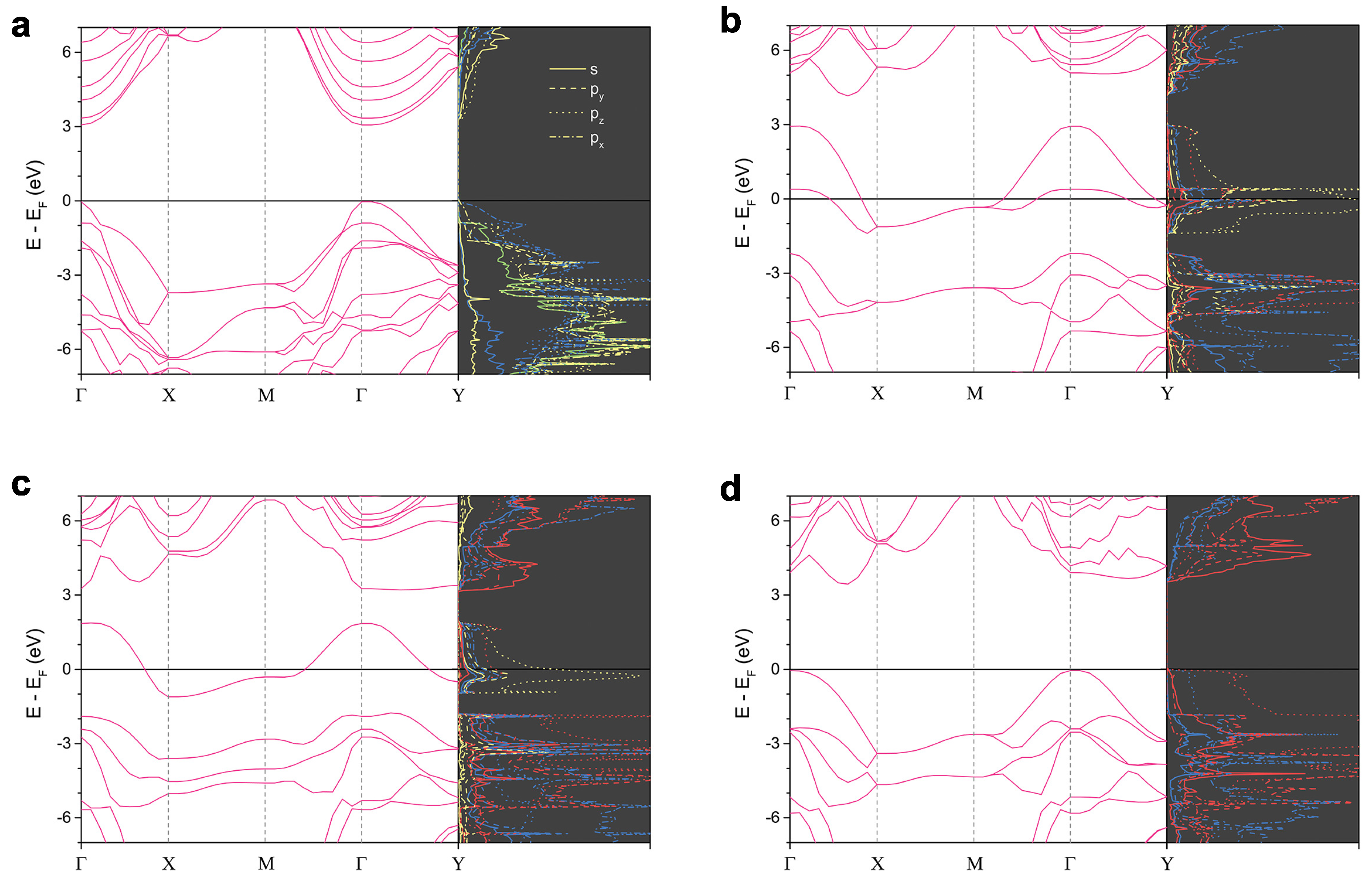
3.2. Electronic Property and Bonding
3.3. Elastic Constants
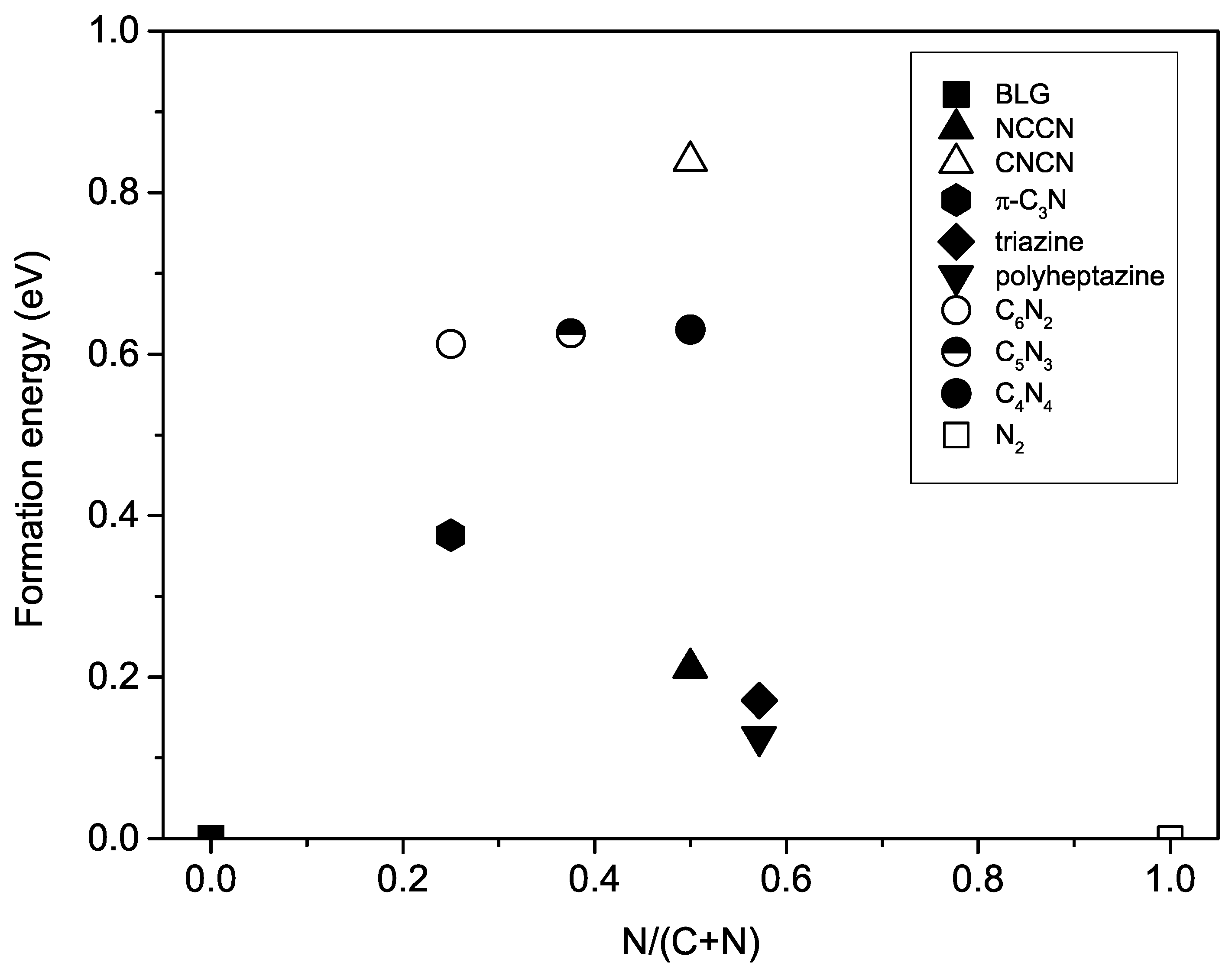
3.4. Formation Energy
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 2D | 2-dimensional |
| PBE | Perdew-Burke-Ernzerhof |
| HSE06 | Heyd-Scuseria-Ernzehof (2006) |
| DOS | density of states |
References
- Lee, C.; Wei, X.; Kysar, J.W.; Hone, J. Measurement of the Elastic Properties and Intrinsic Strength of Monolayer Graphene. Science 2008, 321, 385–388. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.Q.; Ye, H.Q. Ab initio elastic constants for the lonsdaleite phases of C, Si and Ge. J. Phys. Condens. Matter 2003, 15, 5307. [Google Scholar] [CrossRef]
- Németh, P.; Garvie, L.A.J.; Aoki, T.; Dubrovinskaia, N.; Dubrovinsky, L.; Buseck, P.R. Lonsdaleite is faulted and twinned cubic diamond and does not exist as a discrete material. Nat. Commun. 2014, 5, 5447. [Google Scholar] [CrossRef]
- McCulloch, D.G.; Wong, S.; Shiell, T.B.; Haberl, B.; Cook, B.A.; Huang, X.; Boehler, R.; McKenzie, D.R.; Bradby, J.E. Investigation of Room Temperature Formation of the Ultra-Hard Nanocarbons Diamond and Lonsdaleite. Small 2020, 16, 2004695. [Google Scholar] [CrossRef]
- Barboza, A.P.M.; Guimaraes, M.H.D.; Massote, D.V.P.; Campos, L.C.; Neto, N.M.B.; Cancado, L.G.; Lacerda, R.G.; Chacham, H.; Mazzoni, M.S.C.; Neves, B.R.A. Room-Temperature Compression-Induced Diamondization of Few-Layer Graphene. Adv. Mater. 2011, 23, 3014–3017. [Google Scholar] [CrossRef]
- Martins, L.G.P.; Matos, M.J.S.; Paschoal, A.R.; Freire, P.T.C.; Andrade, N.F.; Aguiar, A.L.; Neves, B.R.A.; de Oliveira, A.B.; Mazzoni, M.S.C.; Filho, A.G.S.; et al. Raman evidence for pressure-induced formation of diamondene. Nat. Commun. 2017, 8, 96. [Google Scholar] [CrossRef]
- Leenaerts, O.; Partoens, B.; Peeters, F.M. Hydrogenation of bilayer graphene and the formation of bilayer graphane from first principles. Phys. Rev. B 2009, 80, 245422. [Google Scholar] [CrossRef]
- Samarakoon, D.K.; Wang, X.Q. Tunable Band Gap in Hydrogenated Bilayer Graphene. ACS Nano 2010, 4, 4126–4130. [Google Scholar] [CrossRef]
- Chernozatonskii, L.A.; Sorokin, P.B.; Kvashnin, A.G.; Kvashnin, D.G. Diamond-Like C2H Nanolayer, Diamane: Simulation of the Structure and Properties. JETP Lett. 2009, 90, 134–138. [Google Scholar] [CrossRef]
- Gao, Y.; Cao, T.; Cellini, F.; Berger, C.; de Heer, W.A.; Tosatti, E.; Riedo, E.; Bongiorno, A. Ultrahard carbon film from epitaxial two-layer graphene. Nat. Nanotechnol. 2018, 13, 133–138. [Google Scholar] [CrossRef]
- Pakornchote, T.; Ektarawong, A.; Alling, B.; Pinsook, U.; Tancharakorn, S.; Busayaporn, W.; Bovornratanaraks, T. Phase stabilities and vibrational analysis of hydrogenated diamondized bilayer graphenes: A first principles investigation. Carbon 2019, 146, 468–475. [Google Scholar] [CrossRef]
- Kvashnin, A.G.; Sorokin, P.B. Lonsdaleite Films with Nanometer Thickness. J. Phys. Chem. Lett. 2014, 5, 541–548. [Google Scholar] [CrossRef]
- Kvashnin, A.G.; Avramov, P.P.V.; Kvashnin, D.G.K.; Chernozatonskii, L.A.; Sorokin, P.B. Features of Electronic, Mechanical, and Electromechanical Properties of Fluorinated Diamond Films of Nanometer Thickness. J. Phys. Chem. C 2017, 121, 28484–28489. [Google Scholar] [CrossRef]
- Pimenta Martins, L.G.; Silva, D.L.; Smith, J.S.; Lu, A.Y.; Su, C.; Hempel, M.; Occhialini, C.; Ji, X.; Pablo, R.; Alencar, R.S.; et al. Hard, transparent, sp3-containing 2D phase formed from few-layer graphene under compression. Carbon 2021, 173, 744–757. [Google Scholar] [CrossRef]
- Zhu, L.; Li, W.; Ding, F. Giant thermal conductivity in diamane and the influence of horizontal reflection symmetry on phonon scattering. Nanoscale 2019, 11, 4248–4257. [Google Scholar] [CrossRef]
- Raeisi, M.; Mortazavi, B.; Podryabinkin, E.V.; Shojaei, F.; Zhuang, X.; Shapeev, A.V. High thermal conductivity in semiconducting Janus and non-Janus diamanes. Carbon 2020, 167, 51–61. [Google Scholar] [CrossRef]
- Piazza, F.; Gough, K.; Monthioux, M.; Puech, P.; Gerber, I.; Wiens, R.; Paredes, G.; Ozoria, C. Low temperature, pressureless sp2 to sp3 transformation of ultrathin, crystalline carbon films. Carbon 2019, 145, 10–22. [Google Scholar] [CrossRef]
- Piazza, F.; Monthioux, M.; Puech, P.; Gerber, I. Towards a better understanding of the structure of diamanoïds and diamanoïd/graphene hybrids. Carbon 2020, 156, 234–241. [Google Scholar] [CrossRef]
- Piazza, F.; Cruz, K.; Monthioux, M.; Puech, P.; Gerber, I. Raman evidence for the successful synthesis of diamane. Carbon 2020, 169, 129–133. [Google Scholar] [CrossRef]
- Bakharev, P.V.; Huang, M.; Saxena, M.; Lee, S.W.; Joo, S.H.; Park, S.O.; Dong, J.; Camacho-Mojica, D.C.; Jin, S.; Kwon, Y.; et al. Chemically induced transformation of chemical vapour deposition grown bilayer graphene into fluorinated single-layer diamond. Nat. Nanotechnol. 2020, 15, 59–66. [Google Scholar] [CrossRef]
- Pakornchote, T.; Ektarawong, A.; Busayaporn, W.; Pinsook, U.; Bovornratanaraks, T. Roles of nitrogen substitution and surface reconstruction in stabilizing nonpassivated single-layer diamond. Phys. Rev. B 2020, 102, 075418. [Google Scholar] [CrossRef]
- Pandey, K.C. New dimerized-chain model for the reconstruction of the diamond (111)-(2 × 1) surface. Phys. Rev. B 1982, 25, 4338. [Google Scholar] [CrossRef]
- Iarlori, S.; Galli, G.; Gygi, F.M.C.; Parrinello, M.; Tosatti, E. Reconstruction of the diamond (111) surface. Phys. Rev. Lett. 1992, 69, 2947–2950. [Google Scholar] [CrossRef]
- Pamuk, B.; Calandra, M. Exchange-driven dimerization, magnetism, and insulating state in diamond (111). Phys. Rev. B 2019, 99, 155303. [Google Scholar] [CrossRef]
- Artyukhov, V.I.; Chernozatonskii, L.A. Structure and Layer Interaction in Carbon Monofluoride and Graphane: A Comparative Computational Study. J. Phys. Chem. A 2010, 114, 5389–5396. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef]
- Krukau, A.V.; Vydrov, O.A.; Izmaylov, A.F.; Scuseria, G.E. Influence of the exchange screening parameter on the performance of screened hybrid functionals. J. Chem. Phys. 2006, 125, 224106. [Google Scholar] [CrossRef]
- Togo, A.; Tanaka, I. First principles phonon calculations in materials science. Scr. Mater. 2015, 108, 1–5. [Google Scholar] [CrossRef]
- Blöchl, P.E.; Jepsen, O.; Andersen, O.K. Improved tetrahedron method for Brillouin-zone integrations. Phys. Rev. B 1994, 49, 16223. [Google Scholar] [CrossRef] [PubMed]
- Sofo, J.O.; Usaj, G.; Cornaglia, P.S.; Suarez, A.M.; Hernández-Nieves, A.D.; Balseiro, C.A. Magnetic structure of hydrogen-induced defects on graphene. Phys. Rev. B 2012, 85, 115405. [Google Scholar] [CrossRef]
- Rudenko, A.N.; Keil, F.J.; Katsnelson, M.I.; Lichtenstein, A.I. Exchange interactions and frustrated magnetism in single-side hydrogenated and fluorinated graphene. Phys. Rev. B 2013, 88, 081405. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, S.Y.; Huang, H.; Li, W.T.; Qiao, J.B.; Wang, W.X.; Yin, L.J.; Bai, K.K.; Duan, W.; He, L. Scanning Tunneling Microscopy of the π Magnetism of a Single Carbon Vacancy in Graphene. Phys. Rev. Lett. 2016, 117, 166801. [Google Scholar] [CrossRef]
- Bu, S.; Yao, N.; Hunter, M.A.; Searles, D.J.; Yuan, Q. Design of two-dimensional carbon-nitride structures by tuning the nitrogen concentration. Npj Comput. Mater. 2020, 6, 128. [Google Scholar] [CrossRef]
- Bafekry, A.; Neek-Amal, M.; Peeters, F.M. Two-dimensional graphitic carbon nitrides: Strain-tunable ferromagnetic ordering. Phys. Rev. B 2020, 101, 165407. [Google Scholar] [CrossRef]
- Wei, X.; Fragneaud, B.; Marianetti, C.A.; Kysar, J.W. Nonlinear elastic behavior of graphene: Ab initio calculations to continuum description. Phys. Rev. B 2009, 80, 205407. [Google Scholar] [CrossRef]
- Ding, Y.; Wang, Y. Density Functional Theory Study of the Silicene-like SiX and XSi3 (X = B, C, N, Al, P) Honeycomb Lattices: The Various Buckled Structures and Versatile Electronic Properties. J. Phys. Chem. C 2013, 117, 18266–18278. [Google Scholar] [CrossRef]
- Wei, Q.; Peng, X. Superior mechanical flexibility of phosphorene and few-layer black phosphorus. Appl. Phys. Lett. 2014, 104, 251915. [Google Scholar] [CrossRef]
- Chernozatonskii, L.A.; Sorokin, P.B.; Kuzubov, A.A.; Sorokin, B.P.; Kvashnin, A.G.; Kvashnin, D.G.; Avramov, P.V.; Yakobson, B.I. Influence of Size Effect on the Electronic and Elastic Properties of Diamond Films with Nanometer Thickness. J. Phys. Chem. C 2011, 115, 132–136. [Google Scholar] [CrossRef]
- Mahan, G.D. Condensed Matter in a Nutshell; Princeton University Press: Princeton, NJ, USA, 2011. [Google Scholar]
- Miller, T.S.; Jorge, A.B.; Suter, T.M.; Sella, A.; Corà, F.; McMillan, P.F. Carbon nitrides: Synthesis and characterization of a new class of functional materials. Phys. Chem. Chem. Phys. 2017, 19, 15613–15638. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phase | Space Group | Lattice Parameters | Atomic Species | Wyckoff Sites | Positions |
|---|---|---|---|---|---|
| CH | Å | C | 4 h | (0.000, 0.626, 0.027) | |
| Å | C | 4 h | (0.000, 0.367, 0.079) | ||
| Å | H | 4 h | (0.000, 0.519, 0.120) | ||
| Å | |||||
| CN | Å | C | 2 a | (0.000, 0.616, 0.538) | |
| Å | C | 2 a | (0.000, 0.359, 0.463) | ||
| Å | C | 2 a | (0.000, 0.333, 0.601) | ||
| Å | N | 2 a | (0.000, 0.628, 0.397) | ||
| CN | Å | C | (0.000, 0.126, 0.154) | ||
| Å | C | (0.500, 0.377, 0.155) | |||
| Å | C | (0.500, 0.624, 0.226) | |||
| C | (0.000, 0.881, 0.226) | ||||
| Å | C | (0.000, 0.851, 0.092) | |||
| N | (0.500, 0.636, 0.090) | ||||
| N | (0.500, 0.374, 0.293) | ||||
| N | (0.000, 0.190, 0.292) | ||||
| CN | Å | C | 4 h | (0.000, 0.378, 0.031) | |
| Å | N | 4 h | (0.000, 0.625, 0.089) | ||
| Å | |||||
| Å |
| Phases | |||||||
|---|---|---|---|---|---|---|---|
| CH | 577 | 41 | 22 | 384 | 8 | 451 | 210 |
| CN | 709 | 64 | 27 | 362 | 148 | 300 | 245 |
| CN | 666 | 73 | 26 | 392 | 120 | 335 | 256 |
| CN | 645 | 71 | 17 | 429 | 87 | 375 | 270 |
| NCCN [21] | 568 | 66 | 51 | 217 | 243 | ||
| CNCN [21] | 526 | 61 | 38 | 170 | 220 | ||
| -CN [21] | 595 | 106 | 27 | 510 | 16 | 159 | 244 |
| H-diamane [11] | 487 | 38 |
| Phases | ||||||||
|---|---|---|---|---|---|---|---|---|
| CH | 1036 | 73 | 40 | 688 | 15 | 810 | 378 | 310 |
| CN | 1963 | 177 | 76 | 1002 | 410 | 830 | 678 | 569 |
| CN | 1808 | 199 | 71 | 1062 | 3217 | 909 | 694 | 553 |
| CN | 1715 | 190 | 56 | 1140 | 226 | 996 | 719 | 533 |
| NCCN [21] | 2191 | 253 | 198 | 836 | 939 | 718 | ||
| CNCN [21] | 1968 | 227 | 142 | 635 | 825 | 618 | ||
| -CN [21] | 2030 | 361 | 91 | 1739 | 199 | 541 | 831 | 624 |
| H-diamane [21] | 1026 | 81 |
| Phases | ||||||
|---|---|---|---|---|---|---|
| CH | 102 | 370 | 1003 | 39 | 394 | 692 |
| CN | 147 | 725 | 1666 | 60 | 716 | 1097 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pakornchote, T.; Ektarawong, A.; Pinsook, U.; Bovornratanaraks, T. Modifying Electronic and Elastic Properties of 2-Dimensional [110] Diamond by Nitrogen Substitution. C 2021, 7, 8. https://doi.org/10.3390/c7010008
Pakornchote T, Ektarawong A, Pinsook U, Bovornratanaraks T. Modifying Electronic and Elastic Properties of 2-Dimensional [110] Diamond by Nitrogen Substitution. C. 2021; 7(1):8. https://doi.org/10.3390/c7010008
Chicago/Turabian StylePakornchote, Teerachote, Annop Ektarawong, Udomsilp Pinsook, and Thiti Bovornratanaraks. 2021. "Modifying Electronic and Elastic Properties of 2-Dimensional [110] Diamond by Nitrogen Substitution" C 7, no. 1: 8. https://doi.org/10.3390/c7010008
APA StylePakornchote, T., Ektarawong, A., Pinsook, U., & Bovornratanaraks, T. (2021). Modifying Electronic and Elastic Properties of 2-Dimensional [110] Diamond by Nitrogen Substitution. C, 7(1), 8. https://doi.org/10.3390/c7010008