Boehm Titration Revisited (Part I): Practical Aspects for Achieving a High Precision in Quantifying Oxygen-Containing Surface Groups on Carbon Materials


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials and Solutions
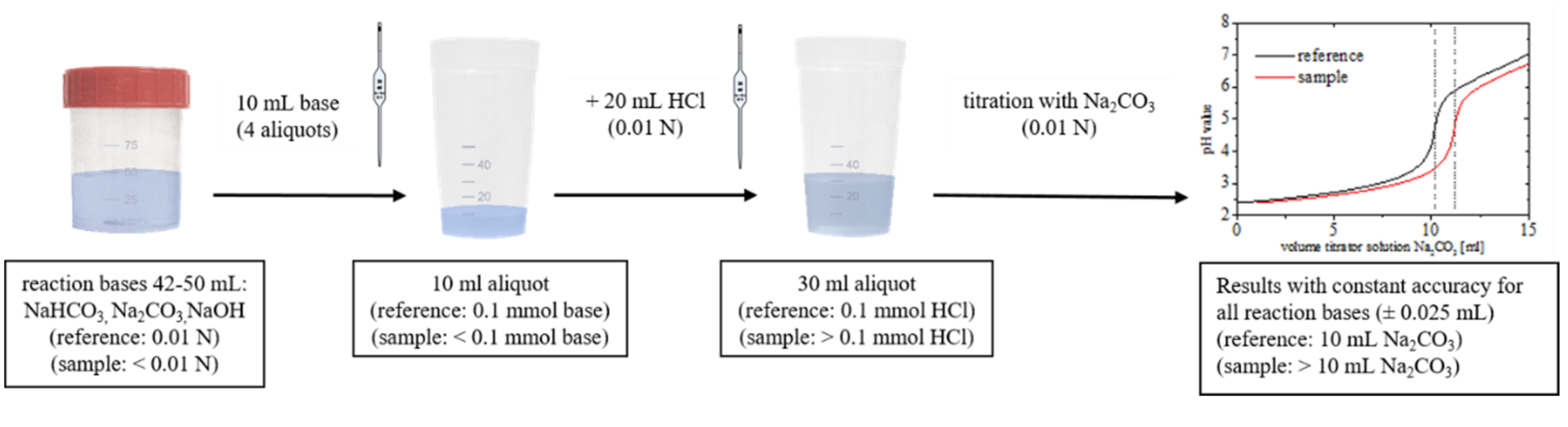
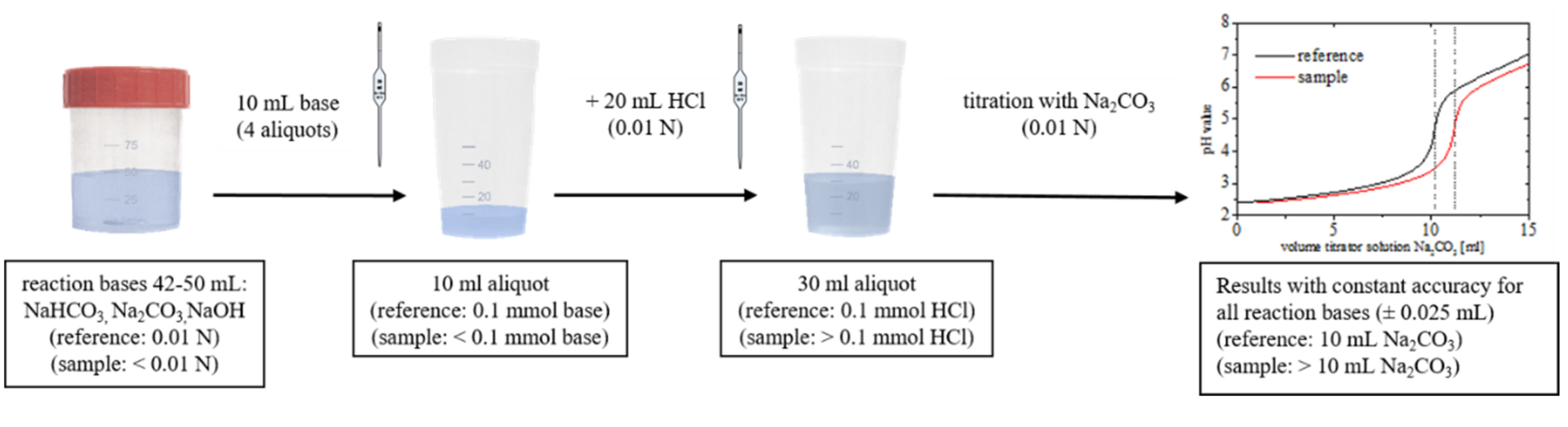
2.2. Titration Experiments
2.3. Experiments with CNTs
2.4. Quantification of Oxygen Groups
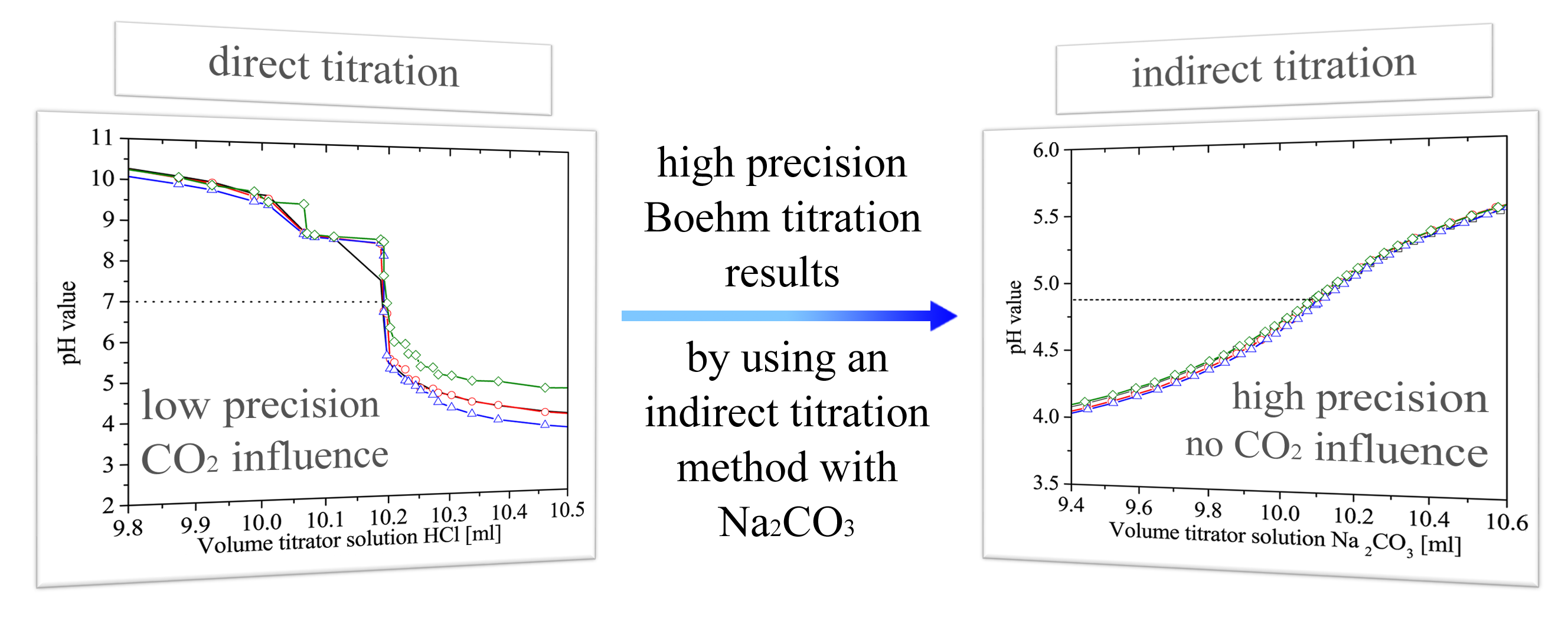
3. Results and Discussion
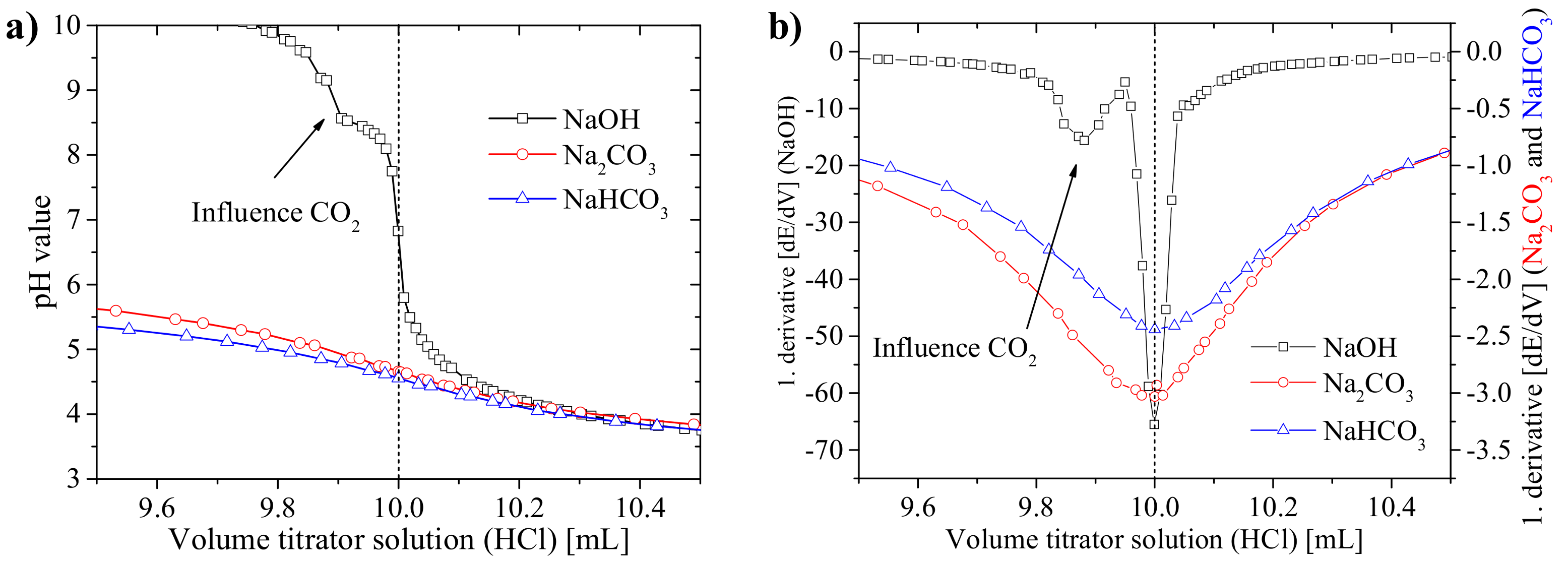
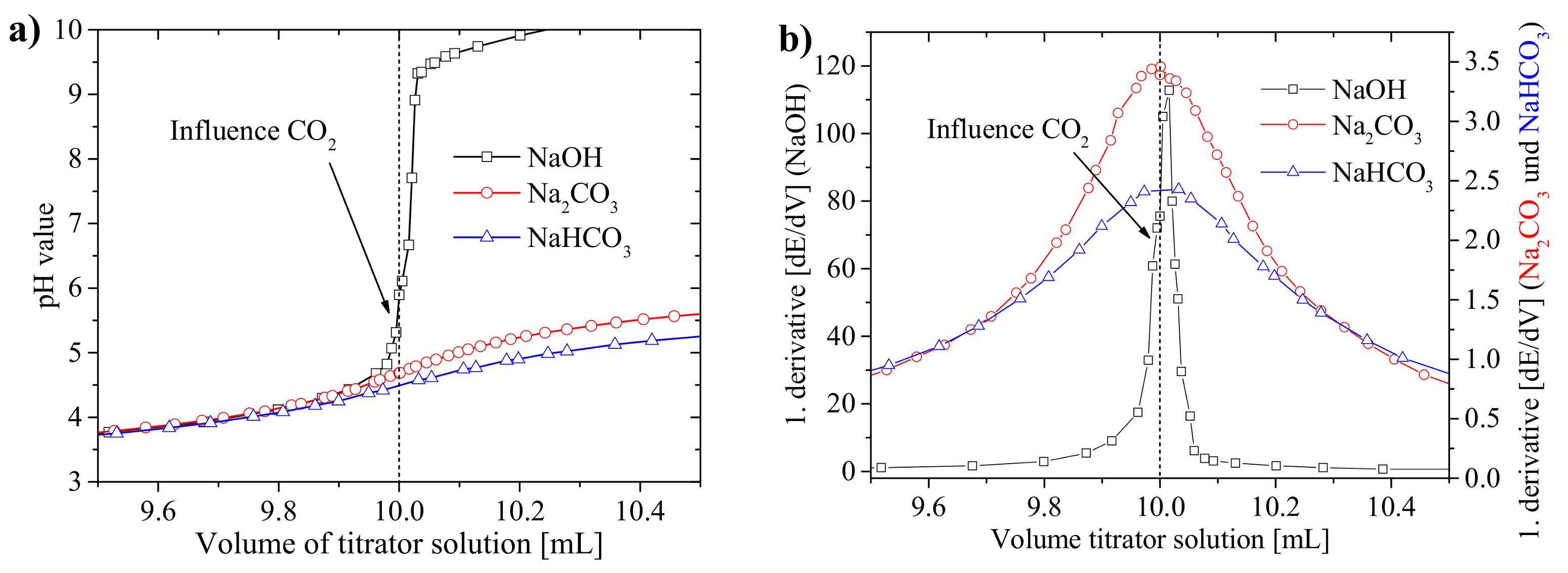
3.1. Practical Aspects of the Acid–Base Titration
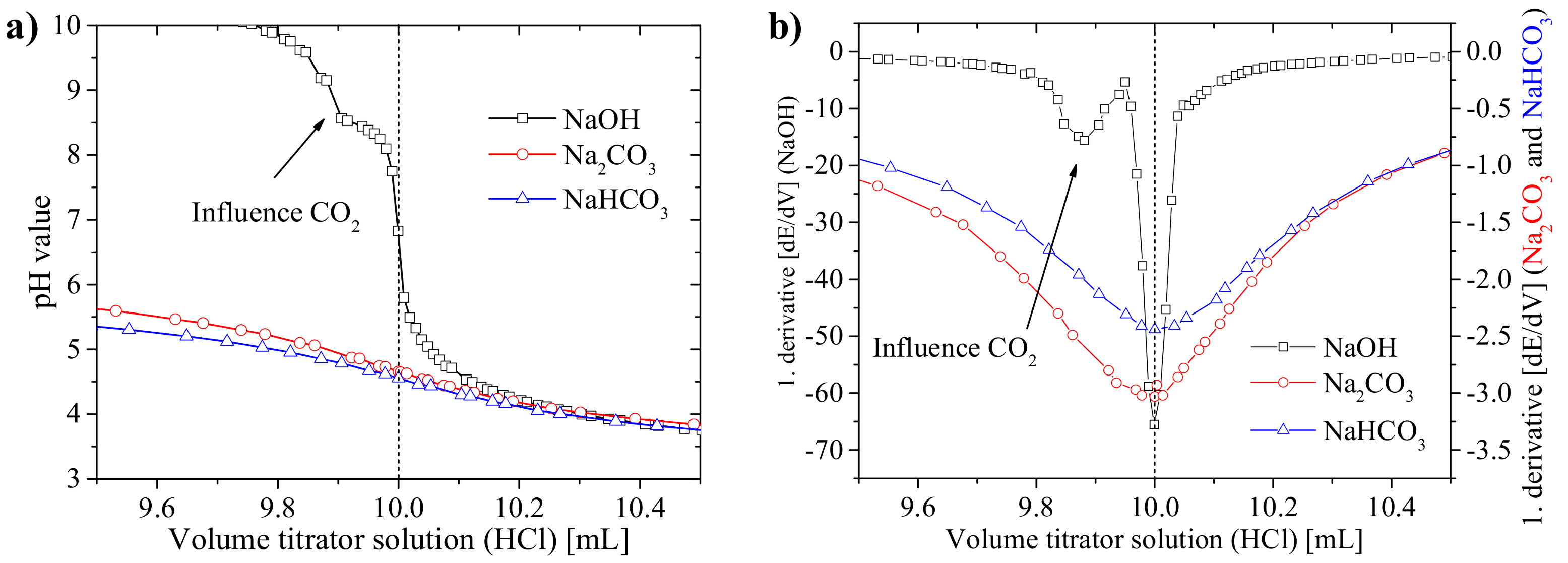
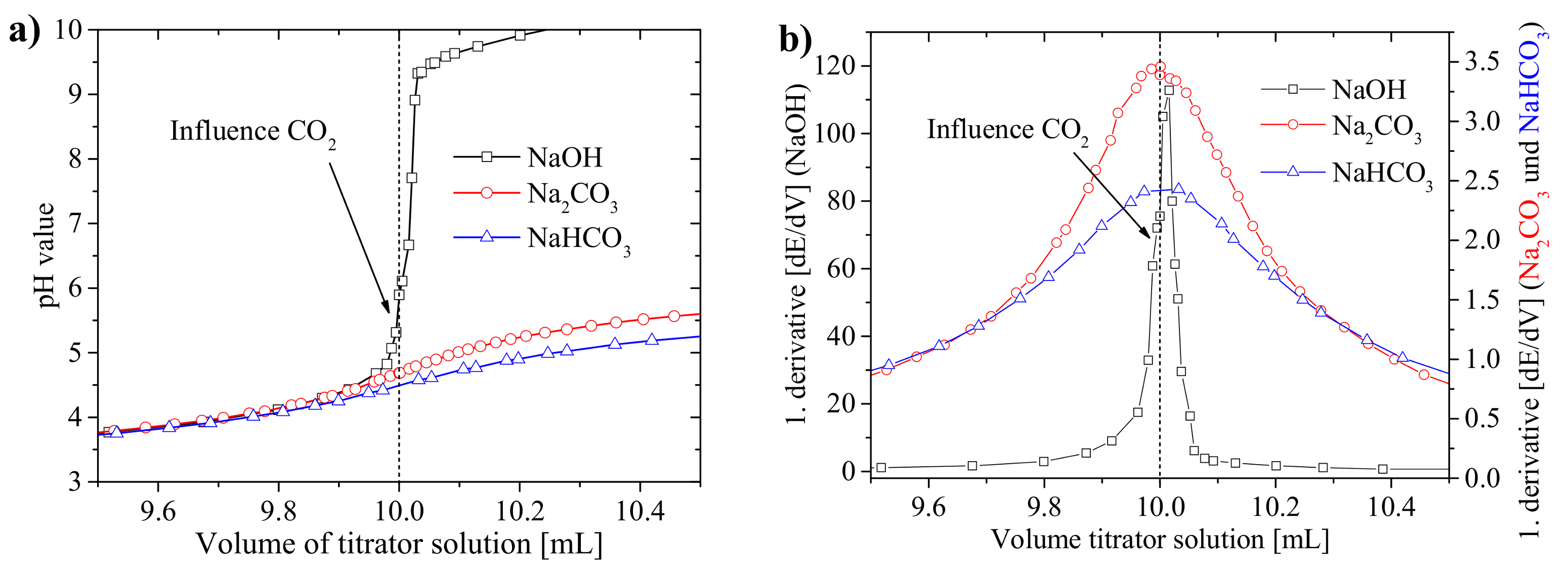
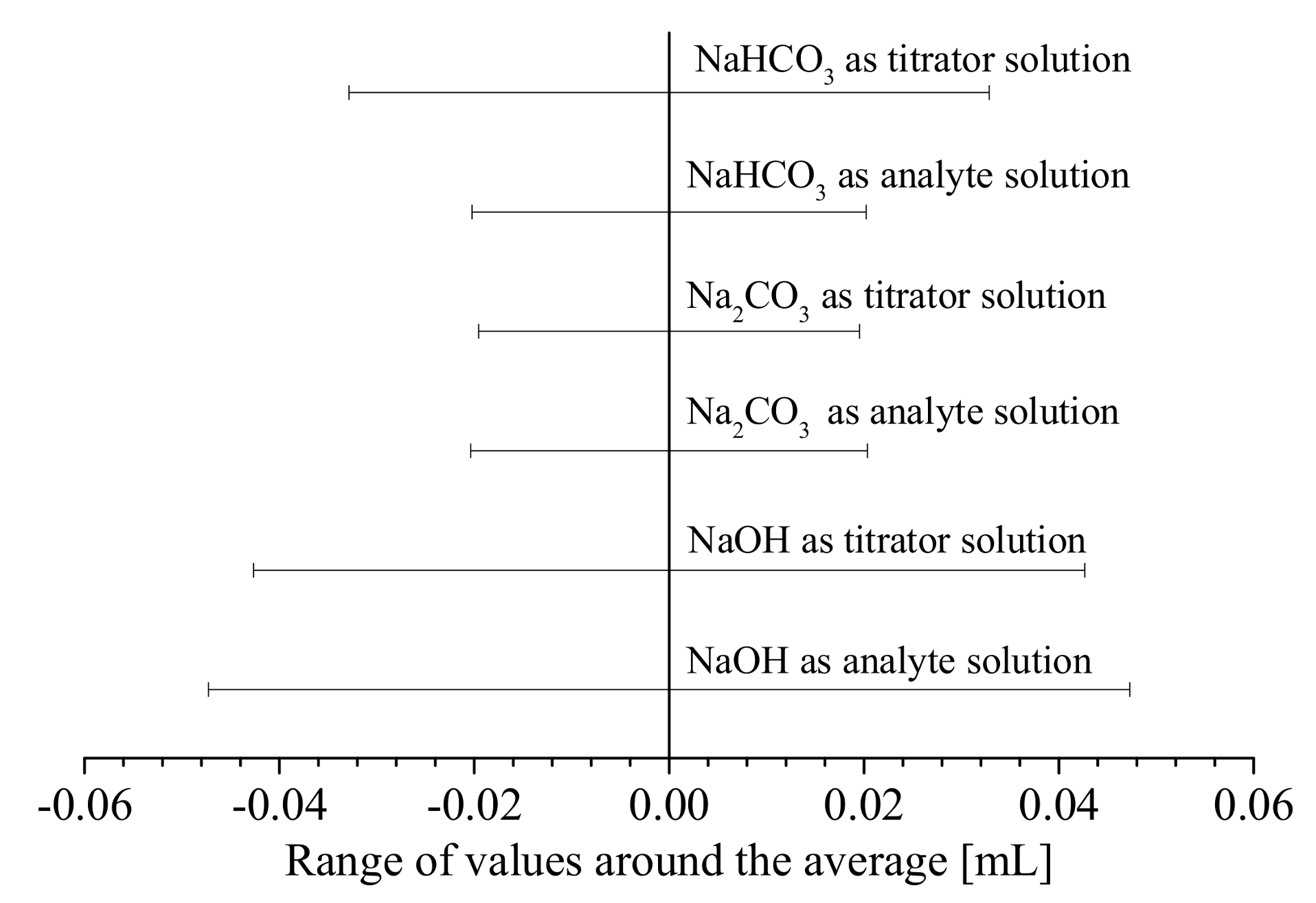
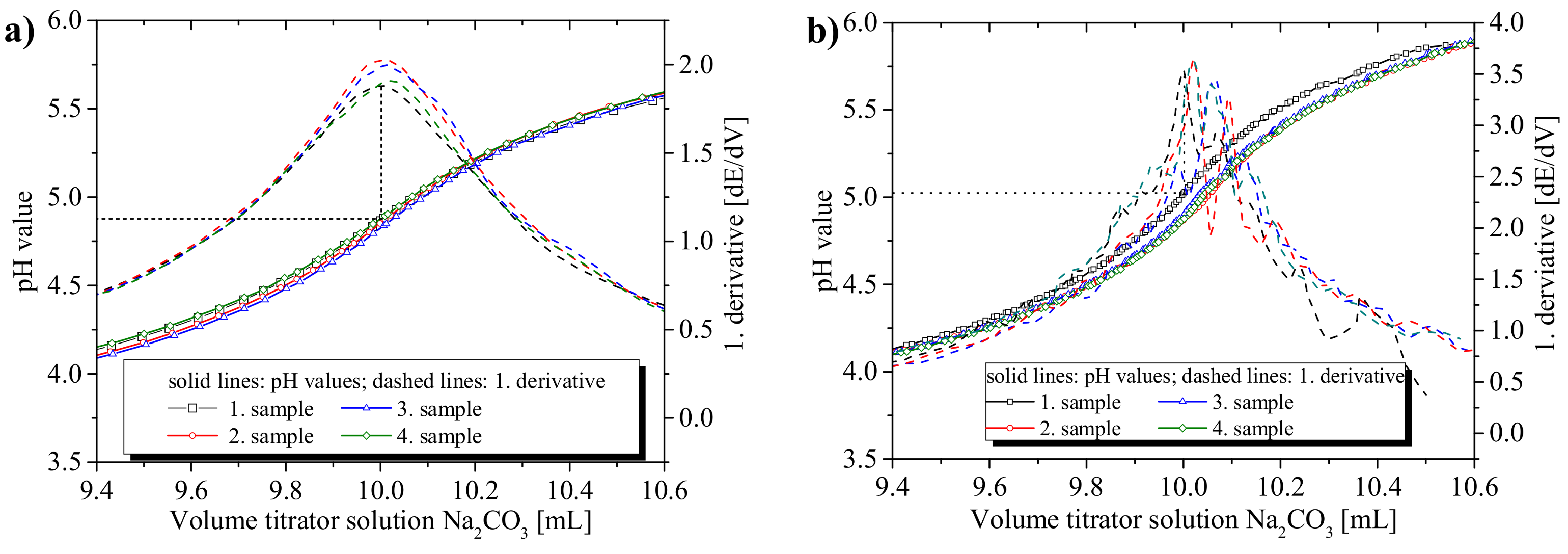
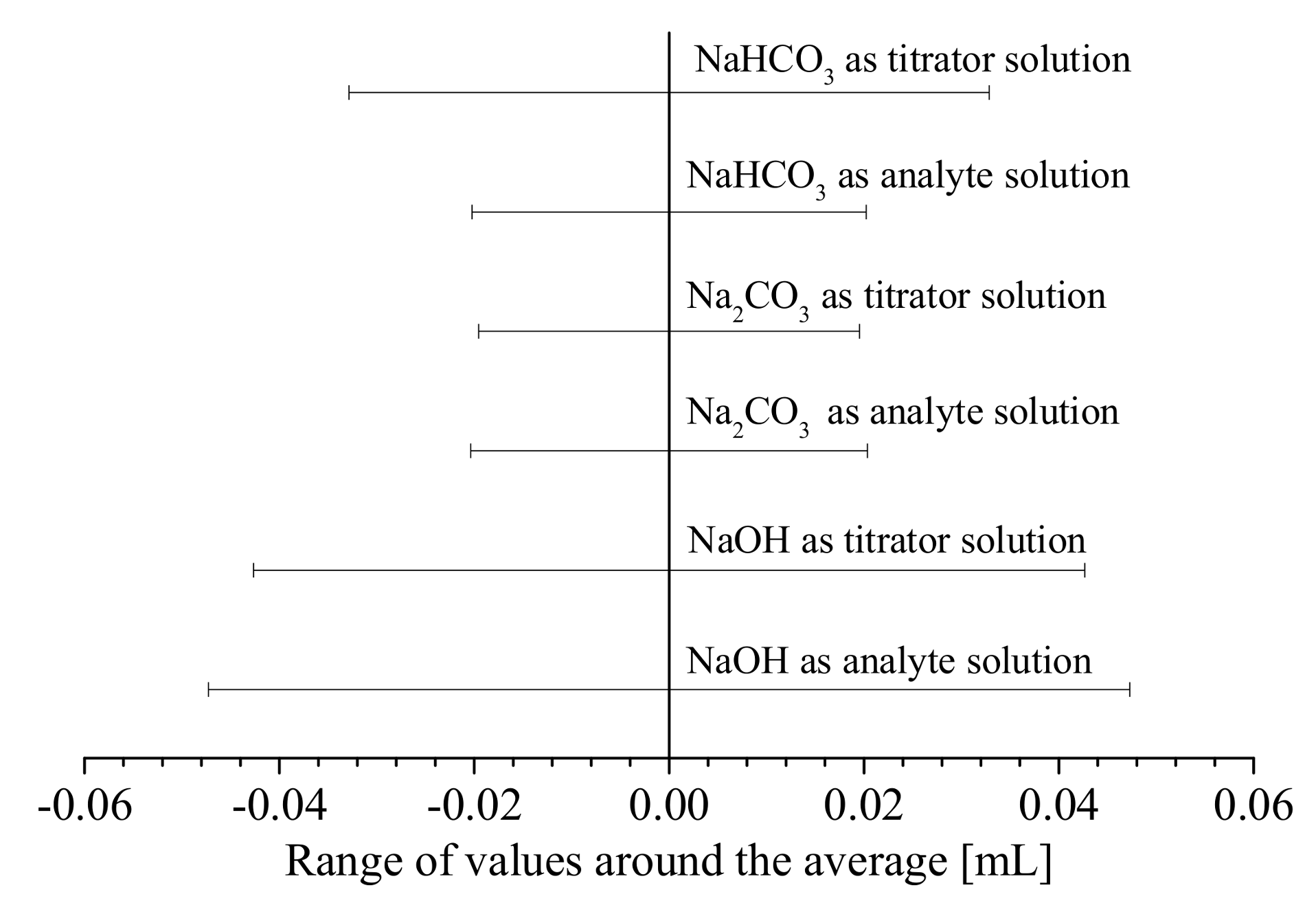
3.1.1. Precision of the Acid–Base Titration
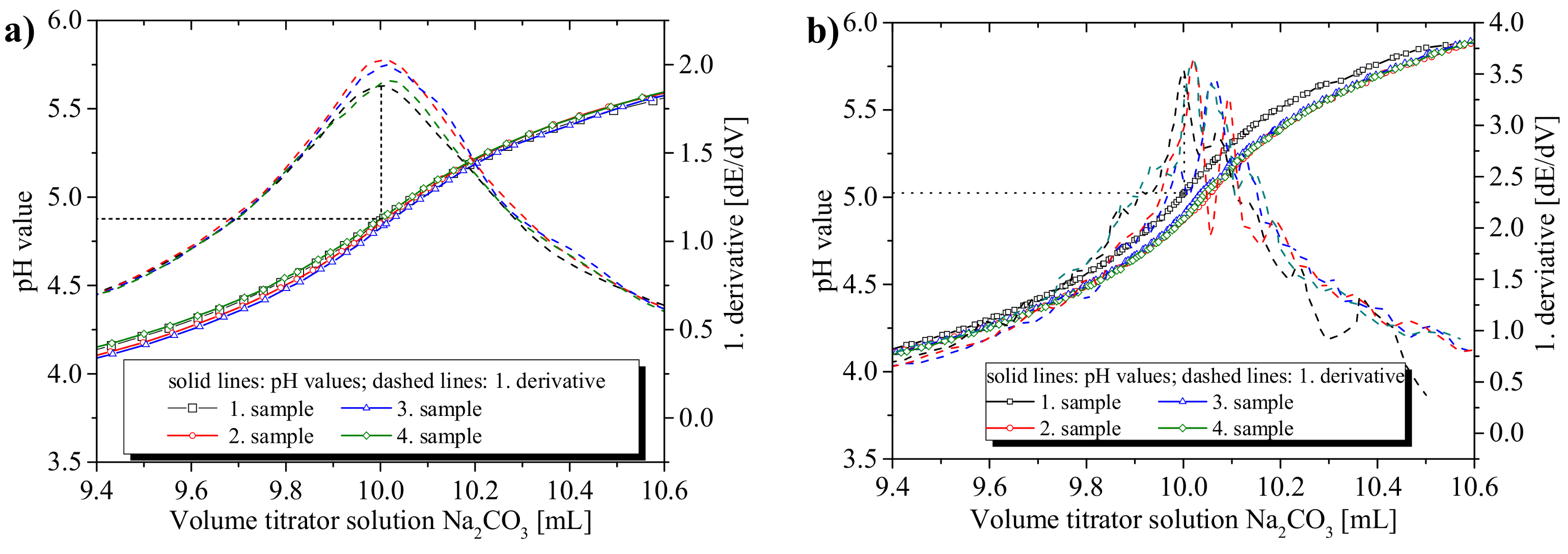
3.1.2. Influence of Titration Time on the Precision and the pH Values at the Equivalence Points
3.1.3. Recommendations for the Titration Method
3.2. Aspects of the Base Treatment and Following Removal of the Carbon Material
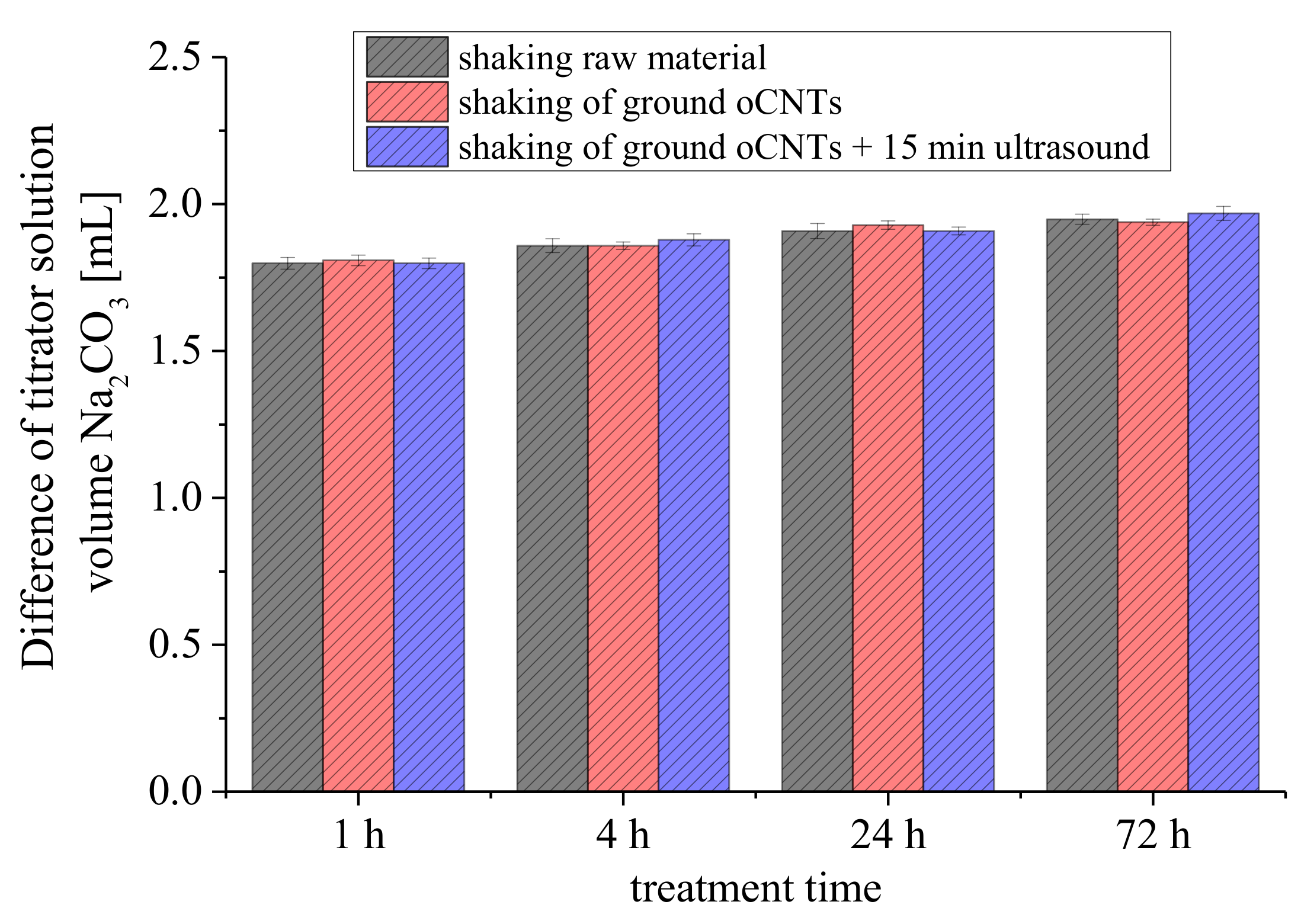
3.2.1. Removal of CNTs
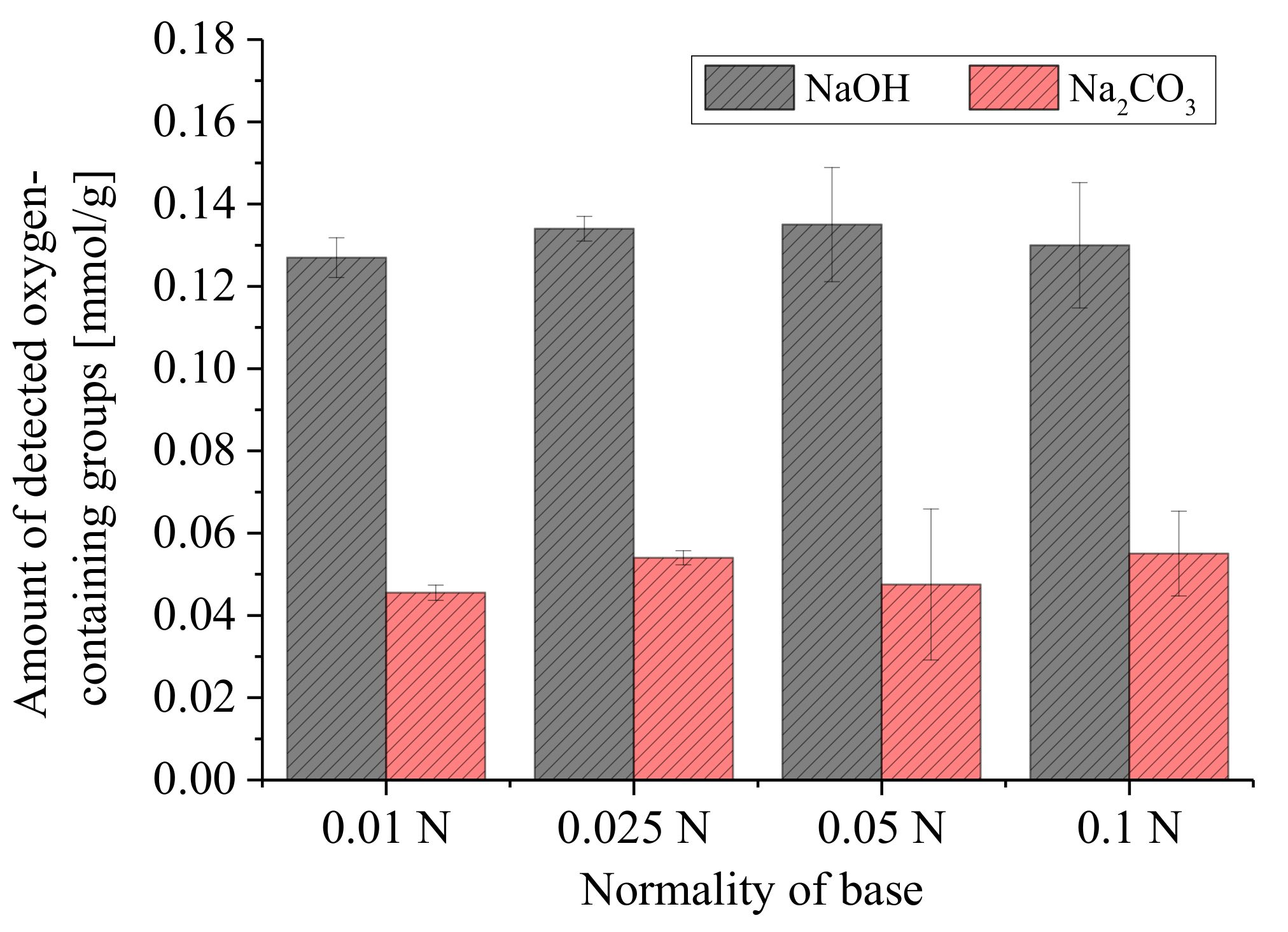
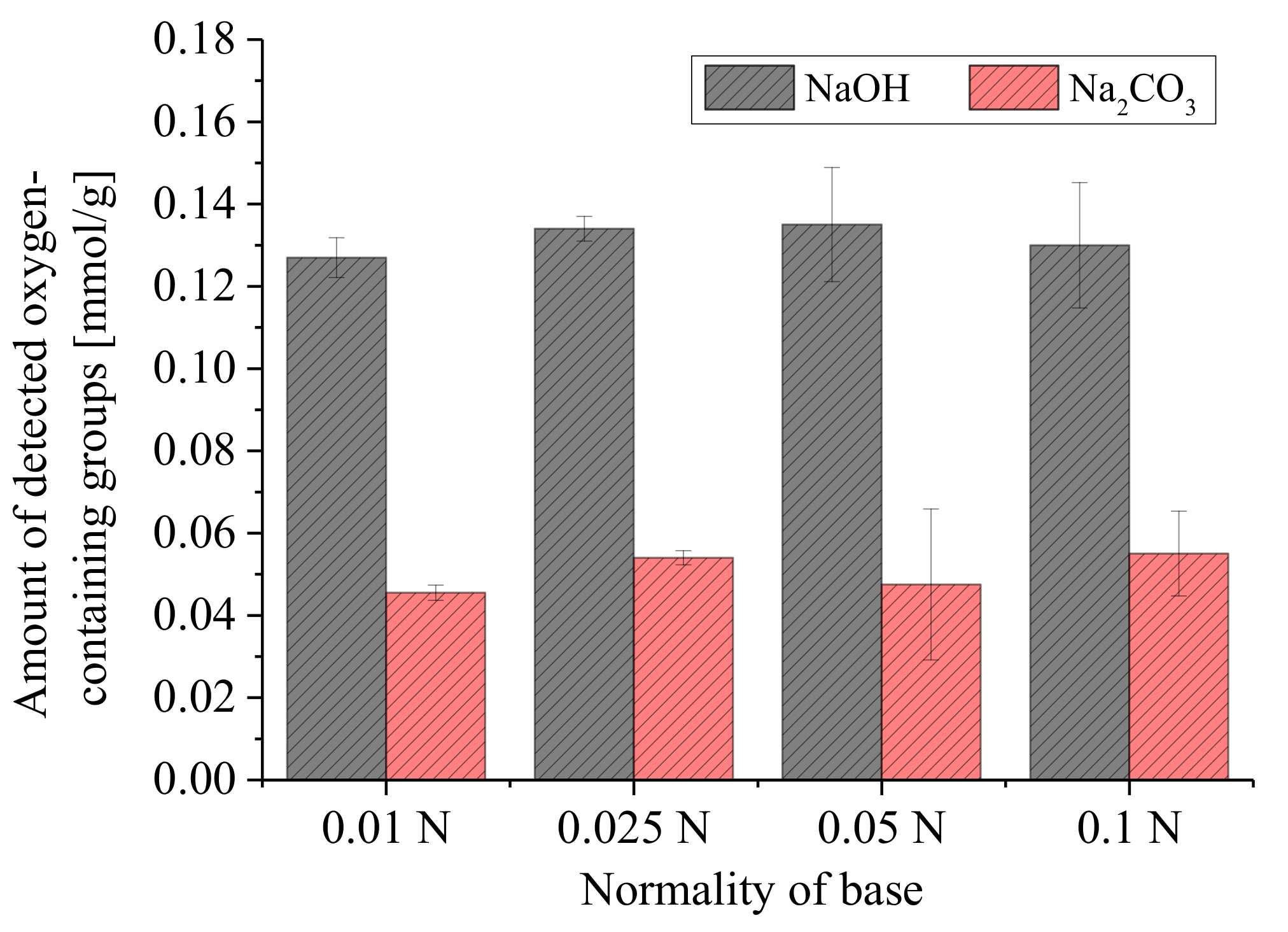
3.2.2. Concentration of Reaction Base and Amount of Carbon
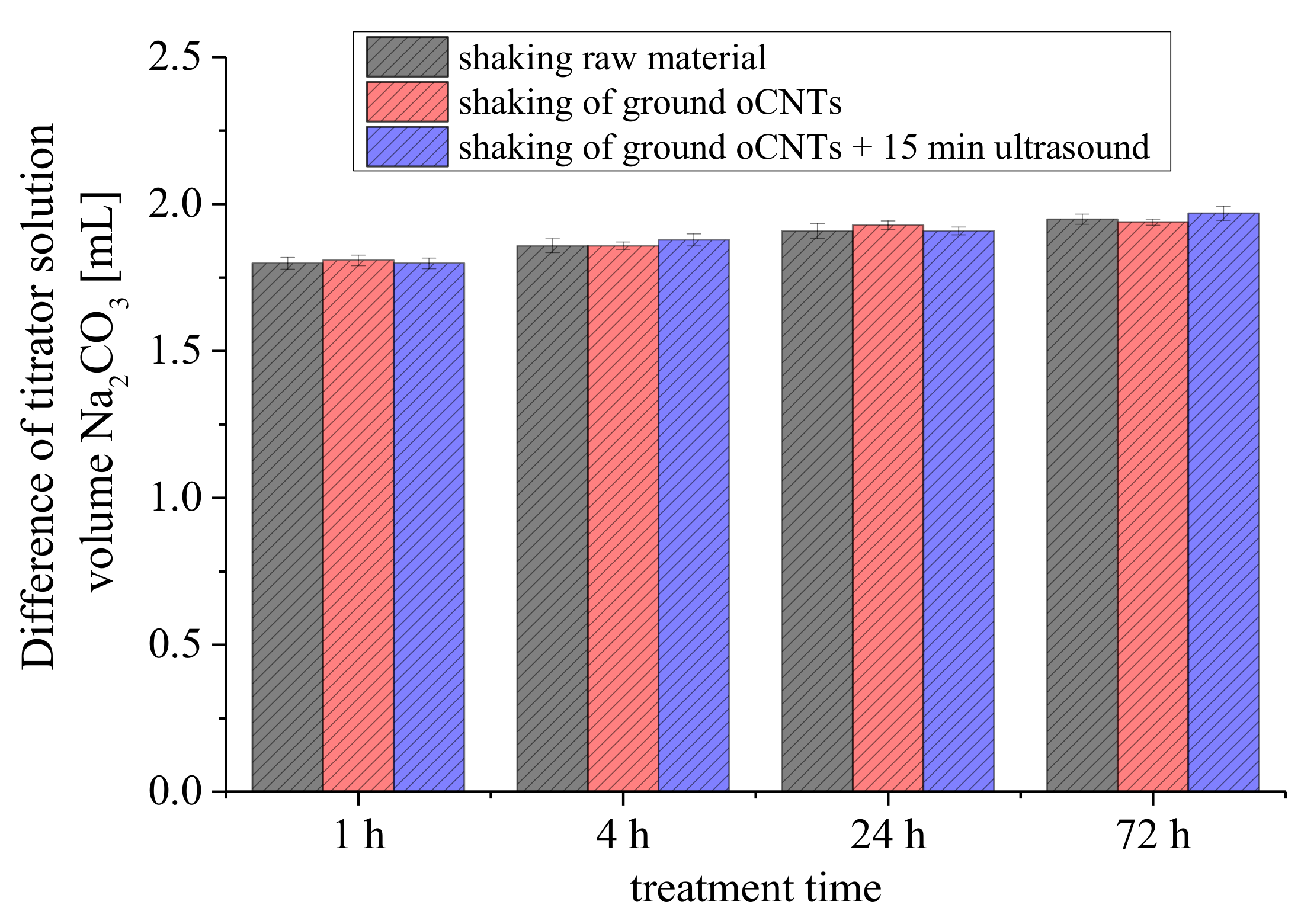
3.2.3. Treatment Time of the Reaction Base with Carbon
3.3. Error Analyses and Quantification of Surface Groups
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Boehm, H.P. Chemical identification of surface groups. Adv. Catal. 1966, 16, 179–274. [Google Scholar] [CrossRef]
- Boehm, H.P.; Diehl, E.; Heck, W.; Sappok, R. Surface oxides of carbon. Angew. Chem. Int. Ed. 1964, 3, 669–677. [Google Scholar] [CrossRef]
- Boehm, H.P. Some aspects of the surface chemistry of carbon blacks and other carbons. Carbon 1994, 32, 759–769. [Google Scholar] [CrossRef]
- Goertzen, S.L.; Thériault, K.D.; Oickle, A.M.; Tarasuk, A.C.; Andreas, H.A. Standardization of the boehm titration. Part I. CO2 expulsion and endpoint determination. Carbon 2010, 48, 1252–1261. [Google Scholar] [CrossRef]
- Oickle, A.M.; Goertzen, S.L.; Hopper, K.R.; Abdalla, Y.O.; Andreas, H.A. Standardization of the boehm titration: Part II. Method of agitation, effect of filtering and dilute titrant. Carbon 2010, 48, 3313–3322. [Google Scholar] [CrossRef]
- Validation of Analytical Procedures: Text and Methodology. Available online: http://www.ich.org/products/guidelines/quality/quality-single/article/validation-of-analytical-procedures-text-and-methodology.html (accessed on 22 February 2018).
- Schönherr, J.; Buchheim, J.; Scholz, P.; Adelhelm, P. Boehm titration revisited (part II): A comparison of boehm titration with other analytical techniques on the quantification of oxygen-containing surface groups for a variety of carbon materials. C 2018, 4. [Google Scholar] [CrossRef]
- Kim, Y.S.; Yang, S.J.; Lim, H.J.; Kim, T.; Park, C.R. A simple method for determining the neutralization point in boehm titration regardless of the CO2 effect. Carbon 2012, 50, 3315–3323. [Google Scholar] [CrossRef]
- Hollemann, A.F.; Wiberg, E.; Wiberg, N. Carbondioxide. In Hollemann-Wiberg, Lehrbuch der Anorganischen Chemie, 102th ed.; Wiberg, N., Ed.; WILEY-VCH Verlag GmbH: Berlin, Germany; New York, NY, USA, 2007; pp. 893–896. [Google Scholar]
- Plonska-Brzezinska, M.E.; Lapinski, A.; Wilczewska, A.Z.; Dubis, A.T.; Villalta-Cerdas, A.; Winkler, K.; Echegoyen, L. The synthesis and characterization of carbon nano-onions produced by solution ozonolysis. Carbon 2011, 49, 5079–5089. [Google Scholar] [CrossRef]
- Kim, Y.S.; Yang, S.J.; Lim, H.J.; Kim, T.; Lee, K.; Park, C.R. Effects of carbon dioxide and acidic carbon compounds on the analysis of boehm titration curves. Carbon 2012, 50, 1510–1516. [Google Scholar] [CrossRef]
- Kim, Y.S.; Park, C.R. One-pot titration methodology for the characterization of surface acidic groups on functionalized carbon nanotubes. Carbon 2016, 96, 729–741. [Google Scholar] [CrossRef]
- Hanelt, S.; Orts-Gil, G.; Friedrich, J.F.; Meyer-Plath, A. Differentiation and quantification of surface acidities on mwcnts by indirect potentiometric titration. Carbon 2011, 49, 2978–2988. [Google Scholar] [CrossRef]
- Feng, X.; Dementev, N.; Feng, W.; Vidic, R.; Borguet, E. Detection of low concentration oxygen containing functional groups on activated carbon fiber surfaces through fluorescent labeling. Carbon 2006, 44, 1203–1209. [Google Scholar] [CrossRef]
- González-Guerrero, A.B.; Mendoza, E.; Pellicer, E.; Alsina, F.; Fernández-Sánchez, C.; Lechuga, L.M. Discriminating the carboxylic groups from the total acidic sites in oxidized multi-wall carbon nanotubes by means of acid–base titration. Chem. Phys. Lett. 2008, 462, 256–259. [Google Scholar] [CrossRef]
- Scheibe, B.; Borowiak-Palen, E.; Kalenczuk, R.J. Oxidation and reduction of multiwalled carbon nanotubes—Preparation and characterization. Mater. Charact. 2010, 61, 185–191. [Google Scholar] [CrossRef]
- Kim, Y.-T.; Mitani, T. Competitive effect of carbon nanotubes oxidation on aqueous edlc performance: Balancing hydrophilicity and conductivity. J. Power Sources 2006, 158, 1517–1522. [Google Scholar] [CrossRef]
- Schmidlin, L.; Pichot, V.; Comet, M.; Josset, S.; Rabu, P.; Spitzer, D. Identification, quantification and modification of detonation nanodiamond functional groups. Diam. Relat. Mater. 2012, 22, 113–117. [Google Scholar] [CrossRef]
- Wang, Z.; Shirley, M.D.; Meikle, S.T.; Whitby, R.L.D.; Mikhalovsky, S.V. The surface acidity of acid oxidised multi-walled carbon nanotubes and the influence of in-situ generated fulvic acids on their stability in aqueous dispersions. Carbon 2009, 47, 73–79. [Google Scholar] [CrossRef]
- Lee, G.-J.; Pyun, S.-I.; Kim, C.-H. Kinetics of double-layer charging/discharging of the activated carbon fiber cloth electrode: Effects of pore length distribution and solution resistance. J. Solid State Electrochem. 2004, 8, 110–117. [Google Scholar] [CrossRef]
- Guedidi, H.; Reinert, L.; Lévêque, J.-M.; Soneda, Y.; Bellakhal, N.; Duclaux, L. The effects of the surface oxidation of activated carbon, the solution ph and the temperature on adsorption of ibuprofen. Carbon 2013, 54, 432–443. [Google Scholar] [CrossRef]
- Contescu, A.; Contescu, C.; Putyera, K.; Schwarz, J.A. Surface acidity of carbons characterized by their continuous pk distribution and boehm titration. Carbon 1997, 35, 83–94. [Google Scholar] [CrossRef]
- Bandosz, T.J.; Buczek, B.; Grzybek, T.; Jagiełło, J. The determination of surface changes in active carbons by potentiometric titration and water vapour adsorption. Fuel 1997, 76, 1409–1416. [Google Scholar] [CrossRef]
- Mestre, A.S.; Pires, J.; Nogueira, J.M.F.; Carvalho, A.P. Activated carbons for the adsorption of ibuprofen. Carbon 2007, 45, 1979–1988. [Google Scholar] [CrossRef]
- Strelko, V.; Malik, D.J. Characterization and metal sorptive properties of oxidized active carbon. J. Colloid Interface Sci. 2002, 250, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Tennant, M.F.; Mazyck, D.W. The role of surface acidity and pore size distribution in the adsorption of 2-methylisoborneol via powdered activated carbon. Carbon 2007, 45, 858–864. [Google Scholar] [CrossRef]
- Kato, Y.; Machida, M.; Tatsumoto, H. Inhibition of nitrobenzene adsorption by water cluster formation at acidic oxygen functional groups on activated carbon. J. Colloid Interface Sci. 2008, 322, 394–398. [Google Scholar] [CrossRef] [PubMed]
- Seredych, M.; Hulicova-Jurcakova, D.; Lu, G.Q.; Bandosz, T.J. Surface functional groups of carbons and the effects of their chemical character, density and accessibility to ions on electrochemical performance. Carbon 2008, 46, 1475–1488. [Google Scholar] [CrossRef]
- Schönherr, J.; Buchheim, J.; Scholz, P.; Stelter, M. Oxidation of carbon nanotubes with ozone and hydroxyl radicals. Carbon 2017, 111, 631–640. [Google Scholar] [CrossRef]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schönherr, J.; Buchheim, J.R.; Scholz, P.; Adelhelm, P. Boehm Titration Revisited (Part I): Practical Aspects for Achieving a High Precision in Quantifying Oxygen-Containing Surface Groups on Carbon Materials. C 2018, 4, 21. https://doi.org/10.3390/c4020021
Schönherr J, Buchheim JR, Scholz P, Adelhelm P. Boehm Titration Revisited (Part I): Practical Aspects for Achieving a High Precision in Quantifying Oxygen-Containing Surface Groups on Carbon Materials. C. 2018; 4(2):21. https://doi.org/10.3390/c4020021
Chicago/Turabian StyleSchönherr, Jan, Johannes R. Buchheim, Peter Scholz, and Philipp Adelhelm. 2018. "Boehm Titration Revisited (Part I): Practical Aspects for Achieving a High Precision in Quantifying Oxygen-Containing Surface Groups on Carbon Materials" C 4, no. 2: 21. https://doi.org/10.3390/c4020021
APA StyleSchönherr, J., Buchheim, J. R., Scholz, P., & Adelhelm, P. (2018). Boehm Titration Revisited (Part I): Practical Aspects for Achieving a High Precision in Quantifying Oxygen-Containing Surface Groups on Carbon Materials. C, 4(2), 21. https://doi.org/10.3390/c4020021